

Abstract
The development of miniaturized cell culture platforms for performing parallel cultures and combinatorial assays is important in cell biology from the single-cell level to the system level. In this paper we developed an integrated microfluidic cell-culture platform, Cell-microChip (Cell-μChip), for parallel analyses of the effects of microenvir-onmental cues (i.e., culture scaffolds) on different mammalian cells and their cellular responses to external stimuli. As a model study, we demonstrated the ability of culturing and assaying several mammalian cells, such as NIH 3T3 fibro-blast, B16 melanoma and HeLa cell lines, in a parallel way. For functional assays, first we tested drug-induced apoptotic responses from different cell lines. As a second functional assay, we performed "on-chip" transfection of a reporter gene encoding an enhanced green fluorescent protein (EGFP) followed by live-cell imaging of transcriptional activation of cyclooxygenase 2 (Cox-2) expression. Collectively, our Cell-μChip approach demonstrated the capability to carry out parallel operations and the potential to further integrate advanced functions and applications in the broader space of combinatorial chemistry and biology.
Keywords: Microfluidic devices, Cell-based assay, Apoptosis, Transfection, Cell culture
1 Introduction
Conventional cell-based experiments are typically performed on a large cell population. Researchers have begun to appreciate that there are local variables associated with the heterogeneous microenvironment in the macroscopic culture setting, which often leads to experimental inconsistency. On the other hand, differences among individual cells are often ignored since the conventional assays (e.g., Weston blots and microarray analysis) are conducted at a collective fashion. As a result, it is challenging to elucidate complex cellular systems and analyze dynamic signaling pathways (Irish et al. 2006) using the conventional experiment systems. To overcome these challenges, it is essential to develop a new technology platform to enable (1) improved control on the cell culture microenvironment, (2) precise cell assays with the single-cell resolution, and (3) sequential and parallel operation by combining those mentioned in (1) and (2). We envision such a technology can be applied for screening drug candidates (Dittrich and Manz 2006; Padron et al. 2000), evaluating biological pathways (Minor 2003), and understanding pharmacological effects (Hill et al. 1998; Umezawa 2005), thus constituting critical technological foundations for a broad spectrum of biomedical research.
Microfluidic devices (Auroux et al. 2002; Dittrich and Manz 2006; Dittrich et al. 2006; Reyes et al. 2002) offer a robust analytical tool that allows rapid analysis of cellular responses to external stimuli in a parallel way. Moreover, microfluidics, with its intrinsic advantages of sample/reagent economy, precise control over physical and chemical micro-environments, high throughput, scalability and digital controllability, whose features allow microfluidics to investigate complex and dynamic biological processes at the single-cell level. In addition, microscale cell culture using microfluidics allows investigating the function of microenvironmental cues at the single-cell level. Especially, compared to static microfluidic cell-culture systems, integrated microfluidics allows for the control of adding and/or removing of biochemical cues at specific temporal as well as spatial points. This unique advantage makes it possible to do novel microenvironmental experiments, such as cell–cell interactions and extracellular matrix (ECM)–cell interactions. However, there are several issues associated with the fabrication and control of integrated microfluidic cell culture systems in order to achieve routine cellular assays of mammalian cells. Even though microfluidic cell culture systems have been developed extensively, they can mainly be described into two ways: what kinds of cell culture platforms were used, and what types of cells and applications were tested. Many different cell culture platforms have been developed, including two-dimensional, three-dimensional (Cartmell et al. 2003; Toh et al. 2007) and co-culture platforms (Sin et al. 2004). Those microfluidic approaches enabled to culture and assay many different cells, such as liver (Kane et al. 2006; Sin et al. 2004; Zhang et al. 2008), muscle (Tourovskaia et al. 2005; Tourovskaia et al. 2006), neural (Millet et al. 2007; Park et al. 2006), and stem cells (Chung et al. 2005; Gomez-Sjoberg et al. 2007; Kim et al. 2006). Despite these recent advances, several critical questions and several challenges should be addressed more explicitly, and optimized to fully achieve the potential of microfluidic cell culture and assays. It requires the abilities: (1) to test a robust and flexible fluidic configuration (e.g., flow through vs. circulation) for cell culture and (2) to fabricate microfluidic network capable of performing sequential and parallel operations such as parallel culture of multiple cell types and subsequent phenotypic and functional assays in the same microfluidic chip. In this context, incorporation of isolation valves (Unger et al. 2000) and peristaltic pumps (Chou et al. 2001) should allow individual addressability and digital controllability (Lee et al. 2005; Wang et al. 2006) of each cell culture chamber embedded on a microfluidic device, which in turn should enable complex manipulations of the culture microenvironments as well as multiple analytical measurements.
Over the years, there have been a variety of microfluidic chips directed for functional biological assays. These include differentiation of cell through different flow rates (Gu et al. 2004), fully automated cell culture system by two-layer PDMS chips (Gomez-Sjoberg et al. 2007), optimization of drug cocktail to regulate cell activities through closed-loop control algorithm and microfluidic platform (Wong et al. 2008), and modelling of galactose pathway in an alternating culture environment (Bennett et al. 2008). All these devices or systems have provided additional modules and thus are superior to the conventional setting on conducting biological research or routine operations.
In this paper, we describe the design and operation of polydimethylsiloxane (PDMS)-based Cell-microChip (Cell-μChip, Fig. 1) envisioned as a digitally controlled platform for performing parallel cell culture and sequential cellular assays. The potential of the Cell-μChip to support the optimal culture and assays of human and murine cell lines was demonstrated. To determine the optimal culture conditions, these cells were cultured in six cell culture chambers embedded on a Cell-μChip under two different medium supply modes in parallel (i.e., circulation and direct feeding). The growth and viability of the micro-cultures were monitored and quantified in real time in the Cell-μChip using an integrated CCD camera. Sequential staining with acridine orange (AO) and propidium iodide (PI) (Hudson et al. 1969; Traganos et al. 1977) allowed the identification of viable and dead cells, respectively. To demonstrate the ability to perform "on chip" functional assays, we analyzed drug-induced apoptotic responses. Furthermore, we showed that the Cell-μChip is amenable to complex sequential operations such as genetic manipulation and monitoring of transcriptional activation of gene expression.
Fig. 1.

(a) Schematic representation of an integrated Cell-microChip (Cell-μChip) for performing multiple cell culture and assays under a digitally controlled interface. Three pairs of parallel-oriented cell culture chambers are incorporated in a Cell-μChip, where multiple cell types can be cultured under two different modes of medium supply, i.e., circulatory (channels i, iii and v) and direct feeding (channels ii, iv and vi). The operation of this microchip is controlled by pressure driven valves with their delegated functions indicated by their colors: red for regular valve (for isolation and gating) and yellow for pumping valve (for fluid transport and circulation). (b) Optical image of the actual device. The microchip was loaded with various colors of food dyes to enhance the visualization of different parts in the entire system: red and yellow as in (a); blue indicates the flow channel and the medium reservoir
2 Experimental
2.1 Microfluidic cell culture
Using the integrated valves and pumps, bovine fibronectin (FN) (Sigma) solution (1.0 mg mL−1) filled in Teflon tubing was introduced into the six cell culture chambers of the Cell-μChip. The Cell-μChip was kept at 37°C for 30 min for FN coating. DMEM was then introduced into the cell culture chambers to extrude the FN solution. The medium reservoirs and medium tubings were then dead-end filled with DMEM at 10 psi for 60 min (Song et al. 2008). Then, individual cell suspensions (NIH 3T3, HeLa and B16) with 2×106 cells mL−1 were sequentially introduced by gravitation into the cell culture chambers. After cell loading, cells located outside the culture chambers were removed by washing with fresh DMEM. The Cell-μChip was placed in an incubator for 6 h. The pump was turned on to introduce the DMEM in a circulating or feed through fashion in the respective culture chambers. The flow rate of the medium was controlled in the range of 0.1–4 nL s−1. Cell growth was monitored by collecting bright field micrographs of cells inside the Cell-μChip at 12-h intervals.
2.2 Immunoassay for fibronectin
To confirm FN coating efficiency in cell culture chambers, immunoassay for FN was performed. After FN coating in a Cell-μChip, blocking solution containing 5% BSA and 0.1% N-dodecyl-β-D-maltoside (DDM) (Pierce) was introduced in a Cell-μChip, and then kept at room temperature for 1 h. Mouse anti-FN (BD Biosciences) was loaded into cell culture chambers, and incubated at room temperature for 2 h. Excess antibody in cell culture chambers were rinsed with PBS with 0.1% Tween 20 (PBS-T) twice. Then secondary goat anti-mouse IgG conjugated with Alexa555 (Invitrogen) were introduced in cell culture chambers and incubated at room temperature for 1 h. After washing with PBS-T twice, fluorescent intensity was measured with a fluorescent microscope, and quantified with MetaMorph.
2.3 AO/PI fluorescence staining
After culturing the cells in the Cell-μChip for 4 days, a solution composed of DMEM, AO and PI in a ratio of 10:1:1 was introduced into the cell culture chambers. After 5 min incubation at 37°C, the cells were imaged under a fluorescence microscope.
2.4 Apoptosis assay
NIH 3T3, HeLa and B16 cells were cultured in the cell culture chambers of the Cell-μChip for 24 h. Staurosporine (0, 0.1, 1 and 10 μM as final concentrations) or actinomycin D (0, 0.1, 1 and 10 μM as final concentrations) in DMEM culture medium were loaded into the cell culture chambers. After 2 h incubation, the medium in all cell culture chambers was replaced by the MitoTracker Red dye (Invitrogen) for staining of viable cells. The Cell-μChip was incubated for 30 min at 5% CO2, 37°C.Sequentially, the MitoTracker Red solution was replaced by a solution containing 100 μL of Annexin binding buffer and 5 μL of Annexin V-Alexa488 for staining the apoptotic cells. Following 15 min incubation at RT, cell culture chambers were flushed with Annexin binding buffer and the cells were imaged using a fluorescence microscope.
2.5 On-chip transfection and reporter gene imaging
NIH 3T3 cells were loaded into the six culture chambers of the Cell-μChip and were allowed to settle for 24 h. Cells were transfected with the pCox2-EGFP plasmid (a kind gift from Prof. Harvey R. Herschman, UCLA), encoding an enhanced green fluorescent protein (EGFP) under a murine Cox-2 promoter (Liang et al. 2004). The transfection mixture containing plasmid (0.5 μg), medium (30 μL) and transfection reagent (2.5 μL, Superfect reagent, Qiagen) was incubated at RT for 10 min. The mixture was further diluted with 150 μL of DMEM and loaded into all culture chambers of the Cell-μChip. After 3-h incubation at 5% CO2, 37°C, the mixture was replaced with serum-free DMEM and cells were incubated for an additional day. To activate the Cox-2 promoter the culture media in three out of six chambers was replaced with media containing the induction agent TPA (50 ng mL−1). Following 7 h incubation at 5% CO2, 37°C, EGFP expression was imaged using a fluorescence microscope.
3 Results and discussion
3.1 Design of the Cell-μChip
The PDMS-based Cell-μChips (Fig. 1) were fabricated by multilayer soft lithography approach (see supplementary information) (Unger et al. 2000; Xia and Whitesides 1998). It is important to note that the biocompatible and gas-permeable properties (Kim et al. 2007; Korin et al. 2007) of PDMS matrices help to retain proper physiological conditions for a wide range of mammalian cells suitable for different screening applications. A fluidic network for individually addressable cell culture chambers and solution/reagent transport was integrated with embedded pneumatic valves and peristaltic pumps. This design enabled to digitally control sequential operations. The chip consisted of three identical pairs of parallel-oriented culture chambers with identical dimensions (3×0.5×0.1 mm3, corresponding to a volume of 150 nL). To allow synchronized pumping, six internally connected peristaltic pumps were incorporated at the ends of the six cell culture chambers.
Each pair of culture chambers was configured to have two types of medium supplies: one allowing media recirculation through the culture chamber for cellular auto-conditioning and the other enabling direct feeding of cells with fresh media. The size of medium reservoir could accommodate about 10 μL of culture media, a volume sufficient to sustain continuous on-chip cell culture for 8 days. In contrast, supply Teflon tubings were utilized to store and deliver fresh culture media. This design allowed us to perform six cell culture experiments in a closely related microenvironment under two different culture media supply modes.
3.2 Surface modification with fibronectin
We initially used bare PDMS surface to seed cells, however, we found that cells either could not attach to the surface well or they detached so easily when new fresh media were supplied. We reasoned that this problem was due to the inherent hydrophobicity of PDMS materials. Thus, we tested to use ECMs in order to make the surface hydrophilic and biocompatible for cell adhesion. In our searching optimal ECMs, FN is effectively coated on the PDMS surface in our Cell-μChip (Fig. 2). Several different conditions, such as different FN concentrations and incubation time, were tested to optimize the FN coating condition. We confirm the efficiency and homogeneity of FN coating on PDMS by immunoassay. As a result, 1 mg mL−1 of FN for 30 min incubation at 37°C is the optimal condition in a Cell-μChip.
Fig. 2.

Fibronectin coating efficiency on the PDMS surface in a Cell-μChip determined by immunofluorescence assay. (a) Fluorescence images of immunostained FN on the PDMS surface. (b) Quantitative analysis of FN coating efficiency determined with fluorescence images shown in (a)
3.3 Cell culture in the Cell-μChip
We initially demonstrated to culture NIH 3T3 cells in all culture chambers of a Cell-μChip (Fig. 3, Supplementary Information and Fig. S1). Generally, following loading on the chip, it took about 1 h for cells to attach and spread onto the FN-coated culture chamber surfaces. NIH 3T3 cells grew to confluence, and the whole microchannel was fully occupied at day 8. During this period, we continuously monitored cell growth inside the Cell-μChip using a CCD camera (see ESM Movie 1). Interestingly, cells in a Cell-μChip grew on the bottom as well as ceiling. This phenomenon is not allowed under conventional culture conditions using Petri dishes. This observation indicated that a Cell-μChip could provide unique and intrinsic characteristics of cell culture manipulations. To determine cell viability we performed AO/PI fluorescence staining (Fig. 3(b)–(e)). AO staining (Fig. 3(c)) indicated that the majority of cells in the chamber were viable. PI staining (Fig. 3(d)) showed a small percentage of dead cells.
Fig. 3.

Long term culture of NIH 3T3 cells in a Cell-μChip. (a) Time lapse images of the NIH 3T3 cell proliferation in the microchip in a duration of 8 days. (b)–(e) Dead (PI)/live (AO) staining of NIH 3T3 cells cultured in a Cell-μChip for 4 days. (b) A bright field micrograph of NIH 3T3 cell morphologies. (c) A green fluorescence micrograph of the live-stained cells. (d) A red fluorescence micrograph of the dead-stained cells. (e) A merged fluorescence image of (b) and (c)
3.4 Parallel culture of multiple cell lines in a single Cell-μChip
To demonstrate this concept, we cultured NIH 3T3, HeLa and B16 cells in a single Cell-μChip (Fig. 4). NIH 3T3, HeLa and B16 cells were sequentially loaded into the pairs of culture chambers; i–ii, iii–iv and v–vi accordingly. After cell adhesion, the six peristaltic pumps were turned on to feed cells with the medium, in either recirculation or direct feeding setting. The results were consistent with those observed for the parallel culture of NIH 3T3 cells. Cell numbers increased significantly for all three cell lines. To examine cell viability in the three reservoir-attached cell culture chambers, we performed sequential PI/AO staining. As a result, most of the cells are viable, and few dead cells were observed (data not shown). To quantify the culture parameters in the Cell-μChip we sought to determine growth rate for the three cell types (Fig. 4(d)). We observed that the curves reflected a linear growth phase until the cells reached confluence followed by a stationary phase. Comparing the cell growth with medium-recirculation or direct feeding setting, first we used NIH 3T3 cells and continuously monitored them. There was no clear difference in NIH 3T3 cell growth between the two settings (Fig. 4(d)). In the case of HeLa and B16 cells, we obtained similar results as NIH 3T3 cells (data not shown). This result indicates that a Cell-μChip serves a platform to perform multiple cell culture in a single device.
Fig. 4.

Demonstration of six parallel cell cultures in a closely related microenvironment. After 3 days culture, the cell morphologies were shown in (a) NIH 3T3, (b) HeLa and (c) B16. (d) Growth curves of chip-cultured NIH 3T3, HeLa and B16 cells were quantified by monitoring the number of cells inside the cell culture chambers over time. After 5 days culture, we could not count cell number precisely due to cell confluence and multiple cell layers in the cell culture chambers
3.5 On-chip apoptosis assay
Apoptosis is not only fundamentally involved in developing cells and maintaining tissue homeostasis, but also can be closely related to several diseases including cancer, autoimmune, and neurodegeneration. Even though, extensive studies have been reported to dissect apoptosis's molecular basis, more system level as well as single-cell level analysis by using integrated microfluidics would bring new insights for the underlying mechanisms of these biological processes. To demonstrate the capability of "on chip" functional analyses with the Cell-μChip, we performed a drug-induced apoptosis assay (Fig. 5). We used two kinds of apoptosis inducers, staurosporine (ST) (Rajotte et al. 1992; Tafani et al. 2001; Wang et al. 1996) and actinomycin D (AD) (Martin et al. 1990). NIH 3T3, HeLa and B16 cells were treated with apoptosis inducers at four concentrations (0, 0.1, 1 and 10 μM) in the Cell-μChips. ST or AD treated cells increased apoptotic cell population with a dose-dependent manner of apoptosis inducers. Although cell viability among untreated and treated cells appeared to be the same based on the MitoTracker staining, the cells underwent the apoptosis process at various speeds according to the drug concentration, as demonstrated by the Annexin V staining. We conclude that the Cell-μChip served as a platform to perform multiparametric functional assays.
Fig. 5.
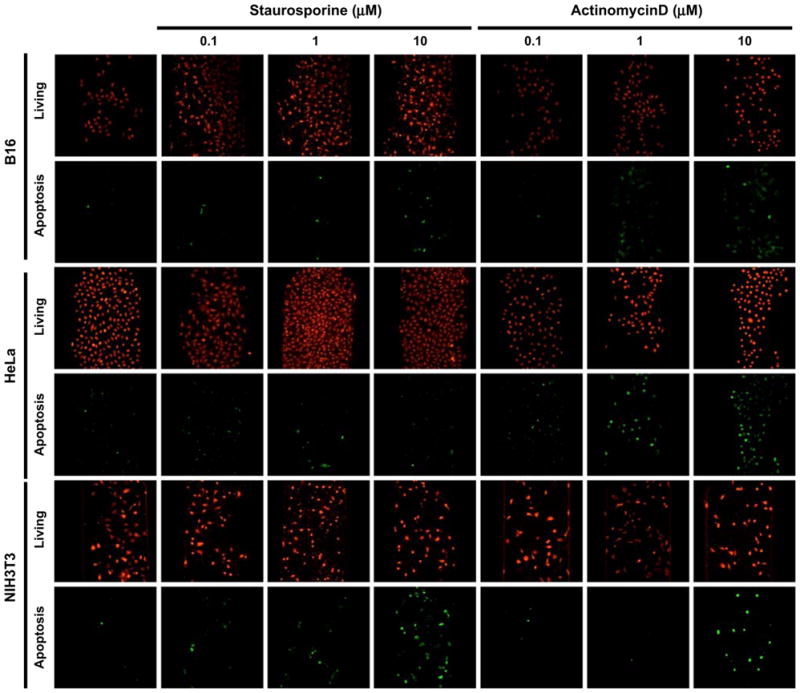
Multiparametric apoptosis assays performed in the Cell-μChips. NIH 3T3, HeLa and B16 cells were treated with either staurosporine or actinomycin D to induce apoptosis. Apoptotic cells were stained with Annexin V conjugated with Alexa488 (green), and living cells were stained with MitoTracker (red)
3.6 On-chip transfection and monitoring of reporter gene expression
Plasmid DNA transfection is one of the common methods to manipulate gene expression in mammalian cells. However, the detailed conditions of optimal transfection for different cell lines are variable. Combinatorial approach from parallel microfluidic cellular assays can help to identify the optimal condition. To demonstrate the feasibility of performing the other assay in the Cell-μChip, gene transfection experiments were carried out (Fig. 6). For proof-of-concept, we used a plasmid vector with an enhanced green fluorescent protein (EGFP) driven by a cyclooxygenase-2 (Cox-2) promoter. Therefore, EGFP expression serves as a reporter of Cox-2 transcription. The basal expression level of Cox-2 in NIH 3T3 cells is very low. Tetradecanoylphorbol acetate (TPA) can activate Cox-2 transcription. EGFP expression in these cells was monitored under a fluorescence microscope. As shown in Fig. 6, even though weak EGFP signals were detected in the negative control chambers, strong EGFP signals were observed in TPA-induced transfected cells in the corresponding cell culture chambers.
Fig. 6.
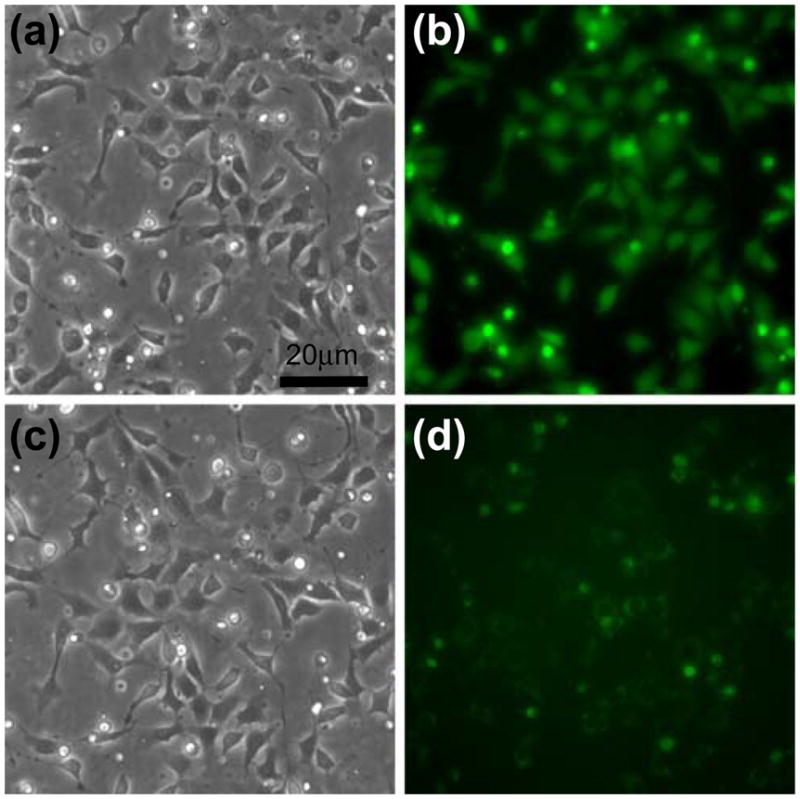
On-chip transfection and EGFP induction in NIH 3T3 cells. The plasmid vector which encodes EGFP driven by a Cox-2 promoter was transfected with NIH 3T3 cells. (a) Bright field and (b) fluorescence images of NIH 3T3 cells stimulated with TPA for 7 h. (c) Bright field and (d) fluorescence images of NIH 3T3 cells without stimulation
4 Conclusion
In summary, we developed a fully digitally controlled microfluidic cell culture and assay platform that could support parallel cell culture and sequential cell assays. Through the integration of isolation valves, murine and human cells lines could be cultured in different cell culture chambers and tested for different conditions of cell culture and assays in a single Cell-μChip. Real-time monitoring of cell morphology and numbers, viability assay, apoptosis assay and transfection to monitor expression of a reporter gene vector were also performed using the same platform. Our results indicate that intrinsic advantages of microfluidic devices enable the execution of complicated and integrated biological operations in stand-alone devices such as the Cell-μChip. We envision that this platform will be further integrated with advanced functions and utilities for more sophisticated cell culture applications.
Supplementary Material
Acknowledgments
This research was supported by the NIH NanoSystems Biology Cancer Center, the DOE-UCLA Institute of Molecular Medicine and the NIH-UCLA Center for In Vivo Imaging in Cancer Biology and Siemens Medical Solutions USA Inc. We thank Stephanie M. Shelly, Dan Rohle, Shirley Quan and Mireille Riedinger for the outstanding technical support with conventional cell culture conditions. ONW is an Investigator of the Howard Hughes Medical Institute. CGR was supported by a Developmental Project Award (ICMIC, NIH/ NCI grant no. CA08630). C.J.S. was supported by a National Institutes of Health (NIH) Research Training in Pharmacological Sciences training grant PHS T32 CM008652.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10544-008-9260-x) contains supplementary material, which is available to authorized users.
Contributor Information
Zeta Tak For Yu, Department of Mechanical and Aerospace Engineering, University of California, Los Angeles, CA 90095, USA. Crump Institute for Molecular Imaging, University of California, Los Angeles, CA 90095, USA. Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, USA.
Ken-ichiro Kamei, Crump Institute for Molecular Imaging, University of California, Los Angeles, CA 90095, USA. Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, USA.
Hiroko Takahashi, Crump Institute for Molecular Imaging, University of California, Los Angeles, CA 90095, USA.
Chengyi Jenny Shu, Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, CA 90095, USA.
Xiaopu Wang, Crump Institute for Molecular Imaging, University of California, Los Angeles, CA 90095, USA. Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, USA.
George Wenfu He, Crump Institute for Molecular Imaging, University of California, Los Angeles, CA 90095, USA. Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, USA.
Robert Silverman, Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, USA.
Caius G. Radu, Email: caiusradu@ucla.edu, Crump Institute for Molecular Imaging, University of California, Los Angeles, CA 90095, USA. Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, USA
Owen N. Witte, Email: owenw@microbio.ucla.edu, Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, USA. Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, CA 90095, USA. The Howard Hughes Medical Institute, University of California, Los Angeles, CA 90095, USA
Ki-Bum Lee, Email: kblee@rci.rutgers.edu, Department of Chemistry and Chemical Biology, Institute for Advanced Materials, Devices and Nanotechnology, The Rutgers Stem Cell Research Center, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA.
Hsian-Rong Tseng, Email: hrtseng@mednet.ucla.edu, Crump Institute for Molecular Imaging, University of California, Los Angeles, CA 90095, USA. Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, USA.
References
- Auroux PA, Iossifidis D, Reyes DR, Manz A. Micro total analysis systems. 2. Analytical standard operations and applications. Anal Chem. 2002;74:2637–2652. doi: 10.1021/ac020239t. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Pang WL, Ostroff NA, Baumgartner BL, Nayak S, Tsimring LS, Hasty J. Metabolic gene regulation in a dynamically changing environment. Nature. 2008;454:1119–1122. doi: 10.1038/nature07211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell SH, Porter BD, Garcia AJ, Guldberg RE. Effects of medium perfusion rate on cell-seeded three-dimensional bone constructs in vitro. Tissue Eng. 2003;9:1197–1203. doi: 10.1089/10763270360728107. [DOI] [PubMed] [Google Scholar]
- Chou HP, Unger MA, Quake SR. A microfabricated rotary pump. Biomed Microdevices. 2001;3(4):323–330. doi: 10.1023/ A:1012412916446. [DOI] [Google Scholar]
- Chung BG, Flanagan LA, Rhee SW, Schwartz PH, Lee AP, Monuki ES, Jeon NL. Human neural stem cell growth and differentiation in a gradient-generating microfluidic device. Lab Chip. 2005;5:401–406. doi: 10.1039/b417651k. [DOI] [PubMed] [Google Scholar]
- Dittrich PS, Manz A. Lab-on-a-chip: microfluidics in drug discovery. Nat Rev Drug Discov. 2006;5:210–218. doi: 10.1038/nrd1985. [DOI] [PubMed] [Google Scholar]
- Dittrich PS, Tachikawa K, Manz A. Micro total analysis systems. Latest advancements and trends. Anal Chem. 2006;78:3887–3907. doi: 10.1021/ac0605602. [DOI] [PubMed] [Google Scholar]
- Gomez-Sjoberg R, Leyrat AA, Pirone DM, Chen CS, Quake SR. Versatile, fully automated, microfluidic cell culture system. Anal Chem. 2007;79:8557–8563. doi: 10.1021/ac071311w. [DOI] [PubMed] [Google Scholar]
- Gu W, Zhu X, Futai N, Cho BS, Takayama S. Computerized microfluidic cell culture using elastomeric channels and Braille displays. Proc Natl Acad Sci U S A. 2004;101:15861–15866. doi: 10.1073/pnas.0404353101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DC, Wrigley SK, Nisbet LJ. Novel screen methodologies for identification of new microbial metabolites with pharmacological activity. Adv Biochem Eng Biotechnol. 1998;59:73–121. doi: 10.1007/BFb0102297. [DOI] [PubMed] [Google Scholar]
- Hudson B, Upholt WB, Devinny J, Vinograd J. The use of an ethidium analogue in the dye-buoyant density procedure for the isolation of closed circular DNA: the variation of the superhelix density of mitochondrial DNA. Proc Natl Acad Sci U S A. 1969;62:813–820. doi: 10.1073/pnas.62.3.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish JM, Kotecha N, Nolan GP. Mapping normal and cancer cell signalling networks: towards single-cell proteomics. Nat Rev Cancer. 2006;6:146–155. doi: 10.1038/nrc1804. [DOI] [PubMed] [Google Scholar]
- Kane BJ, Zinner MJ, Yarmush ML, Toner M. Liver-specific functional studies in a microfluidic array of primary mammalian hepatocytes. Anal Chem. 2006;78:4291–4298. doi: 10.1021/ ac051856v. [DOI] [PubMed] [Google Scholar]
- Kim L, Vahey MD, Lee HY, Voldman J. Microfluidic arrays for logarithmically perfused embryonic stem cell culture. Lab Chip. 2006;6:394–406. doi: 10.1039/b511718f. [DOI] [PubMed] [Google Scholar]
- Kim L, Toh YC, Voldman J, Yu H. A practical guide to microfluidic perfusion culture of adherent mammalian cells. Lab Chip. 2007;7:681–694. doi: 10.1039/b704602b. [DOI] [PubMed] [Google Scholar]
- Korin N, Bransky A, Dinnar U, Levenberg S. A parametric study of human fibroblasts culture in a microchannel bioreactor. Lab Chip. 2007;7:611–617. doi: 10.1039/b702392h. [DOI] [PubMed] [Google Scholar]
- Lee CC, Sui G, Elizarov A, Shu CJ, Shin YS, Dooley AN, Huang J, Daridon A, Wyatt P, Stout D, Kolb HC, Witte ON, Satyamurthy N, Heath JR, Phelps ME, Quake SR, Tseng HR. Multistep synthesis of a radiolabeled imaging probe using integrated microfluidics. Science. 2005;310:1793–1796. doi: 10.1126/science.1118919. [DOI] [PubMed] [Google Scholar]
- Liang Q, Yamamoto M, Curiel DT, Herschman HR. Noninvasive imaging of transcriptionally restricted transgene expression following intratumoral injection of an adenovirus in which the COX-2 promoter drives a reporter gene. Mol Imaging Biol. 2004;6:395–404. doi: 10.1016/j.mibio.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Lennon SV, Bonham AM, Cotter TG. Induction of apoptosis (programmed cell death) in human leukemic HL-60 cells by inhibition of RNA or protein synthesis. J Immunol. 1990;145:1859–1867. [PubMed] [Google Scholar]
- Millet LJ, Stewart ME, Sweedler JV, Nuzzo RG, Gillette MU. Microfluidic devices for culturing primary mammalian neurons at low densities. Lab Chip. 2007;7:987–994. doi: 10.1039/b705266a. [DOI] [PubMed] [Google Scholar]
- Minor LK. Assays to measure the activation of membrane tyrosine kinase receptors: focus on cellular methods. Curr Opin Drug Discov Devel. 2003;6:760–765. [PubMed] [Google Scholar]
- Padron JM, van der Wilt CL, Smid K, Smitskamp-Wilms E, Backus HH, Pizao PE, Giaccone G, Peters GJ. The multilayered postconfluent cell culture as a model for drug screening. Crit Rev Oncol Hematol. 2000;36:141–157. doi: 10.1016/S1040-8428(00)00083-4. [DOI] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. Microfluidic culture platform for neuroscience research. Nat Protoc. 2006;1:2128–2136. doi: 10.1038/nprot.2006.316. [DOI] [PubMed] [Google Scholar]
- Rajotte D, Haddad P, Haman A, Cragoe EJ, Jr, Hoang T. Role of protein kinase C and the Na+/H+ antiporter in suppression of apoptosis by granulocyte macrophage colony-stimulating factor and interleukin-3. J Biol Chem. 1992;267:9980–9987. [PubMed] [Google Scholar]
- Reyes DR, Iossifidis D, Auroux PA, Manz A. Micro total analysis systems. 1. Introduction, theory, and technology. Anal Chem. 2002;74:2623–2636. doi: 10.1021/ac0202435. [DOI] [PubMed] [Google Scholar]
- Sin A, Chin KC, Jamil MF, Kostov Y, Rao G, Shuler ML. The design and fabrication of three-chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol Prog. 2004;20:338–345. doi: 10.1021/bp034077d. [DOI] [PubMed] [Google Scholar]
- Song WH, Kwan J, Kaigala GV, Hoang VN, Backhouse CJ. Readily integrated, electrically controlled microvalves. J Micromechanics Microengineering. 2008;8:1071–1078. doi: 10.1039/b802853b. [DOI] [PubMed] [Google Scholar]
- Tafani M, Minchenko DA, Serroni A, Farber JL. Induction of the mitochondrial permeability transition mediates the killing of HeLa cells by staurosporine. Cancer Res. 2001;61:2459–2466. [PubMed] [Google Scholar]
- Toh YC, Zhang C, Zhang J, Khong YM, Chang S, Samper VD, van Noort D, Hutmacher DW, Yu H. A novel 3D mammalian cell perfusion-culture system in microfluidic channels. Lab Chip. 2007;7:302–309. doi: 10.1039/b614872g. [DOI] [PubMed] [Google Scholar]
- Tourovskaia A, Figueroa-Masot X, Folch A. Differentiation-on-a-chip: a microfluidic platform for long-term cell culture studies. Lab Chip. 2005;5:14–19. doi: 10.1039/b405719h. [DOI] [PubMed] [Google Scholar]
- Tourovskaia A, Figueroa-Masot X, Folch A. Long-term microfluidic cultures of myotube microarrays for high-throughput focal stimulation. Nat Protoc. 2006;1:1092–1104. doi: 10.1038/ nprot.2006.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traganos F, Darzynkiewicz Z, Sharpless T, Melamed MR. Simultaneous staining of ribonucleic and deoxyribonucleic acids in unfixed cells using acridine orange in a flow cytofluorometric system. J Histochem Cytochem. 1977;25:46–56. doi: 10.1177/25.1.64567. [DOI] [PubMed] [Google Scholar]
- Umezawa Y. Genetically encoded optical probes for imaging cellular signaling pathways. Biosens Bioelectron. 2005;20:2504–2511. doi: 10.1016/j.bios.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- Wang X, Zelenski NG, Yang J, Sakai J, Brown MS, Goldstein JL. Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO J. 1996;15:1012–1020. [PMC free article] [PubMed] [Google Scholar]
- Wang J, Sui G, Mocharla VP, Lin RJ, Phelps ME, Kolb HC, Tseng HR. Integrated microfluidics for parallel screening of an in situ click chemistry library. Angew Chem Int Ed Engl. 2006;45:5276–5281. doi: 10.1002/anie.200601677. [DOI] [PubMed] [Google Scholar]
- Wong PK, Yu F, Shahangian A, Cheng G, Sun R, Ho CM. Closed-loop control of cellular functions using combinatory drugs guided by a stochastic search algorithm. Proc Natl Acad Sci U S A. 2008;105:5105–5110. doi: 10.1073/pnas. 0800823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia YN, Whitesides GM. Soft lithography. Annu Rev Mater Sci. 1998;28:153–184. doi: 10.1146/annurev.matsci.28.1.153. [DOI] [Google Scholar]
- Zhang MY, Lee PJ, Hung PJ, Johnson T, Lee LP, Mofrad MR. Microfluidic environment for high density hepatocyte culture. Biomed Microdevices. 2008;10:117–121. doi: 10.1007/s10544-007-9116-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.