

Abstract
A series of monomethoxy poly(ethylene glycol)–poly(lactide) (mPEG–PLA) diblock copolymers were designed according to polymer–drug compatibility and synthesized, and mPEG–PLA micelle was fabricated and used as a nanocarrier for solubilization and oral delivery of Cyclosporine A (CyA). CyA was efficiently encapsulated into the micelles with nanoscaled diameter ranged from 60 to 96 nm with a narrow size distribution. The favorable stabilities of CyA-loaded polymeric micelles were observed in simulated gastric and intestinal fluids. The in vitro drug release investigation demonstrated that drug release was retarded by polymeric micelles. The enhanced intestinal absorption of CyA-loaded polymeric micelles, which was comparable to the commercial formulation of CyA (Sandimmun Neoral®), was found. These suggested that polymeric micelles might be an effective nanocarrier for solubilization of poorly soluble CyA and further improving oral absorption of the drug.
Keywords: Monomethoxy poly(ethylene glycol)–poly(lactide), Polymeric micelles, Cyclosporine A, Solubility parameter, In vitro release, Intestinal absorption
Introduction
The oral route is the most common route of drug administration in view of its convenience and patient acceptance, even more so in the case of chronic therapies [1]. Many existing and new therapeutic entities are characterized by a low degree of water solubility leading to poor and erratic oral bioavailability [2]. In order to overcome this hurdle, several strategies such as micronization [3,4], formation of solid solutions [5], microemulsification [6], and novel drug delivery systems, including nanoparticles [7], lipid-based vesicles [8,9], have been proposed. Among these approaches, polymeric micelles, constituted of amphiphilic block copolymers, have attracted much attention in the decade [10-12]. Generally, block copolymers with concentration above the critical association concentration (CAC) self-assemble into spherical polymeric micelles with a core–shell structure in water: the hydrophobic segments aggregate to form an inner core being able to accommodate hydrophobic drugs with improved solubility by hydrophobic interactions; the hydrophilic shell consists of a brush-like protective corona that stabilizes the micelles in aqueous solution [13-15]. Polymeric micelles as novel drug vehicles present numerous advantages, such as reduced side effects of drugs, selective targeting, stable storage, stable toward dilution, and prolonged blood circulation time [15,16]. Furthermore, polymeric micelles possess a nanoscaled size with a narrow distribution. They can protect drugs against premature degradation in vivo owing to their core–shell architecture [17,18]. More importantly, polymeric micelles are fabricated according to the physicochemical properties of drugs and the compatibility between the core of micelles and drug molecules [15,19]. From the pharmaceutical point of view, these amphiphilic carriers can solubilize more poorly water-soluble drugs within their hydrophobic core than most surfactant micelles. Since most of polymeric micelles are intended to be administered intravenously [20], the development of polymeric micelles via the oral route has been attracted attention [16], [21], [22]. Francis et al. [9] reported that the polymeric micelles exhibited high stability in gastric and intestinal fluids and no significant cytotoxicity toward Caco-2 cells, and the apical-to-basal permeability of Cyclosporine A (CyA) across Caco-2 cells increased significantly when loaded in polymeric micelles compared to free CyA. However, the absorption enhancement of drug loaded in polymeric micelles by oral delivery to rats has remained elusive. These prompted us to investigate the suitability of polymeric micelles as carriers to enhance the oral absorption of BCS Class II (i.e., low solubility–high permeability) drugs.
In the present study, CyA which belongs to BCS Class II was selected as a model drug. CyA is a highly lipophilic cyclic undecapeptide of 11 amino acids, and a highly effective immunosuppressive agent which is widely used in clinic for prevention of allograft rejection after organ transplantation and treatment of autoimmune disease [23-25]. Nevertheless, the oral bioavailability of CyA is low and irregular [26] due to the large molecular weight (1202 Da), low solubility in water (23 μg/mL at room temperature) [27], very high lipophilicity (log P = 2.92) [28], a substrate of P-glycoprotein, and vulnerable to intestinal mucosa and liver P450 3A4. The currently available oral formulation of CyA is in the form of a microemulsion containing a high concentration of Cremophor RH40 which has been reported to induce undesirable side effects, such as nephrotoxicity and induction of anaphylactic reactions in sensitized patients, although the oral absorption of CyA was remarkably enhanced. Consequently, there has been an urgent requirement to design and develop a novel dosage form of CyA aimed at decreasing the side effects of the current formulation while preserving the bioavailability of the drug.
The purposes of this study were to design and evaluate CyA-loaded polymeric micelles in vitro. Therefore, monomethoxy poly(ethylene glycol)–poly(lactide) (mPEG–PLA) with molecular weight of 2500, 5000, 10000, and 15000 Da for PLA block were strategically designed and synthesized by ring-opening polymerization ofd,l-dilactide (d,l-LA) in the presence of mPEG with molecular weight of 5000 Da, respectively. The micelles preparation, CyA solubilization, and micelles properties were investigated by the size measurement, drug loading content (LC), encapsulation efficiency (EE), stability in gastrointestinal tract, and in vitro drug release. Intestinal absorption in situ and ex vivo of CyA-loaded polymeric micelles was assessed, respectively.
Materials and Methods
Materials
Cyclosporine A (CyA) was kindly donated from Hangzhou Zhongmei Huadong Pharmaceutical Co. Ltd., China. Monomethoxy poly(ethylene glycol) with molecular weight of 5000 Da (mPEG) and stannous 2-ethylhexanoate were purchased from Sigma. 3,6-dimethyl-1,4-dioxane-2,5-dione (d,l-lactide) was obtained from Daigang Biological Technology Co. Ltd., China. All other reagents were of analytical grade, except those for HPLC assay which were of HPLC grade.
Animals
Sprague-Dawley (SD) rats were obtained from Animals Center of Peking University Health Science Center. All animals were provided with standard food and water ad libitum and were exposed to alternating 12-h periods of light and darkness. Temperature and relative humidity were maintained at 25 °C and 50%, respectively. All care and handling of animals were performed with the approval of Institutional Authority for Laboratory Animal Care of Peking University.
Prediction of Compatibility Between Polymers and the Drug
Compatibility between polymers and the drug was predicted by interaction parameters calculated from partial solubility parameter [29]. The solubility parameter was obtained by Hansen’s approach, where the partial solubility parameters (δd, δh, and δp) for the drug (CyA) and a range of biodegradable polymers (PLA, PLGA, and PCL) were calculated by the group contribution method (GCM) [30] using Eqs. 12, and 3 presented below.
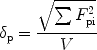 |
where FdiFpi, and Fhi refer to the specific functional group contributions van der Waals dispersion forces (Fdi), dipole–dipole interactions (Fpi), and hydrogen bonding (Fhi), respectively. While the interaction parameter between drug and polymer was calculated according to Eqs. 4 and 5 presented below:
Here, δd,p means the δd of polymer, δd,d the δd of drug (CyA), δp,p the δp of polymer, δp,d the δp of drug (CyA), δh,p the δh of polymer, δh,d the δh of drug (CyA), ϕp and ϕd mean the volume fractions of polymer and drug, respectively.
Synthesis and Characterization of mPEG–PLA Copolymers
mPEG–PLA copolymers were synthesized as previously described [20]. The synthesized diblock copolymers were referred to as mPEG x –PLA y. x and y represented the number-averaged molecular weight of the mPEG and PLA block in kDa. For example, mPEG5–PLA2.5 consisted of a 5-kDa mPEG block connected to a 2.5-kDa PLA block.
The critical aggregation concentration (CAC) of mPEG–PLA copolymer was determined by fluorescence spectroscopy using pyrene (Fluka, >99%) as a hydrophobic probe as previously reported [20]. The fluorescence spectra of pyrene were measured at varying copolymer concentrations using a Shimadzu RF-5301 PC fluorescence spectrometer at 25 °C. The excitation wavelength was adjusted to 392 nm, and the detection of fluorescence was performed at 333 and 335 nm. CAC was measured from the onset of a rise in the intensity ratio of peak at 335 nm to peak at 333 nm in the fluorescence spectra of pyrene plotted versus the logarithm of polymer concentration.
Preparation of mPEG–PLA Polymeric Micelles
mPEG5–PLA2.5 and mPEG5–PLA5 polymeric micelles were prepared by rotary evaporation method [31]. In brief, 25 mg of mPEG–PLA was dissolved in 10 mL of methanol followed by evaporation under vacuum at 60 °C to form a homogeneous film. The resulting film was dispersed in 10 mL of water at 60 °C and then vortexed for 3 min. The mixture was filtered through a 0.45-μm filter (Millex-GV, Millipore, USA) to obtain a clear and homogeneous micellar solution. The CyA-loaded micelles were prepared as described above except 25 mg of mPEG–PLA was replaced by a mixture of 5 mg of CyA and 25 mg of mPEG–PLA.
mPEG5–PLA10 and mPEG5–PLA15 polymeric micelles were prepared by dialysis method [20]. In brief, 25 mg of mPEG–PLA and 5 mg of CyA were dissolved in 3 mL of dimethyl sulfoxide (DMSO). The solution was then introduced into a dialysis bag (Spectrapor, MWCO = 3500) and dialyzed against 1 L of distilled water, which was replaced every 4 h in the course over 48 h. The micellar solution obtained in the dialysis bag was then filtered through a 0.45-μm filter (Millex-GV, Millipore, USA) to remove nonencapsulated CyA. CyA-free micelles were produced by the same method without adding CyA at the first stage of the preparation.
Particle Size Measurement
The average particle size and size distribution of polymeric micelles were determined by dynamic light scattering (DLS) (Zetasizer ZEN 3600, Malvern, UK). All DLS measurements were performed with a scattering angle of 90° at 25 °C after diluting the micellar solution to an appropriate volume with water. The results were the mean values of three samples.
Determination of Encapsulation Efficiency and Drug Loading Content
In order to determine drug loading content (LC, w/w %) and entrapment efficiency (EE, w/w %) of micelles, CyA-loaded polymeric micelles solution was freeze-dried and then dissolved in methanol and CyA content in micelles was determined on a Shimadzu series HPLC system (Shimadzu LC-10AT, Kyoto, Japan) equipped with a UV detector (Shimadzu SPD-10A) and reversed phase column (ODS C18, 5 μm, 4.6 mm × 250 mm, Dikma, China). The mobile phase consisted of acetonitrile/water (90/10, v/v) and was pumped at a flow rate of 1.0 mL/min. The detection wavelength was 210 nm. The column temperature was set to 70 °C. The LC and EE of the micelles were then calculated based on the following formula:
Stability of CyA-Loaded Polymeric Micelles in Simulated Gastric and Intestinal Fluids
The stability of CyA-loaded polymeric micelles in simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) for a period of 12 h was evaluated from the changes of EE and particle size of micelles with time upon incubation of the micelles samples in SGF or SIF at 37 °C, respectively. The EE and particle size of micelles were determined as described earlier.
In Vitro Release of CyA from Micelles
One milliliter of micelles solution with known CyA content was placed into a dialysis bag with molecular weight cut-off of 50 kDa. The dialysis bag was immersed into a flask containing 30 mL of release medium (SGF or SIF) containing 30% (v/v) ethanol (sink condition) which was kept in a constant temperature shaking water bath at 37 °C and 100 rpm. At predetermined time intervals, aliquots (1 mL) of the release medium was taken and replaced by fresh medium. The content of CyA in the medium was measured by HPLC method as described above. The cumulative release percentage of CyA was calculated.
Intestinal Absorption
Male SD rats weighing 200 ± 20 g were used for this research. All animals were housed with free access standard food and tap water, and exposed to alternating 12 h periods of light and darkness. Temperature and relative humidity were maintained at 25 °C and 50%, respectively. After an acclimatization period of 2 days, the rats were fasted for 12 h but allowed free access to water prior to the experiments. Rats were anaesthetized via intraperitoneal injection of 15% w/v urethane at a dose of 1.5 g/kg and a laparotomy was performed.
In situ rat perfusion experiments [32-35]
The abdominal cavity was opened, and a segment of the ileum was exposed. The segment was flushed with 50 ml of warm Krebs–Ringer buffer solution [KRB, pH 7.4, composed of (in mM): NaCl 111.9, KCl 5.0, CaCl21.2, MgCl21.2, NaH2PO40.4, Na2HPO41.6, NaHCO325.0, NaGlutamate 4.9, NaPyruvate 4.9, Na2Fumarate 5.4, glucose 11.5] to remove the luminal contents and then flushed with air to minimize the amount of luminal fluid. These rats were placed in a warm table for the absorption studies after surgery.
The perfusion solution was prepared using 1 mg/mL of CyA-loaded polymeric micelles (mPEG5-PLA5) or Sandimmun Neoral® in KRB containing 3-mg/mL phenol red. Phenol red acted as a nonabsorbable marker in the luminal medium to correct the appreciable influence of the secretion or absorption of water on CyA content during the experiment. A recirculating system was constructed by connecting a small reservoir containing 40 mL of stirring perfusion solution kept at 37 °C with the inlet cannula and by directing the flow from the outlet cannula back to the reservoir. The perfusion pump (BT300-300M, Lange, Co, Ltd. China) was utilized to drive the perfusate at a rate of 2 mL/min through the inlet cannula into the intestinal segment. 0.5 mL of perfusate samples were collected and replaced by fresh KRB containing 3-mg/mL phenol red at 0.25, 0.5, 1, 2, 3, 4, 6, 8 h after perfusion. The CyA concentration in the perfusate samples was measured by HPLC method as described above, and the phenol red concentration was determined using UV spectrophotometer (Agilent 8453, Agilent Technologies, UK) at 558 nm.
Ex vivo experiments [29,36]
A laparotomy was performed and segment of ileum were removed and used for the everted gut sac technique. In brief, the ileum was rinsed twice with saline solution (0.9% NaCl) at room temperature. Then the specimen was put into oxygenated Tyrode solution [pH 7.4, composed of (in mM): NaCl 115, KCl 2.7, CaCl21.8, MgCl21.1, NaH2PO40.4, Na2HPO41.6, NaHCO312.7, glucose 5.6] immediately at 37 °C. Then the intestine was gently everted over a glass rod and divided into 5-cm sacs which were filled with fresh oxygenated Tyrode solution using silk suture. The sacs were incubated in gentle shaking oxygenated Tyrode solution containing CyA-loaded polymeric micelles or Sandimmun Neoral® for 120 min. Thereafter, the Tyrode solution with and without sacs was collected. The content of remaining CyA in the medium was measured by HPLC method as described above. The mass of the bound CyA was determined by subtraction of the mass of remaining CyA in the medium from the initial mass of CyA and reported as percent binding:
The commercial formulation of CyA, Sandimmun Neoral®, was also tested as control.
Statistical Analysis
All data were expressed as mean standard deviation (SD) unless particularly outlined. The statistical significance of differences among more than two groups was determined by one-way ANOVA by the software SPSS 13.0. A value of p < 0.05 was considered to be significant.
Results and Discussion
Compatibility Between Polymers and the Drug
The compatibility between a drug and polymer is known to be one of the key factors in determining the effectiveness of polymeric delivery systems [30]. In general, the better the compatibility between a drug and polymer, the higher the loading content and the slower the drug release as well. Thus, the compatibility between a drug and polymer which was referred to miscibility and interaction has been shown to be of importance in the design of a wide range of delivery systems including polymeric micelles.
Thermodynamic criteria for the mutual miscibility of two substances is based on the Gibbs energy of mixing (Δ GM) which is defined by the following equation: Δ GM = ΔHM – TΔSM. The two substances are said to be mutually soluble if ΔGM is negative. Equation 5was used to calculate ΔHM considering it takes into account all forces: Van der Waals dispersion, dipole–dipole, and hydrogen bonding. A smaller ΔHM indicated a stronger interaction between two substances. As shown in Eq. 5, the value of ΔHM mainly depends on that of ∆, indicating that the compatibility between a drug and polymer can be evaluated by ∆ or ΔHM which is referred as interaction parameter. The interaction parameters of CyA with various polymers calculated by GCM were listed in Table 1. The results showed that the interaction parameter of CyA with PLA was lower than those of other drug–polymer pairs, suggesting that PLA was identified to be more compatible to CyA among the three polymers. Based on the results calculated, PLA was therefore selected as the hydrophobic segments of the amphiphilic copolymer for the subsequent studies.
Table 1.
The interaction parameters for CyA and various polymers
| Polymer | ∆ (J/cm3) | ∆HM(J) |
|---|---|---|
| PLA | 4.03 | 0.78 |
| PLGA (LA/GA, 50:50) | 5.37 | 2.16 |
| PCL | 5.68 | 2.97 |
Synthesis and Characterization of mPEG–PLA Copolymers
mPEG–PLA copolymers were synthesized by ring-opening polymerization ofd,l-dilactide by using mPEG as initiator. Various chain lengths of PLA in the copolymers were obtained by modulating the feed ratio of mPEG andd,l-dilactide. Figure 1 showed the 1H-NMR spectrum of mPEG5–PLA2.5 which was representative for all synthesized mPEG–PLAs: the peak at 3.6 ppm corresponded to methylene protons (–OCH2CH2–) in mPEG blocks, signals at 1.5 ppm could be attributed to the hydrogen atoms of CH3O-groups for PLA segments, respectively [33]. The ratio of number-averaged molecular weight (Mn) of the mPEG to PLA blocks of each copolymer was calculated from the 1H-NMR data. In brief, the ratio of the number of methylene protons (–OCH2CH2–) in PEG and (–OCH3) in PLA was equally as the ratio of integral value of H in 3.6 and 1.5 ppm. The molecular weight of mPEG was known as 5000 and the molecular weight of PLA linked to mPEG would be known as listed in Table 2. It could be seen that the copolymers could be synthesized reliably.
Figure 1.
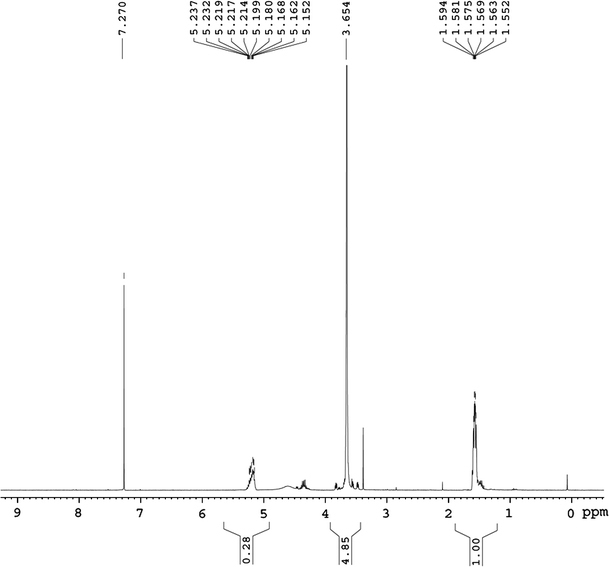
1H-NMR spectrum of mPEG5–PLA2.5 copolymer
Table 2.
Characterization of mPEG5–PLAy copolymers and polymeric micelles
| Copolymers | Mn(PLA) | CAC (10−7 mol/L) | CyA loaded | LC (%) | EE (%) | |
|---|---|---|---|---|---|---|
| d(nm) | PDI | |||||
| mPEG5–PLA2.5 | 2249 | 3.07 | 60.1 ± 8.2 | 0.23 | 12.1 ± 0.8 | 44.0 ± 3.0 |
| mPEG5–PLA5 | 4802 | 1.99 | 71.9 ± 0.4 | 0.19 | 15.2 ± 1.1 | 77.9 ± 4.1 |
| mPEG5–PLA10 | 9247 | 1.57 | 89.0 ± 0.5 | 0.20 | 10.7 ± 1.5 | 33.1 ± 2.1 |
| mPEG5–PLA15 | 15825 | 1.08 | 95.2 ± 1.2 | 0.23 | 9.7 ± 0.2 | 10.9 ± 0.2 |
d and PDI represent the average diameter and the polydispersity index (PDI) of micelles in aqueous solution, respectively
Micelle formation requires the balance between the attractive interactions of the insoluble PLA moieties and the repulsive interactions of the soluble PEG segments. In effect, PEG moderates the association of PEG–PLA molecules, leading to micelle formation. When the PEG proportion in the copolymer chains is too low, PEG segments cannot moderate the association of the separating PLA–PEG molecules, and macroscopic agglomerates are formed. In line with these observations, Shin et al. reported that micelles could not be formed by a poly(ethylene glycol)/epsilon-caprolactone copolymer with a too-high caprolactone content (70.7% by weight) [37]. Moreover, a low CAC is an important feature for the application of these micelles in drug delivery because it assures that micelles will be stable in vivo, where considerable dilution takes place. Polymeric micelles can be formed only when the block copolymer concentration is higher than CAC which characterizes the micelle stability. The micellization behavior of the synthesized mPEG5–PLAy copolymers was therefore investigated. Table 2 summarized the CAC values of various synthesized mPEG–PLA diblock copolymers ranging from 1.08 × 10−7 to 3.07 × 10−7 mol/L. These values appeared much lower than those of low molar mass surfactants, indicating that micelles formed from mPEG–PLA copolymers as drug carriers could preserve stability without dissociation after dilution [13,38], which was of major interest for oral administration. Moreover, the hydrophilicity of mPEG–PLA copolymers mainly depending on the mass ratio of mPEG/PLA or mPEG content had much influence on the CAC value [39]. As shown in Table 2, the CAC values of copolymers appeared to decrease with increasing of PLA block length. mPEG5–PLA15 had longer hydrophobic PLA block and, thus, could self-assemble more easily to form micelles, leading to lower CAC value. This was consistent with our previous report [20].
Characterization of mPEG–PLA Polymeric Micelles
The polymeric micelles were evaluated by particle size, encapsulation efficiency, and drug loading content. The mean particle size and size distribution of CyA-loaded polymeric micelles were determined by DLS. The mean diameter ranged from 60 to 96 nm for CyA-loaded micelles with narrow distributions (Table 2). It appeared that the micelle size gradually increased with increasing of the length of PLA chains, which was consistent with our previous report [20]. This result was also in agreement with the characteristic of amphiphilic copolymeric micelles, i.e., the shorter the hydrophobic block length, the smaller the micelles. It might be attributed to the fact that it is difficult to form compact polymeric micelles for amphiphilic copolymers with longer hydrophobic chain length.
The encapsulation efficiency (EE) and loading content (LC) of micelles were presented in Table 2. It could be found that the LC and EE of CyA-loaded mPEG5–PLA5 polymeric micelles were significantly higher than those of others (p < 0.05 for LC and p < 0.01 for EE, respectively). Notably, the results were not the general trend that LC and EE of polymeric micelles increased with increasing hydrophobic chain length. This finding may be assigned to the factors contributing to LC and EE. In general, LC and EE depend on the composition of the copolymers [38], [40,41], the length of hydrophobic block [42], initial diblock copolymeric concentration or the feed weight ratio of the drug to the copolymer, the solvent used in formulation process[41,43] and micelle preparation method [44,45], and so on. In the case of our experiment, mPEG5–PLA2.5 and mPEG5–PLA5 polymeric micelles were prepared by rotary evaporation method, whereas mPEG5–PLA10 and mPEG5–PLA15 polymeric micelles were prepared by dialysis method. In general, the LC and EE of polymeric micelles prepared by rotary evaporation method are higher than those of polymeric micelles prepared by dialysis method [45-47]. The results in current study were in agreement with this trend. Moreover, for mPEG5–PLA2.5 and mPEG5–PLA5 polymeric micelles, the LC and EE were the general trend, i.e., increased with increasing hydrophobic chain length, however, the results were opposite for mPEG5–PLA10 and mPEG5–-PLA15 polymeric micelles, as reported that LC and EE decreased with increasing hydrophobic chain length when polymeric micelles were prepared by dialysis method due to enhanced water-insolubility of copolymers [48].
Stability Studies of CyA-Loaded Polymeric Micelles in Simulated Gastric and Intestinal Fluid
The particle size and entrapment efficiency of micelles incubated in SGF and SIF at 37 °C were determined at predetermined time intervals. As shown in Fig. 2, there was no significant change in either EE or particle size of micelles with time from 0 h until 12 h, indicating that the CyA-loaded polymeric micelles were stable in SGF and SIF at 37 °C for a long enough time. Further, the micelles appeared equally stable in these two media for the time period studied.
Figure 2.
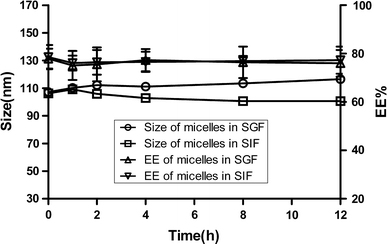
Variation of size (a) and entrapment efficiency (EE) (b) of CyA-loaded polymeric micelles with incubation time in SGF and SIF at 37 °C (n = 3)
In addition, in the case of release experiment, it was found that no CyA was detected in SGF or SIF at the end of 24 h, which might be due to the fact that the amount of released drug was probably below the detection limit of HPLC method. This might be advantageous in micelle stability aspect [49]. Since micelles retained almost all the incorporated CyA, the formulations had high stability, and drug in micelle core would be protected from biological degradation in GI before reaching absorption site. Thus, from a stability perspective, these micelles appear to be suitable candidates as an oral dosage form of CyA.
In Vitro Release of CyA from Micelles
When developing oral colloidal delivery systems for highly hydrophobic drugs such as CyA, it is important to adequately control the release rate to avoid precipitation upon dilution in the stomach and maximize the absorption in the small bowel. Therefore, the in vitro release of CyA from polymeric micelles at different release medium was evaluated. Prior to conducting these release assays, it was verified that CyA could freely diffuse through the dialysis membrane (Fig. 3), and that sink condition was respected by addition of 30% (v/v) ethanol in the release medium while preserving the stability of polymeric micelles (data not shown). In addition, the release property of CyA from polymeric micelles in SGF did not differ from that in SIF (data not shown), indicating that the release media had no effect on the release of CyA from micelles. Consequently, SIF was chosen as the release medium for the subsequent release studies.
Figure 3.
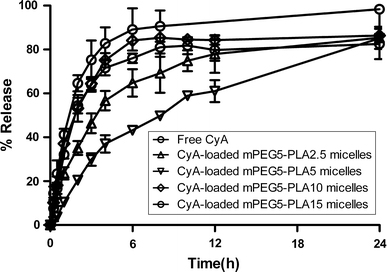
Release profiles of CyA from mPEG–PLA polymeric micelles in SIF at 37 °C, respectively (n = 3)
As shown in Fig. 3, 85% of CyA was released from polymeric micelles within 24 h. The comparison of the profiles of CyA release from the micelles and the solution showed that the entrapment of CyA in the mPLA–PEG micelles could significantly retard its in vitro release [16,50]. Assuming that similar sustained release also occurs in vivo, this would provide the micelles the necessary time required to exert their influence on drug bioavailability. In addition, the release rate of CyA from mPEG5–PLA10 micelles was comparable to that from mPEG5–PLA15 micelles and faster than that from mPEG5–PLA2.5 and mPEG5–PLA5 micelles, and remarkable burst release was observed in the release profiles of the former. These might be attributed to the fact that it is difficult to form compact polymeric micelles for amphiphilic copolymers with longer hydrophobic chain length. Furthermore, polymeric micelles prepared by dialysis method were looser than those prepared by other methods. In addition, as compared to the CyA-loaded mPEG5–PLA2.5 micelles, mPEG5–PLA5 micelles released markedly slower, which was consistent with previous report [51]. This might be assigned to the relatively bigger size of mPEG5–PLA2.5 micelles [52] and stronger interaction between CyA and longer hydrophobic chain of PLA. As reported earlier, the release of a drug from polymeric micelles could be affected by the level of drug encapsulation, its physical state, the nature of the polymer and the level of polymer-drug compatibility [30,53,54].
Based on the results of CyA-loading and CyA-release experiments, mPEG5–PLA5 polymeric micelles were therefore selected for the subsequent studies.
Intestinal Absorption
The absorption profiles of CyA-loaded mPEG5–PLA5 polymeric micelles and commercial formulation (iSandimmun Neoral® ) were evaluated, respectively. As shown in Fig. 4, the cumulative absorption percentage of CyA-loaded polymeric micelles was higher than that of Sandimmun Neoral® at each determined time point. For example, the cumulative absorption percentage of CyA-loaded polymeric micelles was higher than 65% within the first 2 h while it was lower than 60% for Sandimmun Neoral® . This absorption enhancement of CyA-loaded polymeric micelles compared with Sandimmun Neoral® was further confirmed by the everted sac experiment which showed that the binding percentage (59.93 ± 2.17%) of CyA-loaded polymeric micelles was significantly higher than that of Sandimmun Neoral® (51.24 ± 5.79) (p < 0.05). The absorption enhancement of CyA by polymeric micelles could be attributed to the fact that the solubility of CyA which belongs to BCS class II drug (i.e., low solubility–high permeability) according to BCS was enhanced and the nanoscaled size of micelles would make the micelles adhere to the intestinal mucosal epithelial to get a longer absorbed time.
Figure 4.
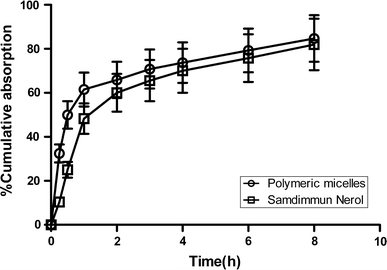
In situ intestinal absorption profiles of CyA-loaded in mPEG5–PLA5 polymeric micelles and Sandimmun Neoral® in rats (n = 3)
Earlier investigation demonstrated that polymeric micelles transported Caco-2 cell monolayers neither by the paracellular route nor by M-cells. One possible mechanism was that polymeric micelles might undergo an active transport that probably involved endocytosis, which was confirmed by using FAE cell model which mimics M-cells and are specialized in the uptake of macromolecules and particles via transcytosis [55]. Further experiments should be conducted using Caco-2 cell model due to the fact Caco-2 cells were considered to be a representative model for assessing intestinal uptake of drugs, which are in progress by our group.
Conclusions
mPEG–PLA micelles were designed and prepared to encapsulate CyA, a highly lipophilic drug. The results of the present study demonstrated that polymeric micelles sustained the drug release, and the CyA-loaded mPEG–PLA micelles were stable in gastrointestinal tract. As anticipated, the intestinal absorption of CyA was significantly improved by polymeric micelles and comparable to that of Sandimmun Neoral® . These findings indicated that polymeric micelles would be a promising nanocarrier for improving the oral absorption of poorly absorbable drugs although the mechanism of polymeric micelles to cross intestinal barrier is still uncertain.
Acknowledgments
We would like to acknowledge the support of this work by the National Development of Significant New Drugs (New Preparation and New Technology, 2009zx09310-001) and the National Basic Research Program of China (973 program, 2009CB930300).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Lavelle SSEC, Thomas NW, Holland J, Davis SS. Adv. 1995. p. 5. COI number [1:CAS:528:DyaK28Xns1OktA%3D%3D] [DOI]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. 2001. p. 3. COI number [1:CAS:528:DC%2BD3MXitVOhs7o%3D] [DOI] [PubMed]
- Guay DR. Ann. 1999. p. 1083. COI number [1:CAS:528:DyaK1MXntlSktb0%3D] [DOI] [PubMed]
- Strickley RG. Pharm. 2004. p. 201. COI number [1:CAS:528:DC%2BD2cXhtlWqt7o%3D] [DOI] [PubMed]
- Serajuddin AT. J. 1999. p. 1058. COI number [1:CAS:528:DyaK1MXltl2jtbk%3D] [DOI] [PubMed]
- Kawakami K, Yoshikawa T, Hayashi T, Nishihara Y, Masuda K. J. Control. Release. 2002. p. 75. COI number [1:CAS:528:DC%2BD38XjtFyhsbc%3D] [DOI] [PubMed]
- Muller RH, Mader K, Gohla S. Eur. 2000. p. 161. COI number [1:CAS:528:DC%2BD3cXjslyisrg%3D] [DOI] [PubMed]
- Porter CJ, Charman WN. Adv. 2001. p. S127. Suppl. 1. [DOI] [PubMed]
- Francis MF, Cristea M, Yang Y, Winnik FM. Pharm. 2005. p. 209. COI number [1:CAS:528:DC%2BD2MXitlCltrk%3D] [DOI] [PubMed]
- Torchilin VP. Adv. 2002. p. 235. COI number [1:CAS:528:DC%2BD38XhvVWnsr0%3D] [DOI] [PubMed]
- Rosler A, Vandermeulen GW, Klok HA. Adv. 2001. p. 95. COI number [1:CAS:528:DC%2BD3MXovVeqtrY%3D] [DOI] [PubMed]
- Jones M, Leroux J. Eur. 1999. p. 101. COI number [1:CAS:528:DyaK1MXmtVymurk%3D] [DOI] [PubMed]
- Shuai X, Merdan T, Schaper AK, Xi F, Kissel T. Bioconjug. 2004. p. 441. COI number [1:CAS:528:DC%2BD2cXjsFGgsrk%3D] [DOI] [PubMed]
- Riley T, Heald CR, Stolnik S, Garnett MC, Illum L, Davis SS. Langmuir. 2003. p. 8428. COI number [1:CAS:528:DC%2BD3sXmsFOisLo%3D] [DOI]
- Liu L, Li C, Li X, Yuan Z, An Y, He B. J. 2001. p. 1976. COI number [1:CAS:528:DC%2BD3MXisVCku7g%3D] [DOI]
- Pierri E, Avgoustakis K. J. 2005. p. 639. COI number [1:STN:280:DC%2BD2MrmtVWrsg%3D%3D] [DOI] [PubMed]
- Wilhelm M, Zhao CL, Wang YC, Xu RL, Winnik MA, Mura JL, Riess G, Croucher MD. Macromolecules. 1991. p. 1033. COI number [1:CAS:528:DyaK3MXhtVehtb8%3D]; Bibcode number [1991MaMol..24.1033W] [DOI]
- Barreiro-Iglesias R, Bromberg L, Temchenko M, Hatton TA, Concheiro A, Alvarez-Lorenzo C. J. Control. Release. 2004. p. 537. COI number [1:CAS:528:DC%2BD2cXkvF2itb0%3D] [DOI] [PubMed]
- Li PZ, Li XR, Zhou HX, Zhang YH, Wang F, Liu Y. Chin. J. New Drug. 2009. p. 262. COI number [1:CAS:528:DC%2BD1MXkt12mtr8%3D]
- Yang ZL, Li XR, Yang KW, Zhou YX, Chen XW, Zhang YH, Wang F, Ren LJ. Nanoscale Res. 2009. p. 1502. [DOI] [PMC free article] [PubMed]
- Tobio M, Sanchez A, Vila A, Soriano II, Evora C, Vila-Jato JL, Alonso MJ. Colloids Surf. B. 2000. p. 315. COI number [1:CAS:528:DC%2BD3cXltVOltbs%3D] [DOI] [PubMed]
- Yin H, Bae YH. Eur. 2009. p. 223. COI number [1:CAS:528:DC%2BD1MXitVShsLk%3D] [DOI] [PMC free article] [PubMed]
- Noble S, Markham A. Drugs. 1995. p. 924. COI number [1:CAS:528:DyaK28Xit12nuw%3D%3D] [DOI] [PubMed]
- Richardson C, Emery P. Drugs. 1995. p. 26. COI number [1:CAS:528:DyaK28Xht12gs7s%3D] [DOI] [PubMed]
- Dai J, Nagai T, Wang X, Zhang T, Meng M, Zhang Q. Int. 2004. p. 229. COI number [1:CAS:528:DC%2BD2cXlslGhsLY%3D] [DOI] [PubMed]
- Lindholm A, Henricsson S, Lind M, Dahlqvist R. Eur. 1988. p. 461. COI number [1:CAS:528:DyaL1cXkvVGlurY%3D] [DOI] [PubMed]
- Miyake K, Hirayama F, Uekama K. J. 1999. p. 39. COI number [1:CAS:528:DyaK1cXns1ylurs%3D] [DOI] [PubMed]
- Woo JS, Piao MG, Li DX, Ryu DS, Choi JY, Kim JA, Kim JH, Jin SG, Kim DD, Lyoo WS, Yong CS, Choi HG. Int. 2007. p. 134. COI number [1:CAS:528:DC%2BD2sXhtlWnt7zO] [DOI] [PubMed]
- Barthe L, Woodley J, Houin G. Fundam. 1999. p. 154. COI number [1:CAS:528:DyaK1MXitlGlsr0%3D] [DOI] [PubMed]
- Liu J, Xiao Y, Allen C. J. 2004. p. 132. COI number [1:CAS:528:DC%2BD2cXjvFWnsw%3D%3D] [DOI] [PubMed]
- Zhang X, Jackson JK, Burt HM. Int. 1996. p. 195. [DOI]
- Chen SM, Liao JF, Kuo CD, Ho LT. Nephron. 2004. p. 113. [DOI] [PubMed]
- Deferme S, Mols R, Van Driessche W, Augustijns P. J. 2002. p. 2539. COI number [1:CAS:528:DC%2BD38XptVeksLc%3D] [DOI] [PubMed]
- Van Gelder J, Deferme S, Annaert P, Naesens L, De Clercq E, Van den Mooter G, Kinget R, Augustijns P. Drug Metab. 2000. p. 1394. [PubMed]
- Annaert P, Tukker JJ, van Gelder J, Naesens L, de Clercq E, Van den Mooter G, Kinget R, Augustijns P. J. 2000. p. 1054. COI number [1:CAS:528:DC%2BD3cXmtVWmsLY%3D] [DOI] [PubMed]
- Santos CA, Jacob JS, Hertzog BA, Freedman BD, Press DL, Harnpicharnchai P, Mathiowitz E. J. Control. Release. 1999. p. 113. COI number [1:CAS:528:DyaK1MXlsFOku7Y%3D] [DOI] [PubMed]
- Shin IG, Kim SY, Lee YM, Cho CS, Sung YK. J. Control. Release. 1998. p. 1. COI number [1:CAS:528:DyaK1cXptFSgsw%3D%3D] [DOI] [PubMed]
- Kim SY, Shin IG, Lee YM, Cho CS, Sung YK. J. Control. Release. 1998. p. 13. COI number [1:STN:280:DyaK1czltVGitQ%3D%3D] [DOI] [PubMed]
- Li S, Vert M. Macromolecules. 2003. p. 8008. COI number [1:CAS:528:DC%2BD3sXnsFyqtbc%3D]; Bibcode number [2003MaMol..36.8008L] [DOI]
- Hu Y, Jiang X, Ding Y, Zhang L, Yang C, Zhang J, Chen J, Yang Y. Biomaterials. 2003. p. 2395. COI number [1:CAS:528:DC%2BD3sXivVCqsb0%3D] [DOI] [PubMed]
- Pade V, Stavchansky S. Pharm. 1997. p. 1210. COI number [1:CAS:528:DyaK2sXmsFehurY%3D] [DOI] [PubMed]
- Cheon Lee S, Kim C, Chan Kwon I, Chung H, Young Jeong S. J. Control. Release. 2003. p. 437. [DOI] [PubMed]
- Dong Y, Feng SS. Biomaterials. 2005. p. 6068. COI number [1:CAS:528:DC%2BD2MXlt1Wgtro%3D] [DOI] [PubMed]
- Sant VP, Smith D, Leroux JC. J. Control. Release. 2004. p. 301. COI number [1:CAS:528:DC%2BD2cXkslKmtbY%3D] [DOI] [PubMed]
- Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. J. Control. Release. 2005. p. 169. COI number [1:CAS:528:DC%2BD2MXht12jt7nM] [DOI] [PubMed]
- Kohori F, Yokoyama M, Sakai K, Okano T. J. Control. Release. 2002. p. 155. COI number [1:CAS:528:DC%2BD3MXptlaitr0%3D] [DOI] [PubMed]
- Lavasanifar A, Samuel J, Kwon GS. J. Control. Release. 2001. p. 155. COI number [1:CAS:528:DC%2BD3MXnvVyhsLo%3D] [DOI] [PubMed]
- Ge H, Hu Y, Jiang X, Cheng D, Yuan Y, Bi H, Yang C. J. 2002. p. 1463. COI number [1:CAS:528:DC%2BD38XktlWrs7o%3D] [DOI] [PubMed]
- Gao Z, Lukyanov AN, Singhal A, Torchilin VP. Nano Lett. 2001. p. 979. Bibcode number [2002NanoL...2..979G] [DOI]
- Yang ZL, Li XR, Yang KW, Liu Y. J. 2008. p. 539. [DOI] [PubMed]
- Zhang Y, Jin T, Zhuo RX. Colloids Surf. B. 2005. p. 104. COI number [1:CAS:528:DC%2BD2MXntVCqur0%3D] [DOI] [PubMed]
- Yang KW, Li XR, Yang ZL, Li PZ, Wang F, Liu Y. J. 2009. p. 140. [DOI] [PubMed]
- Jeong YI, Cheon JB, Kim SH, Nah JW, Lee YM, Sung YK, Akaike T, Cho CS. J. Control. Release. 1998. p. 169. COI number [1:CAS:528:DyaK1cXot12lsA%3D%3D] [DOI] [PubMed]
- Lee J, Cho EC, Cho K. J. Control. Release. 2004. p. 323. COI number [1:CAS:528:DC%2BD2cXmtFCruw%3D%3D] [DOI] [PubMed]
- Mathot F, des Rieux A, Arien A, Schneider YJ, Brewster M, Preat V. J. Control. Release. 2007. p. 134. COI number [1:CAS:528:DC%2BD2sXhtlWmsbrO] [DOI] [PubMed]