

Abstract
Iron fatty acid complexes (IFACs) are prepared via the dissolution of porous hematite powder in hot unsaturated fatty acid. The IFACs are then decomposed in five different organic solvents under reflux conditions in the presence of the respective fatty acid. The XRD analysis results indicate that the resulting NPs comprise a mixture of wustite, magnetite, and maghemite phases. The solvents with a higher boiling point prompt the formation of larger NPs containing wustite as the major component, while those with a lower boiling point produce smaller NPs with maghemite as the major component. In addition, it is shown that unstable NPs with a mixed wustite–magnetite composition can be oxidized to pure maghemite by extending the reaction time or using an oxidizing agent.
Keywords: Iron fatty acid complexes, Iron oxide, Magnetic nanoparticles, Monodisperse
Introduction
Many studies have shown that long-chain carboxylates, or soaps, of polyvalent metal ions are effective precursors for the large-scale synthesis of metal oxide or metal nanoparticles (NP) via a thermal decomposition process utilizing organic solvents [1-8]. Of these various soaps, those containing iron have attracted particular attention due to the magnetic properties bestowed in the products. For example, iron (ferrous or ferric) fatty acid complexes (IFACs) have been used to synthesize monodisperse iron oxides and iron NPs at different decomposition temperatures using various types of solvent [9-15] and without the presence of any form of solvent [16-18], respectively. The NPs produced using these techniques have a uniform size and are widely applied in a broad range of fields such as magnetic storage media, drug carriers, magnetic resonance imaging (MRI) agents, and hyperthermia [19,20]. Moreover, the size of the NPs can easily be tuned by adjusting the reaction parameters, e.g. the reaction temperature (as determined by the solvent b.p.) or the concentration of the free fatty acid within the reactant. Polyvalent metal soaps are generally prepared via one of three different routes, namely, (1) metathesis of the corresponding soap of Na or K with metal salts (such as chloride or acetate) in aqueous or nonaqueous polar solvents; (2) fusion (or dissolution) of metal oxides, hydroxides, oxy-hydroxides, hydroxycarbonates, or carbonates in hot fatty acids; and (3) direct reaction of metals with hot fatty acids [21]. The metal soap precursors used for the synthesis of iron oxide or iron NPs are generally prepared using the route 1 method [1-18]. However, it has also been shown that the route 2 method is well suited to the preparation of soaps containing elements such as alkaline, alkaline earth metal, Zn, Cd, and so on [22-26]. Jana et al. [27] demonstrated the use of iron oxides as precursors in the synthesis of metal oxide NPs. However, they did not provide any experimental details due to the poor reproducibility of the resulting precipitates. Yu et al. [28] proposed a one-pot process for the preparation of monodisperse magnetite NPs, in which an amorphous iron oxyhydroxide, known as 2-line ferrihydrite, is fully dissolved in a mixture of oleic acid and 1-octadecene solvent at a temperature of 320 °C. In a recent study, the current group demonstrated the feasibility of dissolving hematite in hot long-chain fatty acids such as oleic acid or linoleic acid to produce IFACs for the synthesis of monodisperse magnetic iron oxide NPs via a controlled dissolution–recrystallization process [29]. However, the preparation of IFACs using the route 2 method requires an elevated reaction temperature, which causes the IFACs to decompose virtually as soon as they are formed. Therefore, in synthesizing IFACs using this route, the challenge lies in finding a temperature that is sufficiently high to ensure the complete dissolution of the hematite powder in the hot fatty acid while simultaneously avoiding the decomposition of the resulting IFACs. Assuming that such a temperature can be found, the possibility then exists to synthesize IFACs in a cheap and efficient manner using readily available hematite powder. Furthermore, having prepared the IFACs, iron oxide NPs of a specified size can easily be synthesized via a decomposition process performed using a solvent with an appropriate boiling point.
It is well known that the rate of dissolution of a solid in a liquid is proportional to its surface area. It is also known that the topotactic goethite-to-hematite transformation generates porous hematite [30]. The voids within this porous hematite provide an effective increase in the surface area, and thus hematite produced in this way is expected to have good dissolution properties. This study demonstrates the feasibility of preparing sodium-free IFACs via the dissolution of porous hematite in hot oleic or linoleic acid at a temperature just below the decomposition temperature of the corresponding acid. It is important in view of that the presence trace amount of sodium salts will affect the morphology of iron oxide nanoparticles obtained [9]. Moreover, monodisperse magnetic iron oxide NPs of various dimensions are then synthesized by decomposing the IFACs in a variety of high b.p organic solvents.
Experimental Details
Materials
The synthesis and decomposition experiments were performed using the following chemicals: goethite (α-FeO(OH); 99.9%, Strem), oleic acid (OA, 90%, Showa), oleyl alcohol (85%, Alfa Aesar), benzyl ether (99%, Acros), 1-octadecene (ODE, 90%, Acros), tri-n-octylamine (90%, Kanto), 1-eicosane (99%, TCI), and trimethylamine N-oxide dehydrated (98%, Alfa Aesar). Note that all of the chemicals were used in an as-received condition without further purification.
Synthesis of IFACs from Porous Hematite Powder
The porous hematite was prepared via the thermal decomposition of commercially available goethite powder at a temperature of 300 °C for 2 h. In the IFAC synthesis process, porous hematite was mixed with oleic or linoleic acid in a molar ratio of 1:9 and was then loaded into a three-necked round-bottom flask and heated to a temperature of 290 °C at a rate of ~15 °C/min under a flow of Argon. During the reaction process, the reactant bubbled vigorously and spilled out of the flask as a result of the generation of water. Moreover, the original red color of the hematite gradually changed to brown as the reaction process proceeded. The change in color was consistent with the disappearance of the hematite diffraction peaks observed in the sampled aliquot. After 4 h, XRD and IR analyses showed that the hematite was completely dissolved within the fatty acid. The resulting brown grease-like product was allowed to cool to room temperature and was then diluted with a mixture of hexane and acetone (1:20) and centrifuged at a speed of 5,000 rpm. Finally, the precipitate was washed with acetone and dried at a temperature of 60 °C. The products were then characterized by XRD, FTIR, EA, DSC, TGA, XPS, and SQUID, respectively.
Thermal Decomposition of IFACs in Various Solvents
The oleic or linoleic IFACs were used as precursors in the synthesis of iron oxide NPs utilizing five different solvents with different boiling points (see note c beneath Table 1). Note that for convenience, the different reaction processes are denoted in Table 1as run 1 ~ run 6, respectively, where each run varies in terms of either the precursor, the surfactant, the surfactant concentration, the organic solvent or the reaction time and temperature, respectively. Taking the run 1 reaction process for illustration purposes, a mixture of IFAC (iron–oleate, 6 mmol), oleic acid (9 mmol), and oleyl alcohol solvent (18 mL) was loaded into a three-necked round-bottom flask and was then heated to the b.p. of the solvent (300 °C) at a rate of 15 °C/min under a flow of argon and maintained at this temperature for 60 min. The reaction was performed under reflux conditions with the temperature controlled via a heating mantel and thermocouple positioned above the magnetic stirrer. During the reaction process, the color of the reactant changed from brown to black as a result of the decomposition of IFACs and the formation of NPs. The resulting product was diluted with hexane and acetone and was then centrifuged at a speed of 5,000 rpm. Finally, the precipitate was washed twice by a mixed solvent of hexane/acetone (v/v in 1:4, total volume ~90 mL) to produce a black powder. The phase of the precipitate was then checked using XRD. TEM samples were prepared by dropping a hexane suspension containing the precipitated NPs onto a copper grid (200 mesh) coated with a carbon film. The size of the NPs was determined by averaging the lengths of the major and minor axes of a minimum of 500 different particles using Sigmascan Pro 5 software.
Table 1.
Reaction parameters used in synthesis of monodisperse iron oxide NPs
| Run | Precursora(mmol) | Surfactantb(mmol) | Solventc(mL) | Temperature (°C) | Time (min) | NP diameter (nm) |
|---|---|---|---|---|---|---|
| 1 | 6 | 9 | 18 | 300 | 60 | 4.5 ± 0.4 |
| 2 | 6 | 9 | 18 | 290 | 120 | 8.1 ± 0.5 |
| 3 | 6 | 9 | 18 | 320 | 120 | 10.1 ± 1.0 |
| 4 | 6 | 18 | 18 | 320 | 150 | 13.1 ± 0.8 |
| 5 | 6 | 9 | 18 | 360 | 60 | 15.3 ± 0.9 |
| 6 | 6 | 18 | 18 | 340 | 120 | 20.4 ± 1.1 |
aPrecursors used: runs 1–5, iron–oleate complex and run 6, iron-linoelate complex
bSurfactant used: runs 1–5, oleic acids and run 6, linoleic acids
cSolvent used: run 1, Oleylacohol; run 2, Benzyl ether; runs 3–4, ODE; run 5, Tri-octylamine; and run 6, 1-eicosane
Oxidation of NPs from FeO/Fe3O4to γ-Fe2O3
The NP products containing a mixture of wustite (FeO) and inverse spinel (Fe3O4or γ-Fe2O3) were transformed to pure γ-Fe2O3via treatment with an organic oxidizing agent. In the oxidation process, trimethylamine N-oxide dehydrate was added to the reactant in a flask, and the mixture was then heated at a temperature of 140 °C for 2 h under a flow of argon.
Instrumentation
The phases of the various products were characterized using an X-ray powder diffractometer (Shimadzu XRD-6000) with Cu Kα radiation. The TEM samples were observed using a transmission electron microscope (JEOL JEM 1200EX) with an accelerating voltage of 80 kV and a high-resolution transmission electron microscope (Philips Tecnai G2 F20 or JEOL JEM 2010) with an accelerating voltage of 200 kV. SEM micrographs of the hematite powder were obtained using an FE-SEM (JEOL JSM 7000). The surface area of the hematite powder was determined in accordance with the BET method at a temperature of 77 K using a Micromeritics ASAP 2010 (Accelerated Surface Area and Porosimetry) system. The IR absorbance was analyzed using a Fourier transform infrared spectrometer (FTIR, Nicolet 5700) with a resolution of 4 cm−1. Finally, the amount of iron oxide in the dried precipitates was measured using a thermogravimetric analyzer (TGA) under a constant flow of nitrogen gas.
Results and Discussion
Dissolution of Hematite in Oleic Acid
In this study, the iron soap precursors used to synthesize the NPs were deliberately prepared using oleic or linoleic acid since these acids are known to provide a good protecting surfactant for oxide-based NPs [29] and are in a liquid form at room temperature, and are, therefore, easy to handle in the purification process. Furthermore, the use of iron oxide powder as a precursor is beneficial because such powders are generally stable in air and are extremely cost-effective compared to their organometallic compounds such as Fe(CO)5. The iron oxide IFACs were prepared using two different forms of hematite powder in order to evaluate their relative rates of dissolution in hot fatty acids, namely, commercially available sub-micrometer hematite with a BET surface area of 13.4 m2/g and porous hematite powder produced via the thermal decomposition of goethite. In both cases, the IFACs were synthesized at a temperature of 290 °C, i.e. just below the IFAC decomposition temperature (300 °C). It was found that the sub-micrometer hematite powder had a relatively slow dissolution rate and required approximately 9 h to become completely dissolved in the hot oleic acid. Figure 1 presents a TEM image of the hematite powder produced via the thermal decomposition of goethite. As shown, the hematite has a highly porous structure as a result of the topotactic transformation of goethite (α-FeOOH) to hematite (α-Fe2O3), which induces the formation of slit-shaped micropores via the removal of water droplets generated during the reaction process. Thus, compared to the commercial sub-micrometer hematite powder, the porous hematite has a far higher surface area, i.e. 108.1 cm2/g. As a result, in the IFAC synthesis process, the porous hematite was fully dissolved in the hot oleic acid after just 4 h.
Figure 1.

TEM micrograph of porous hematite produced via topotactic thermal decomposition of goethite. Note the slit-shaped micropores and unchanged acicular shape (inset)
Characterization of Iron–Oleate Complexes
Figure 2 presents the FTIR spectra of oleic acid and the corresponding IFAC, respectively. Comparing the two spectra, it can be seen that the intensity of the C–O stretching at 1,709 cm−1 is significantly reduced in the IFAC, and a new peak centered at 1,551 cm−1 is formed. This result is consistent with the formation of iron carboxylate bonds in the IFAC. The IFAC spectrum also shows evidence of vinyl C–H stretching at 3,010 cm−1, indicating that the oleate ligand does not transform to stearate during the synthesis process. Finally, the broad band around 600 cm−1 is attributed to a stretching of the Fe–O bonds. It is known that iron carboxylates tend to form μ3-oxo-triiron clusters, which show one Fe–O stretching band in the Fe3IIIO case (D3h) and two Fe–O bands in the mixed-valence Fe2IIIFeIIO case (C2v) [31]. Thus, the results presented in Fig. 2 suggest that the iron of the IFAC is not reduced in the current case. The XRD pattern of the iron–oleate complex presented in Fig. 3 shows that the IFAC has a nearly amorphous structure with a broad basal spacing of 25.01 Å. Meanwhile, the elemental analysis results show that the IFAC has a composition of C, 70.52%; H, 10.61%; and Fe, 7.35, which is close to that of [Fe3IIIO(oleate)6(oleate) (Cald.: C, 70.27%; H, 10.81%; Fe, 7.79%). It is well known that the bonding modes of carboxylate ligands are very flexible and may, therefore, produce metal soaps with many different formulae, e.g. a basic form (M(OH)x(RCOO2)y) or acidic form (M(RCOO2)x(RCOOH)y), or with different degrees of hydration (or solvation). Also, part of the iron in the IFAC may be reduced to valence II due to a reaction with the organic solvent. In this study, repeated experiments showed that the composition of the iron–oleate complex was highly sensitive to the processing parameters, e.g. the solvents used to wash the precipitates, the drying temperature, and so forth. However, the variable compositions of the synthesized IFACs were found to have no obvious effect on the composition and quality of the NPs formed in the decomposition reactions. Thus, it is surmised that the presence of free oleic acid in the preparation of the NPs restores the iron–oleate complex to its stable state.
Figure 2.
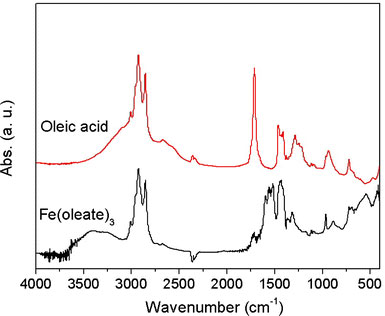
FTIR spectrum of iron–oleate complex produced via dissolution of porous hematite in hot oleic acid. Note that the FTIR spectrum of oleic acid is also presented for comparison purposes
Figure 3.
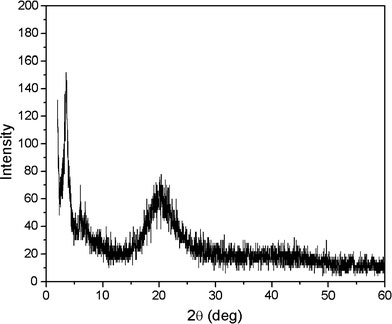
XRD pattern of iron–oleate complex showing 2.51 nm basal spacing and poor crystallinity
Synthesis of Monodisperse Iron Oxide NPs with Tunable Dimensions
The as-formed iron–oleate complexes were decomposed to form monodisperse iron oxide NPs in a mixture of the corresponding fatty acids (i.e. oleic acid or linoleic acid) and various high b.p. organic solvents under reflux. As shown in Table 1, a total of six different NP samples were synthesized by varying the reaction parameters. The TEM images presented in Fig. 4show that the NPs have a uniform size in each of the different samples. Furthermore, the data presented in Table 1show that the size of the NP precipitates is dependent on both the choice of solvent (i.e. the boiling point of the solvent) and the ratio of the IFAC to the free oleic acid. Specifically, the size of the NPs increases with an increasing boiling point and a higher concentration of free acid (see TEM images of run 3 and 4 samples, respectively). However, as the concentration of the free acid is increased, nucleation becomes more difficult, and thus a longer reaction time is required (e.g. 120 min for the run 3 sample, but 150 min for the run 4 sample). In a separate series of experiments (results not shown in Table 1), it was found that the decomposition process could be completed within just 30 min when performed without the addition of free oleic acid. However, the resulting NPs were of a variable size rather than a uniform size. This is ascribed to the hindrance of the Ostwald-ripening process due to the lack of free acid available to dissolve the smaller particles.
Figure 4.

TEM micrographs of NPs obtained from decomposition of IFAC in different high-boiling-point solvents. Note that micrographs 1–6 correspond to runs 1–6 in Table 1. Note also the uniform size of the NPs in each sample. (Inset: typical electron diffraction pattern and high-resolution TEM showing consistent lattice fringe.)
Phase Composition of Monodisperse Iron Oxide NPs
Although the NPs obtained in the six samples described in Table 1 were all spherical and uniform in size, their compositions were notably different. As shown in the XRD profiles presented in Fig. 5, the larger NPs contain wustite (Fe1-xO) as the major component and inverse spinel (Fe3O4 or γ-Fe2O3) as the minor component, while the smaller NPs are comprised virtually entirely of γ-Fe2O3. Wustite is a nonstoichiometric and metastable phase under ambient conditions [32]. Figure 6a–c shows the compositional variation of the run 6 sample (dimension: 20.4 nm) over the course of a prolonged reaction time, i.e. 6 h. It is observed that as oxidation reaction proceeds, the wustite content decreases while the magnetite content increases. In addition, we also carry out the oxidation reaction in a more efficient way, under air at 200 °C after the formation of NPs. It is found that the product is pure maghemite, Fig. 6d. We also found that the long shelf time also make the NPs of run 6 oxidized. Following exposure to ambient conditions for 40 days, the NP sample of run 6 is still a stable colloid, but its composition is fully changed to maghemite, Fig. 6(e). Magnetite and maghemite share the same inverse spinel oxygen lattice and virtually the same cell parameters. Thus, it is difficult to distinguish between them using diffraction methods. However, the Fe2+–Fe3+ intervalence charge transfer associated with magnetite can be detected from a hump in the absorption spectrum in the near IR range [33], while maghemite shows no absorption in this particular range. Furthermore, NPs with high wustite content exhibit an absorption edge in the visible range [34]. Figure 7 shows the absorption spectra of an as-prepared run 6 NP sample and an oxidized run 6 NP sample following exposure to ambient conditions for 40 days. The spectrum corresponding to the as-prepared sample has a broad hump in the IR range attributed to the intervalence charge transfer of magnetite, and the absorption in the range 600–800 nm attributed to the absorption edge that is characteristic of NPs with high wustite content. By contrast, the absorption spectrum of the oxidized sample shows virtually no adsorption at wavelengths greater than 600 nm, indicating the complete oxidation of the Fe3O4 and wustite contents, respectively. The FeO/Fe3O4 composition of the large NPs (e.g. run 6, dimension: 20.4 nm) is also confirmed by the hysteresis loops presented in Fig. 8 recorded at a temperature of 100 K in a magnetic field up to 1 T. Here, the shift in the two hysteresis loops recorded after zero field cooling (ZFC) and field cooling (FC), respectively, can be attributed to a coupling effect at the ferromagnetic/anti-ferromagnetic interface. For the absorption spectra of the other five samples, run 1 and 2 show virtually no adsorption at wavelengths greater than 600 nm, while runs 3–5 show a broad hump attributed to magnetite. A representative spectra of run 2 (8.1 nm, benzyl ether) and run 4 (13.1 nm, ODE) are shown in Fig. 9.
Figure 5.
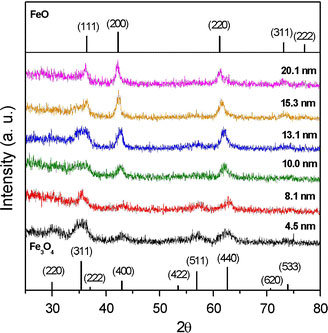
XRD patterns of as-formed NPs produced via thermal decomposition of IFAC in different solvents
Figure 6.
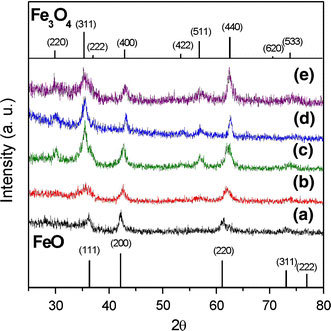
XRD patterns of NPs produced in run 6 sample (20.4 nm) at different reaction times:a2 h,b4 h, andc6 h showing different degrees of oxidation;dcolloidal sample produced in run 6 sample following oxidation in air at 200 °C for 2 h showing the conversion from wustite to a spinel structure (magnetite or maghemite), andecolloidal sample produced in run 6 sample following 40-day shelf time showing the conversion from wustite to a spinel structuree(magnetite or maghemite)
Figure 7.
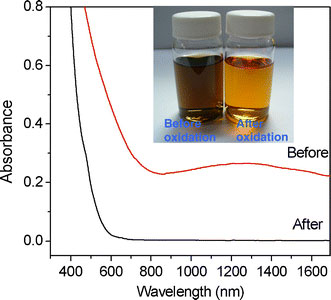
Absorption spectra of NPs produced in run 6 sample (20.4 nm) before and after oxidation, respectively. Before: the NPs are brown-black in color and show a hump in the IR range due to the intervalence charge transfer of magnetite and an absorption edge in the range 600–800 nm due to the presence of wustite. After: the oxidized NPs areredin color and have zero absorbance above 600 nm due to the oxidization of the Fe3O4and wustite contents
Figure 8.
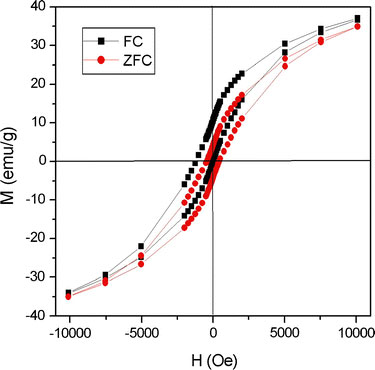
Hysteresis loops of FeO/Fe3O4NPs at temperature of 100 K after zero field cooling (ZFC) and field cooling (FC) in magnetic field up to 1 T. Note that the shifted ZFC loop is the result of a coupling effect at the ferromagnetic and anti-ferromagnetic interface
Figure 9.
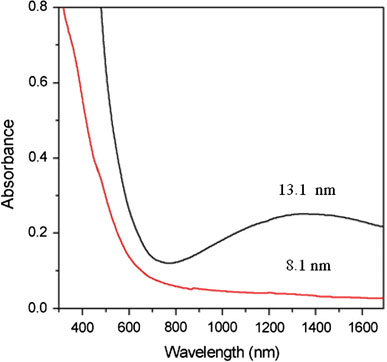
Absorption spectra of as-prepared NPs produced in run 2 (8.1 nm, benzyl ether) and run 4 (13.1 nm, ODE). Note that thehumpin the ODE spectrum is the result of the magnetite content
On the basis of the evidence presented above, it is concluded that the as-prepared NP samples with larger size such as run 6 has a mixed structure comprising both magnetite and wustite. However, with an increasing oxidation time, the wustite transforms initially to magnetite, and then finally to maghemite. For smaller NPs obtained such as run 1, the smaller size is beneficial to the wustite–magnetite–maghemite conversion process, the as-formed wustite change efficiently to magnetite during the work-up process.
Magnetic Properties
As described in the previous section, the as-prepared NP products comprising a mixture of wustite and magnetite can be transformed to pure γ-Fe2O3via the use of an organic oxidizing agent or following prolonged exposure to air. The size-dependent magnetic properties of these pure γ-Fe2O3NPs were evaluated using a superconducting quantum interference device (SQUID). The ZFC curves were obtained by cooling the γ-Fe2O3samples from 300 to 5 K in the absence of an external magnetic field and then measuring the magnetization under a magnetic field of 100 Oe as the temperature was increased to room temperature. Meanwhile, the FC curves were obtained simply by measuring the magnetization of the samples under an external magnetic field of 100 Oe at temperatures ranging from 5 to 300 K. Figure 10presents the corresponding results for NPs with dimensions of 4.5, 8.1, and 20.4 nm, respectively. Based on the maximum values of the ZFC curves, the blocking temperatureTBis determined to be 20 K for the 4.5-nm NPs, 103 K for the 8.1-nm NPs, and 300 K for the 20.4-nm NPs. These results are consistent with Stoner–Wohlfarth theory, which states that the blocking temperature increases as the volume (V) of the NPs increases in accordance with the formulationTB = KV/(25 kB), whereKis the anisotropy constant of the NP andkBis the Boltzmann constant. Figure 11presents the hysteresis loops of the three γ-Fe2O3NP samples as measured at a temperature of 300 K in a magnetic field with an intensity of 2 T. It can be seen that all of the samples exhibit a super-paramagnetic behavior. From inspection, the saturation magnetizations of the three NPs are found to be 52.9 emu/g, (4.5 nm), 62.8 emu/g (8.1 nm), and 67.4 emu/g (20.4 nm), respectively.
Figure 10.
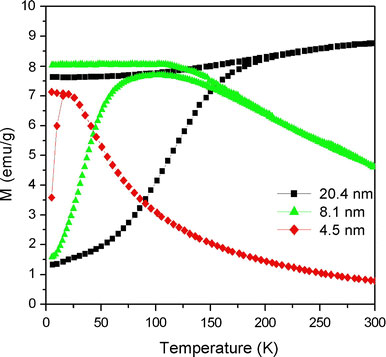
Variation of magnetization (M) with temperature (T) in ZFC and FC modes for maghemite (γ-Fe2O3) NPs with diameters of 4.5, 8.1, and 20.4 nm, respectively
Figure 11.
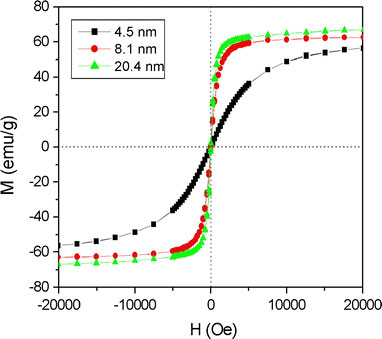
Variation of magnetization (M) with magnetic field (H) at temperature of 300 K for maghemite NPs with diameters of 4.5, 8.1, and 20.4 nm, respectively
Conclusions
A simple method has been presented for the synthesis of sodium-free iron(III) oleate complexes via the direct dissolution of porous hematite powder in unsaturated fatty acids (oleic or linoleic). It has been shown that these complexes represent suitable precursors for the subsequent preparation of iron oxide NPs using an organic solvent under reflux conditions and the corresponding fatty acid as a surfactant. The results have shown that the size of the NP precipitates increases with an increasing reaction temperature (as governed by the boiling point of the organic solvent) and an increasing concentration of free acid in the reactant. Moreover, it has been shown that the larger NPs are comprised primarily of wustite. However, these NPs undergo a wustite–magnetite–maghemite conversion following oxidation. By contrast, the smaller NPs are comprised principally of maghemite, indicating that a smaller NP dimension is beneficial in prompting the wustite–magnetite–maghemite transformation process. Finally, it has been shown that the as-prepared NP products with a mixed wustite–magnetite composition can be transformed to pure γ-Fe2O3products with a super-paramagnetic behavior via the use of an organic oxidizing agent or following prolonged exposure to air.
Acknowledgments
The authors gratefully acknowledge the financial support provided to this study by the National Science Council of Taiwan.
References
- Park J, An K, Hwang Y, Park JG, Noh HJ, Kim JY, Park JH, Hwang N, Hyeon T. Nat. 2004. p. 891. COI number [1:CAS:528:DC%2BD2cXhtVehtrjM]; Bibcode number [2004NatMa...3..891P] [DOI] [PubMed]
- Kim CW, Kim YH, Cha HG, Kwon HW, Kang YS. J. Phys. Chem. B. 2006. p. 24418. COI number [1:CAS:528:DC%2BD28XhtFClt7nI] [DOI] [PubMed]
- Gu H, Soucek MD. Chem. 2007. p. 1103. COI number [1:CAS:528:DC%2BD2sXhsFOktrk%3D] [DOI]
- Zhao SY, Qiao R, Zhang XL, Kang YS. J. Phys. Chem. C. 2007. p. 7875. COI number [1:CAS:528:DC%2BD2sXlt1Wgs7Y%3D] [DOI] [PubMed]
- Bao N, Shen L, Wang Y, Padhan P, Gupta AJ. Am. 2007. p. 12374. COI number [1:CAS:528:DC%2BD2sXhtVGmtbvK] [DOI] [PubMed]
- An K, Lee N, Park J, Kim SC, Hwang Y, Park JG, Kim JY, Park JH, Han MJ, Yu J, Hyeon T. J. 2006. p. 9753. COI number [1:CAS:528:DC%2BD28XmvFWitLk%3D] [DOI] [PubMed]
- Choi SH, Kim EG, Park J, An K, Lee N, Kim SC, Hyeon T. J. Phys. Chem. B. 2005. p. 14792. COI number [1:CAS:528:DC%2BD2MXmtV2kt7o%3D] [DOI] [PubMed]
- Narayanaswamy A, Xu H, Pradhan N, Kim M, Peng X. J. 2006. p. 10310. COI number [1:CAS:528:DC%2BD28XmvFCqu70%3D] [DOI] [PubMed]
- Kovalenko MV, Bodnarchuk MI, Lechner RT, Hesser G, Schaffler F, Heiss W. J. 2007. p. 6352. COI number [1:CAS:528:DC%2BD2sXkslyrsbc%3D] [DOI] [PubMed]
- Bronstein LM, Huang X, Retrum J, Schmucker A, Pink M, Stein BD, Dragnea B. Chem. 2007. p. 3624. COI number [1:CAS:528:DC%2BD2sXntVyjsrg%3D] [DOI]
- Wang SB, Min YL, Yu SH. J. Phys. Chem. C. 2007. p. 3551. COI number [1:CAS:528:DC%2BD2sXhsFOku7w%3D]; Bibcode number [2007JPhD...40.3551W] [DOI]
- Kwon SG, Piao Y, Park J, Angappane S, Jo Y, Hwang NM, Park JG, Hyeon T. J. 2007. p. 12571. COI number [1:CAS:528:DC%2BD2sXhtVKmsbfI] [DOI] [PubMed]
- Kim D, Park J, An K, Yang NK, Park JG, Hyeon T. J. 2007. p. 5812. COI number [1:CAS:528:DC%2BD2sXkt1Shsb8%3D] [DOI] [PubMed]
- Ahnlyaz A, Sakamoto Y, Bergstrom L. Proc. Natl Acad. Sci. USA. 2007. p. 17570. Bibcode number [2007PNAS..10417570A] [DOI] [PMC free article] [PubMed]
- Shavel A, Rodrigue-Gonzalez B, Spasova M, Farle M, Liz-Marzan LM. Adv. 2007. p. 3870. COI number [1:CAS:528:DC%2BD1cXosVSk] [DOI]
- Cha HG, Lee DK, Kim YH, Kim CW, Lee CS, Kang YS. Inorg. 2008. p. 121. COI number [1:CAS:528:DC%2BD2sXhtl2itb%2FI] [DOI] [PubMed]
- Lee DK, Kim YH, Kim CW, Cha HG, Kang YS. J. Phys. Chem. B. 2007. p. 9288. COI number [1:CAS:528:DC%2BD2sXns12lur0%3D] [DOI] [PubMed]
- Han YC, Cha HG, Kim CW, Kim YH, Kang YS. J. Phys. Chem. C. 2007. p. 6275. COI number [1:CAS:528:DC%2BD2sXjvFSrsLY%3D] [DOI]
- Corr SA, Rakovich YP, Gun’ko YK. Nanoscale Res. 2008. p. 87. COI number [1:CAS:528:DC%2BD1cXlslejt7k%3D]; Bibcode number [2008NRL.....3...87C] [DOI]
- Wu W, He Q, Jiang C. Nanoscale Res. 2008. p. 397. COI number [1:CAS:528:DC%2BD1cXhsVyhtrvP] [DOI] [PMC free article] [PubMed]
- Akanni MS, Okoh EK, Burrow HD, Ellis HA. Thermochim. 1992. p. 1. COI number [1:CAS:528:DyaK3sXnt1ejuw%3D%3D] [DOI]
- Chen HS, Lo B, Hwang JY, Chang GY, Chen CM, Tasi SJ, Wang SJ. J. Phys. Chem. B. 2004. p. 17119. COI number [1:CAS:528:DC%2BD2cXot1WmsL4%3D] [DOI]
- Liu H, Owen JS, Alivisatos AP. J. 2007. p. 305. COI number [1:CAS:528:DC%2BD28Xhtleiu7nF] [DOI] [PubMed]
- Murphy JE, Beard MC, Norman AG, Aphrenkiel SP, Johnson JC, Yu P, Micic OI, Ellingson RJ, Nozik AJ. J. 2006. p. 3241. COI number [1:CAS:528:DC%2BD28XhsVSqt7c%3D] [DOI] [PubMed]
- Golnen M, Balkose D, Inal F, Ulku S. Ind. 2005. pp. 1627–1628. [DOI]
- Barman S, Vasudevan S. J. Phys. Chem. B. 2006. p. 651. COI number [1:CAS:528:DC%2BD2MXhtlajtrvE] [DOI] [PubMed]
- Jana NR, Chen Y, Peng X. Chem. 2004. p. 3931. COI number [1:CAS:528:DC%2BD2cXnsVaitb8%3D] [DOI]
- W.W. Yu, J.C. Falkner, C.T. Yavuz, V.L. Colvin, Chem. Commun. 2306 (2004) [DOI] [PubMed]
- Chen CJ, Lin CC, Chiang RK, Lin CR, Lyubutin IS, Alkaev EA, Lai HY. Cryst. 2008. p. 877. [DOI]
- Naono H, Nakai K, Sueyoshi T, Yagi H. J. 1987. p. 439. [DOI]
- Oh SM, Hendrickson DN, Hassett KL, Davis RE. J. 1985. p. 8009. COI number [1:CAS:528:DyaL28Xislynuw%3D%3D] [DOI]
- Minervini L, Grimes RW. J. 1999. p. 235. COI number [1:CAS:528:DyaK1MXhtVKnsrk%3D]; Bibcode number [1999JPCS...60..235M] [DOI]
- Tang J, Myers M, Bosnick JA, Brus LE. J. Phys. Chem. B. 2003. p. 7501. COI number [1:CAS:528:DC%2BD3sXivFantro%3D] [DOI]
- Liu P, Cai W, Zeng H. J. Phys. Chem. C. 2008. p. 3261. COI number [1:CAS:528:DC%2BD1cXhslKns70%3D] [DOI]