

Abstract
Ribbon-containing neurons represent a subset of neural cells that undergo graded membrane depolarizations rather than Na+-channel evoked action potentials. Bipolar cells of the retina are one type of ribbon-containing neuron and extensive research has demonstrated kinetically distinct pools of vesicles that are released and replenished in a calcium-dependent manner. In this study, we look at the properties of the fastest pool of releasable vesicles in these cells, often referred to as the immediately releasable pool (IRP), to investigate the relationships between vesicle release and calcium channels in these terminals. Using whole cell capacitance measurements, we monitored exocytosis in response to different magnitude and duration depolarizations, with emphasis on physiologically relevant step depolarizations. We find that release rate of the IRP increases superlinearly with membrane potential and that the IRP is sensitive to elevated EGTA concentrations in a membrane-potential–dependent manner across the physiological range of membrane potentials. Our results are best explained by a model in which multiple Ca2+ channels act in concert to drive exocytosis of a single synaptic vesicle. Pooling calcium entering through many calcium channels may be important for reducing stochastic noise in neurotransmitter release associated with the opening of individual calcium channels.
INTRODUCTION
A select group of cells in the vertebrate CNS are specialized for continuous exocytosis. These cells include: the photoreceptors and bipolar neurons of the eye, inner hair cells of the ear and lateral line organs, electroreceptors of some fish, and pinealocytes. It is most likely due to the continuous nature of neurotransmission in these cells that their synapses contain large numbers of synaptic vesicles (Lagnado et al. 1996; Mennerick and Matthews 1996; von Gersdorff and Matthews 1994a; von Gersdorff et al. 1996, 1998), specialized slowly inactivating L-type Ca2+ channels (Heidelberger and Matthews 1992; Hudspeth and Lewis 1988), and a number of osmiophillic, proteinaceous organelles called synaptic ribbons, which appear to tether vesicles by fine filaments, and are located directly adjacent to the voltage-gated Ca2+ channels (Issa and Hudspeth 1996; Zenisek et al. 2003). This article focuses on one such ribbon-containing neuron—the retinal bipolar cell of the goldfish.
In relation to the large number of vesicles present throughout the bipolar neuron's presynaptic terminal, a large body of research has parsed out discrete subsets within this overall pool (Lagnado et al. 1996; Mennerick and Matthews 1996; Singer and Diamond 2003; von Gersdorff and Matthews 1994a; von Gersdorff et al. 1998; Zenisek et al. 2000). A small fraction of vesicles (herafter termed as the immediately releasable pool [IRP]) appears to be docked at the plasma membrane and primed for release within a few milliseconds (Gomis et al. 1999; Lagnado et al. 1996; Mennerick and Matthews 1996; Midorikawa et al. 2007; von Gersdorff and Matthews 1994a; von Gersdorff et al. 1996, 1998; Zenisek et al. 2000). A larger fraction of the total vesicle pool has shown the ability to exocytose during longer depolarizations (≤500 ms) to membrane potentials that near maximally activate the calcium current. We will use the term readily releasable pool (RRP) throughout to describe these vesicles. This pool consists of 0.5–1% of the vesicles observed in a bipolar neuron (von Gersdorff et al. 1996). Last, the remaining fraction of synaptic vesicles constitutes, at least in part, a reserve for later release (Lagnado et al. 1996; Mennerick and Matthews 1996).
Investigation into other ribbon synapses has suggested a roughly linear relationship between membrane potential and exocytosis (Brandt et al. 2005; Thoreson et al. 2004). For cells with nonlinear calcium sensors for exocytosis, such results are consistent with release being driven by “nanodomains” of calcium through only a few calcium channels located near each release site (Augustine et al. 1991; Brandt et al. 2005; Mintz et al. 1995). This runs in contrast to what has been observed in nonribbon-type synapses (Augustine et al. 1991) and we tested whether the same holds true for the IRP of the retinal bipolar cell of the goldfish. In contrast to murine hair cells and salamander photoreceptors, our results provide evidence for the summation of calcium entering through many channels at each release site in goldfish bipolar neurons.
METHODS
Cell preparation
All procedures for animal care were carried out according to Yale Animal Care and Use Committee. Bipolar neurons were prepared as previously described (Zenisek et al. 2002). Briefly, adult goldfish (10–15 cm in length) were placed in low-light conditions for 20–25 min and then killed by decapitation. Next, eyes were removed and hemisected and lenses were removed from the eyecup. Eyecups were incubated for 20 min in hyaluronidase (Sigma type V; 1,100 units ml−1) in a saline solution containing (in mM): 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose, and 10 Hepes (pH 7.4 with NaOH). After incubation, retinas were then removed from eyecups, cut into four to eight pieces each, and incubated for 30–35 min in a solution of 30–35 units ml−1 papain (lyophilized powder; Sigma), 0.5 mg ml−1 cysteine, and (in mM): 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose, and 10 Hepes (pH 7.4 with NaOH). After enzymatic treatment, tissue was washed in enzyme-free solution and stored at 12°C for ≤5 h. The pieces of retina were dissociated into single cells by mechanical trituration through a fire-polished Pasteur pipette.
Electrophysiology
Bipolar cells were recognized by their distinct morphologies. To record whole cell currents and to introduce reagents into cells, a high-resistance seal was formed using a glass pipette and the patch of membrane beneath the pipette was ruptured using gentle suction. Isolated terminals were whole cell voltage-clamped using an EPC-10 amplifier (Heka Elektronik, Lambrecht/Pfalz, Germany) controlled by Pulse (InstruTech, Port Washington, NY) acquisition and recording software. The pipette solution for most experiments contained (in mM): 120 Cs-glutamate, 4 Na2ATP, 0.5 GTP, 4 MgCl2, 10 TEA-Cl, and either 0.5 or 10 EGTA (pH 7.3 with NaOH). Electrodes were typically 10–15 MΩ with seal resistances >2 GΩ. For experiments designed to clamp the intracellular [Ca2+] to levels reasonably close to expected under native conditions we used 15 mM ethylene glycol tetraacetic acid (EGTA) and 7.5 mM Ca2+. Using data from Invitrogen (Kd EGTA of ∼100 nM for pH 7.3 and 20°C) we calculated an expected intracellular [Ca2+] of about 100 nM. Comparison of fluorescence intensities of fluo-4 (Invitrogen) containing intracellular solution with buffered calcium/EGTA standard solutions (Invitrogen) indicated that our [Ca2+] was around 300 nM. Recordings had access resistances typically between 16 and 28 MΩ. Using these pipette solutions, potassium channels were blocked, leaving a slowly inactivating L-type calcium current as the predominant voltage-gated conductance under these conditions (Heidelberger and Matthews 1992; Tachibana et al. 1993a).
To measure isolated terminal capacitance, a sinusoidal voltage command (30 mV peak-to-peak at either 800 Hz or 1 kHz) was superimposed on our holding potential of −60 mV. Capacitance was calculated using the Lindau–Neher method incorporated into Pulse software (Lindau and Neher 1988). Capacitance recordings were performed on synaptic terminals, which had become isolated from the soma during the cell isolation procedure. After rupture of the membrane, cells were not stimulated for ≥60 s to allow for diffusional exchange of pipette solution throughout the terminal. Capacitance jumps from synaptic terminals stimulated to 0 mV were included in the average only if calcium currents were >75 pA, leak current at our holding potential of −60 mV was <25 pA, and the terminal demonstrated >10 fF of capacitance increase. Capacitance jumps from synaptic terminals stimulated between −25 and −35 mV were included only if the terminal demonstrated a capacitance increase to a subsequent 250-ms depolarization to 0 mV, that was within 3SE measurements of the mean capacitance increase to a 250-ms depolarization to 0 mV (>20 fF; see Fig. 1). A typical terminal had a resting capacitance of about 2.2 pF and demonstrated 180 pA of peak inward calcium current on depolarization to 0 mV.
FIG. 1.

Exocytosis as a function of depolarization step length. A, left, raw current signal from a bipolar cell terminal loaded with 0.5 mM ethylene glycol tetraacetic acid (EGTA) and depolarized from −60 to 0 mV for 250 ms. A, right: raw capacitance signal from the same terminal as at left. B, left, raw current signal from a bipolar cell terminal loaded with 10 mM EGTA and depolarized from −60 to 0 mV for 250 ms. B, right: the raw capacitance signal from the same terminal as at left. C, left: composite data from all terminals. Number of terminals loaded with 0.5 mM EGTA (black squares) by stimulus duration: 5 ms (n = 8), 10 ms (n = 6), 15 ms (n = 19), 30 ms (n = 7), 60 ms (n = 7), and 120 ms (n = 6). Number of terminals loaded with 10 mM EGTA (red circles) by stimulus duration: 5 ms (n = 8), 10 ms (n = 8), 15 ms (n = 10), 30 ms (n = 10), 60 ms (n = 8), and 120 ms (n = 6). C, right: composite data from terminals loaded with 0.5 mM EGTA and depolarized for indicated times. Red curve shows a single-exponential fit to these data using OriginLab 7.5. The curve had R2 = 0.96 and τ = 2.9 ms.
Changes in membrane capacitance—capacitance jumps—were measured by subtracting an average capacitance signal before stimulation from an average capacitance signal after stimulation. The capacitance signal before stimulation was averaged over a period of 20 ms immediately prior to depolarization. The capacitance signal after stimulation was averaged over a period of 20 ms beginning 20 ms after the termination of the depolarization to allow for conductance changes to cease (Gomis et al. 1999). For depolarizations >120 ms to 0 mV, a slowly decreasing inward current was often observed (Okada et al. 1995; Tachibana and Okada 1991; Tachibana et al. 1993a,b) immediately after repolarization and therefore we waited 250 ms before averaging a 20-ms period to minimize this conductance confounding the capacitance signal while realizing that endocytosis would also be small after 250 ms (von Gersdorff and Matthews 1994a,b) but nonnegligible. Based on the approximately 1-s exponential time constant of the return of the capacitance trace to baseline (Gomis et al. 1999; von Gersdorff and Matthews 1994a), reflecting endocytosis, we estimate that our capacitance jumps underestimate exocytosis by about 22%. Thus our calculated capacitance jumps for 250–500 ms can be considered slight underestimates.
For most experiments (paired-pulse experiments excluded), we included as data only the first capacitance response for a given cell and averaged these individual responses. We performed depolarizations only once per given cell to remove any confounding effects of replenishment of vesicle pools (Gomis et al. 1999; von Gersdorff and Matthews 1994a; Zenisek et al. 2000), capacitance rundown (Neves and Lagnado 1999), or other nonspecific mechanisms leading to variability in amounts of exocytosis (Burrone and Lagnado 2000). Therefore each observation is due to cells tested only once and only for that observation. As a check that our methodology did not introduce any additional variability, we performed experiments testing repeated submaximal depolarizations given to a single cell (250-ms depolarizations to −30 mV with a 5-ms prepulse applied immediately before; >100 s between stimulations) and measured the variability by either 1) using the first capacitance jump from each cell only, averaging across cells and calculating the between cell variance, or 2) averaging the capacitance response from a single cell using three capacitance jumps produced by that cell, calculating the within-cell variance, and averaging this variance across all cells. We found that the variances were the same: ±0.0003 pF2 using either method, whereas the magnitude of the average capacitance response was smaller when averaging within cells (on average, third capacitance jump was ∼61% of the first). This result is consistent with repeated depolarizations producing smaller capacitance jumps over time (a trend we observed in 12 of 15 cells after three depolarizations; ≥60 s between depolarizations), which is most likely due to a combination of the factors mentioned earlier.
Fluorescent microscopy with ribbon-specific fluorescent peptide
Microscopy and introduction of a synaptic ribbon-binding peptide were performed as previously described (Zenisek et al. 2004). N-terminally fluorescein-conjugated peptides with the sequence EQTVPVDLSARDR were synthesized and purified at the W. M. Keck facility at Yale University. Bipolar cells were whole cell voltage-clamped with the same pipette solution as cited earlier with the addition of 42 μM peptide. Cells were observed through an inverted microscope (Zeiss Axiovert 135 or Olympus IX-70) modified for objective-type evanescent field illumination (Axelrod 2001), referred to hereafter as total internal reflection fluorescent microscopy (TIRFM), using a 1.65 numerical aperture objective and an argon laser. The laser beam left the objective at an angle of 65.6–68.0°. Given the refractive indices of cytosol (n = 1.37) and glass coverslip (n = 1.80) and the 65.6–68.0° angle, the intensity of the evanescent field is expected to decline e-fold every 41–43 nm. TIRFM images were captured using an intensified CCD camera (Cascade 512B, Roper Scientific). Images of 30 or 200 ms in duration were recorded at 0.5 Hz. Ribbon locations were observed as bright fluorescent puncta within the plasma membrane area adhered to the glass coverslip. The membrane area from a single image from each individual cell was added together to give total membrane area (n = 19 cells). All ribbons from the same, single image were added to give total synaptic ribbons and this value was divided by total membrane area to give an estimate of ribbon density (means ± SE).
Data analysis
All data are presented as means ± SE unless otherwise noted. The amount of calcium current was determined as the peak current during a depolarization. All experiments were carried out at room temperature (21–24°C).
For judging goodness-of-fit of data to exponential functions with one or more time constants, we calculated adjusted R2 to determine whether adding additional time constants to our model resulted in a better fit to our data. Adjusted R2 is calculated according to the following formula
 |
(1) |
where R2 is the unadjusted coefficient of determination provided by our model, n is the sample size, and p is the number of regressors.
To judge whether data sets using different buffering conditions were statistically significant we used a Student's t-test implemented in OriginLab 7.5. We assumed that P values <0.05 indicated significance unless specified otherwise.
Modeling of relationship between release and calcium channels
Calculations for the model were performed using Microsoft Excel. The methods for calculating release rate as a function of open probability and distance from the channels for various numbers of channels are described in results.
RESULTS
Immediately releasable pool in response to maximal calcium channel activation
Since a large body of experiments looking at the properties of exocytosis and endocytosis in retinal bipolar cells show variability in response using step depolarizations designed to maximally activate Ca2+ current, we chose to first repeat earlier experiments defining the size of different vesicle pools with known concentrations of the slow calcium buffer EGTA to provide a baseline and comparison for our experiments using physiological depolarization potentials (Gomis et al. 1999; Mennerick and Matthews 1996). Previous results have indicated that concentrations of EGTA >5 mM restrict exocytosis to the IRP and block subsequent release from the remainder of the RRP, presumably by restricting the elevation of calcium to a region proximal to calcium channels (Adler et al. 1991; Burrone et al. 2002; Roberts 1993; Zucker and Fogelson 1986).
Figure 1 shows an experiment where we voltage-clamped dissociated synaptic terminals and held the membrane potential at −60 mV, then gave step depolarizations to 0 mV for varied lengths of time. Figure 1, A and B shows raw traces from some of our experiments. Figure 1A shows the current and capacitance signals from a representative terminal loaded with 0.5 mM EGTA and stimulated for 250 ms. Figure 1B shows current and capacitance signals from a representative terminal loaded with 10 mM EGTA and stimulated for 250 ms. Figure 1C, left shows the average capacitance data across all our experiments, whereas Fig. 1C, right shows the short-duration data for terminals loaded with 0.5 mM EGTA from Fig. 1C, left and a single-exponential function fit to the data (see following text).
In 0.5 mM EGTA-loaded terminals stepped to 0 mV, capacitance jumps increased with step duration ≤10 ms but showed little additional exocytosis until durations were increased to >60 ms. Of note, the size of this pool was about 70 fF, considerably larger than most previous reports of the IRP size tested using the whole cell capacitance technique with 0.5 mM exogenous EGTA added (Burrone et al. 2002; Mennerick and Matthews 1996) or with the perforated patch technique (Gomis et al. 1999; Neves and Lagnado 1999), but similar to that recorded from bipolar cells in a retinal slice preparation (Palmer et al. 2003). This is possibly attributable to our use of only the first depolarization in each cell or the higher adenosine 5′-triphosphate concentrations that were used in our experiments. Previous studies using TIRF microscopy to visualize exocytosis estimate that roughly 92% (Zenisek et al. 2000) of all fusion events within the first 30 ms of a step to 0 mV are near active zones, thought to be ribbons. Similarly, nearly 88% of all fusion events within the first 100 ms (Midorikawa et al. 2007) step to 0 mV are at ribbons; thus this IRP likely represents a pool of vesicles near ribbons. A slower rate of increase in the size of the capacitance jump is observable for depolarizations >60 ms. Of note, the effect of a quick capacitance increase followed by a less synchronous secondary burst is qualitatively similar to measurements of glutamate release (Sakaba et al. 1997) and single-vesicle imaging of exocytosis in goldfish bipolar cells (Midorikawa et al. 2007; Zenisek et al. 2000).
With 10 mM EGTA, the sustained rise in capacitance for depolarizations >60 ms in duration, previously observed using 0.5 mM EGTA, appears to be blocked (Gomis et al. 1999; Mennerick and Matthews 1996; Neves and Lagnado 1999) for pulse durations ≤500 ms. Under these conditions, the size of the capacitance jump appears to increase as depolarizations lengthen ≤10 ms as with 0.5 mM EGTA. Unlike with the lower EGTA concentrations, however, longer depolarizations fail to increase the size of the capacitance jump beyond about 35 fF. Our results replicate experiments performed by Burrone et al. (2002).
Under stimulation conditions designed to release the entire IRP, the time course has been described as a simple exponential (Gomis et al. 1999; Mennerick and Matthews 1996)
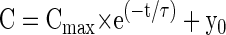 |
(2) |
where Cmax is the size of the IRP, t is time, and τ is the time constant. Using Eq. 2 and constraining the fit for zero capacitance jump at t = 0 s, our 0.5 mM EGTA data for durations ≤60 ms is best fit by a Cmax value of 72 fF with τ = 3.0 ms (see Fig. 1C, right, R2 = 0.78). For the 10 mM EGTA condition, Cmax was 28 fF and τ = 2.3 ms. Our data were well fit by single-exponential functions and using a two-exponential function did not provide a better fit, as judged by an adjusted R2 (see methods). Importantly, the magnitude of our Cmax in terminals loaded with 10 mM EGTA is quantitatively similar to previous research that 1) used simple step depolarizations to −10 mV using the perforated patch technique (Gomis et al. 1999) or 2) using step depolarizations to 0 mV preceded by 1-ms prepulses to +180 mV to maximally activate L-type Ca2+ channels (Mennerick and Matthews 1996).
The fact that the amount of release as a function of depolarization length exhibited a roughly exponential release pattern, followed by a plateau after 10 ms, suggests that these vesicles represent a single distinct pool of different magnitudes under different calcium buffering conditions. To test this idea in a different manner, we gave pairs of depolarizations of different durations in short succession and asked the extent to which the second depolarization was depressed relative to the first. If there is little pool refilling between depolarizations and the single exponential is representative of release of a single stochastic pool, then we expect that the response to the second depolarization (C2) will be governed by the following equation
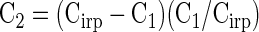 |
(3) |
where Cirp is the size of the IRP, C1 is the size of the first jump in a pair, and C2 is the second. We depolarized a voltage-clamped terminal to 0 mV for a set length of time, measured capacitance for the next 20 ms at the resting potential, waited an additional 30 ms, and then stimulated again for the same depolarization length. Thus we gave two 2.5-ms depolarizations, two 5-ms depolarizations, or two 10-ms depolarizations separated by 50 ms between successive stimuli to individual terminals. The capacitance jump due to the second depolarization was divided by the jump due to the first depolarization to detail the percentage of the IRP remaining after the first stimulation.
Figure 2 A, right shows the size of the second pulse in pairs of depolarizations relative to the first of each pair for both 0.5 mM EGTA (filled squares) and 10 mM EGTA (open triangles). From Fig. 2A, right it is apparent that the time course of depletion at 0.5 and 10 mM EGTA mirror one another and, in both conditions, the second of each pair of 10-ms steps fails to elicit exocytosis, consistent with the idea that 10-ms depolarizations to 0 mV are sufficient to deplete the entire IRP. Our result is in agreement with previous work that demonstrates a fast component of exocytosis depleted with a time constant of about 3.6 ms using stimulation to −10 mV (Gomis et al. 1999). For two depolarizations of 2.5 ms, the second 2.5-ms depolarization was roughly 17% as large as the first for terminals loaded with 0.5 mM EGTA and about 11% as large as the first for terminals loaded with 10 mM EGTA. Equation 3 can be rearranged to calculate the value of Cirp from the values of C1 and C2
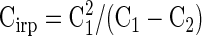 |
(4) |
This equation predicts an IRP size of 53 ± 8, 54 ± 12, and 76 ± 10 fF for depolarizations of 2.5, 5, and 10 ms, respectively, for 0.5 EGTA and 27 ± 5, 27 ± 3, and 33 ± 5 fF for 10 mM EGTA. Together these results indicate that despite the very different sizes of the IRP under these different buffering conditions, the IRP behaves as a single component that is depleted over essentially the same time course, suggesting that we have effectively restricted the size of the IRP depending on the concentration of EGTA, similar to work by Burrone et al. (2002).
FIG. 2.

Exocytosis as a function of short-duration step depolarizations and total internal reflection fluorescent microscopy (TIRFM) imaging of synaptic ribbon density. A, left: composite data of exocytosis from terminals loaded with 0.5 mM EGTA (filled circles) or 10 mM EGTA (open triangles) and depolarized to 0 mV for the indicated time (same data as in Fig. 1). A, right: experiment with 2 step depolarizations of the same duration given 50 ms apart. This experiment is designed to determine the length of time required for a single pulse to deplete the immediately releasable pool (IRP). Ordinate represents the capacitance jump due to the 2nd depolarization as a percentage of the capacitance jump due to the 1st depolarization; 0.5 mM EGTA is represented by filled circles, whereas 10 mM EGTA terminals are represented by open triangles. B, left: brightfield microscopy image of the synaptic terminal of a whole bipolar neuron. B, middle: TIRFM image of the same cell as on left. B, right: TIRFM image of the synaptic terminal of a different whole bipolar neuron. The scale bars for all images indicate 1.70 μm in both axes. C: histogram of number of synaptic ribbons compared with the full-width at half-maximum (FWHM) determined from fitting fluorescent intensity with a Gaussian function (Coggins et al. 2007).
Correlating ribbon number to vesicle pools
Since different buffering conditions give rise to different size IRPs, we next sought to compare ribbon number to the size of the IRP to estimate the number of vesicles released in this kinetic component. Estimates of ribbon number have varied from 36 (Zenisek et al. 2003) to 65 (von Gersdorff et al. 1996) in previous studies, possibly reflecting subtle differences in experimental or cell preparation conditions and/or differences in cell size (von Gersdorff et al. 1996). Of note, ribbons have been described to change in number (von Gersdorff et al. 1996) and location (Vollrath and Spiwoks-Becker 1996; Zenisek et al. 2004) dependent on activity state (Spiwoks-Becker et al. 2004), time of day (Hull et al. 2006; Spiwoks-Becker et al. 2004; Vollrath and Spiwoks-Becker 1996), or dark/light adaptation, so we thought it important to obtain estimates of ribbon numbers in cells treated the same way and in parallel with our capacitance experiments. To determine the number of ribbons, we used a fluorescent peptide that binds synaptic ribbons (Zenisek et al. 2004). Figure 2B shows brightfield and TIRFM images of two bipolar cell terminals loaded with this peptide. The cell shown on the left in brightfield is the same as that shown in the middle image using TIRFM. The cell on the right is a different cell. Quantification of the number of spots/μm2 gives a value of 0.152 ± 0.019. Our mean resting capacitance of all terminals used in our capacitance experiments is 2.2 pF (mean value for both 0.5 and 10 mM EGTA conditions; both conditions displayed similar mean resting capacitance individually and were pooled; data not shown) and, if we assume a mean specific capacitance of 0.9 μF/cm2 or 0.009 pF/μm2 (Gentet et al. 2000), we would expect 37 spots per terminal.
Our method of counting fluorescent spots to estimate ribbon number potentially underestimates the number of ribbons due to our inability to resolve closely spaced objects. Although we may not be able to resolve two ribbons within the diffraction limit of the microscope, we should be able to detect the broadening of the observed spots associated with multiple ribbons, since the size of each ribbon is similar to the diffraction limit of the microscope. Figure 2C is a histogram detailing the full-width at half-maximum (FWHM) of the fluorescent spots. By comparison, we determined the FWHM of a diffraction-limited 40-nm fluorescent bead to be 110 ± 1 nm on our microscope. The peak of our histogram of FWHM was 175 nm and using our point spread function this corresponds to an object of about 136 nm if one assumes a Gaussian ribbon profile and Gaussian point spread function (Zenisek et al. 2002). If this represents the fluorescent size of our average ribbon, two ribbons touching each other would create an object of 272 nm along the long axis and 136 along the short axis. Once blurred by diffraction, it would be 293 nm on one axis, 175 nm on the other. If one assumes that a Gaussian function fit to such an object would have a FWHM halfway between the long and short axes, then two ribbons would have a minimum FWHM of about 232 nm. Of 31 spots, we found two that had a FWHM of >232 nm and, based on the sizes, we estimate that one of those spots is likely two ribbons, whereas the other is likely to be three ribbons (or perhaps two ribbons separated by a greater distance). Assuming that our counting underestimates the number of ribbons by approximately 3/31 ≈ 10% we find that there is, on average, 0.167 ribbon/μm2, corresponding to 41 ribbons in our average cell.
These results suggest that the capacitance response is 0.68 fF per ribbon for a 10-ms depolarization with 10 mM EGTA or about 27 vesicles per ribbon if we assume a mean outer vesicle diameter of 38 nm per vesicle (Coggins et al. 2007), a membrane thickness of about 4 nm (Mitra et al. 2004), and a specific capacitance of 0.9 μF/cm2 (providing an estimate of 25.4 aF per vesicle). By contrast, a 10-ms depolarization to 0 mV elicited an equivalent of about 68 vesicles per ribbon in the 0.5 mM EGTA condition using the same assumptions. The number of vesicles released under elevated buffering conditions is similar to the number of vesicles observed at the base row of the ribbon in electron microscopy images (von Gersdorff et al. 1996). Depolarizing cells under 0.5 mM EGTA conditions, however, cause additional vesicles to be released with similar kinetics, either at extra-ribbon sites (Midorikawa et al. 2007), docked vesicles adjacent to the synaptic ribbon (Lenzi et al. 1999), or at distal locations on the synaptic ribbon, possibly due to compound exocytosis (Edmonds et al. 2004; Glowatzki and Fuchs 2002; Heidelberger et al. 1994; Matthews and Sterling 2008). Even using conservative estimates for ribbon number and specific capacitance (1.0 μF/cm2, giving 28.3 aF per vesicle), we estimate an IRP almost threefold greater than the number of vesicles at the base of the ribbon under low buffering conditions (61 estimated vs. 22 vesicles determined from electron microscopy; von Gersdorff et al. 1996).
Release at physiologically relevant potentials
Previous research into carp, salamander, and goldfish on-type bipolar neuron responses to light stimuli indicates that when cells are initially exposed to polychromatic light, transient voltage changes of 5–10 mV (10–20 mV in salamander) are observed and these transient changes are often followed by a slow decline with a plateau of 0- to 5-mV depolarization above resting potential (Joselevitch and Kamermans 2007; Kaneko 1973; Lasansky 1992; Werblin and Dowling 1969). Thus although standard experimental protocols use depolarizations to 0 mV, bipolar cells are unlikely to reach this level under physiological conditions. Instead, bipolar cells operate over a narrow range of membrane potentials more negative than the peak activation of calcium current. Therefore we revisited the properties of the IRP at more physiological membrane potentials.
Figure 3 shows the results of experiments on cells loaded with 0.5 mM EGTA using step depolarizations to different membrane potentials. We chose to restrict this analysis to membrane potentials ranging from −35 to −25 mV, since 1) these potentials are within the physiological range for bipolar neurons and 2) depolarizations to membrane potentials more negative than −35 mV failed to evoke a measurable capacitance jump. Figure 3A, left shows the simple step depolarization used initially. Steps to −35, −30, −28, and −25 mV for 250 ms gave rise to capacitance jumps of 8, 8, 17, and 48 fF, respectively (see Fig. 3B, left), or 11, 11, 24, and 68% of the IRP measured at 0 mV.
FIG. 3.

Exocytosis due to step depolarizations to physiological membrane potentials using 0.5 mM EGTA. A, left: pulse protocol (P1) used to probe the amount of exocytosis elicited by 250-ms step depolarizations to various membrane potentials (−XX mV) from a holding potential of −60 mV. A, right: pulse protocol (P2) designed to probe IRP size and terminal capacity to exocytose the remainder of the IRP—using a 15-ms pulse to 0 mV—if a portion had been predepleted by a 235-ms step depolarization to a physiological membrane potential. B, left: composite data for all terminals. Bars, from leftmost to rightmost, represent terminals depolarized for −35, −30, −28, and [minus[25 mV. B, right: comparison of capacitance jump for terminals depolarized to −28 mV using P1 (leftmost bar), depolarized to −28 mV using P2 (middle bar), and depolarized to 0 mV for 15 ms (rightmost bar). The P2 and 15 ms to 0 mV protocols are significantly different (P < 0.05) from the P1 protocol. C: average charge for terminals loaded with 0.5 mM EGTA and step depolarized to −35, −30, −28, and −25 mV (from left to right, respectively). The number of terminals stimulated at each condition is: −35 mV (n = 4), −30 mV (n = 4), −28 mV (n = 6), and −25 mV (n = 11).
Recent studies using TIRF microscopy indicate that long (>100 ms) depolarizations to 0 mV evoke extra-ribbon release of synaptic vesicles (Midorikawa et al. 2007; Zenisek 2008; Zenisek et al. 2004) and endosomes (Coggins et al. 2007) in goldfish bipolar neurons. To test whether prolonged depolarizations to physiological potentials cause substantial release outside of the vesicles comprising the IRP, we subjected cells to the protocol shown on the right-hand side of Fig. 3A. For this protocol, cells were given 235-ms step depolarizations to −28 mV followed immediately by a 15-ms step to 0 mV [protocol 2 (P2) shown in Fig. 3A, right]. If long depolarizations released vesicles outside of the IRP, then one would expect that the capacitance jump elicited by P2 would be >15-ms depolarizations to 0 mV, which depletes only the IRP. Instead, we found that the magnitude of the capacitance jump was indistinguishable between cells stimulated with this protocol compared with cells stimulated with a simple step depolarization to 0 mV for 15 ms. These results are consistent with a model in which 250-ms depolarizations to physiological membrane potentials tap vesicles within the IRP only for release.
EGTA selectively blocks exocytosis at more negative membrane potentials
We tested the sensitivity to EGTA of release in response to depolarizations between −35 and −25 mV. Figure 4 summarizes the results. Bipolar cells were depolarized to −35, −30, −28, and −25 mV for 250 ms and capacitance was measured in cells loaded with 10 mM EGTA. We found that, under these conditions, we failed to observe capacitance changes in response to depolarizations of −35 or −30 mV, but did see significant release at more positive potentials (Fig. 4A, left). For depolarizations to −28 and −25 mV, terminals loaded with 10 mM EGTA showed capacitance jumps of 7 and 15 fF, respectively. Figure 4B shows the charge carried as inward current during these depolarizations. Since our pipette solution was designed to block potassium channels, the current measured is predominantly through calcium channels.
FIG. 4.

Exocytosis due to step depolarizations to physiological membrane potentials using 10 mM EGTA. A: composite data for all terminals. Bars, from leftmost to rightmost, represent terminals depolarized for −35, −30, −28, and −25 mV. B: comparison of capacitance jump for terminals depolarized to −28 mV using P1 (leftmost bar), depolarized to −28 mV using P2 (middle bar), and depolarized to 0 mV for 15 ms (rightmost bar). The P2 and 15 ms to 0 mV protocols are significantly different (shown by stars; P < 0.05) from the P1 protocol. C: average charge for terminals loaded with 0.5 mM EGTA and step depolarized to −35, −30, −28, and −25 mV (from left to right, respectively). The number of terminals stimulated at each condition is: −35 mV (n = 4), −30 mV (n = 4), −28 mV (n = 8), and −25 mV (n = 11).
As with terminals loaded with 0.5 mM EGTA, additional exocytosis could be elicited by a 0-mV pulse at the end of a 235-ms depolarization to −28 mV. Similar to cells dialyzed with 0.5 mM EGTA, the total amount of exocytosis elicited by the 235-ms depolarization to −28 mV and 15-ms step to 0 mV was not different from the size of the IRP (Fig. 4A, right). If we divide the capacitance jump for terminals loaded with 10 mM EGTA by the capacitance jump for terminals loaded with 0.5 mM EGTA elicited by the same step depolarization, we find that EGTA has a suppressive effect that is greatest in response to the weakest depolarizations. For terminals stimulated to −35, −30, −28, or −25 mV (using protocol 1 only), we find 10 mM EGTA suppresses exocytosis to −17 ± 7, −15 ± 6, 39 ± 29, and 32 ± 11% of the amount in terminals loaded with 0.5 mM EGTA, respectively.
Figure 5 A, left shows the charge carried through channels in bipolar cells under different conditions. For some cells, the charge was larger for terminals loaded with 10 mM EGTA than that for terminals under the low buffering condition, but not significantly so, except for the 250-ms depolarization to −28 mV, possibly a result of calcium-dependent inactivation of calcium currents previously described in these cells (von Gersdorff and Matthews 1996). Figure 5A, right compares the capacitance jump experienced by terminals loaded with 0.5 or 10 mM EGTA with the membrane depolarization. At each membrane potential except −28 mV (P > 0.28), the capacitance jump was significantly (P < 0.05) greater with 0.5 mM EGTA than that with 10 mM EGTA.
FIG. 5.

Comparison of physiological membrane depolarizations using 0.5 or 10 mM EGTA. A, left: charge introduced during depolarizations as a function of the step depolarization protocol. Black bars (leftmost within protocol) represent terminals loaded with 0.5 mM EGTA; white bars (rightmost within protocol) represent terminals loaded with 10 mM EGTA. Comparing terminals within protocols shows that for −28-mV P1 step depolarizations the charge is significantly larger for 10 mM EGTA-loaded terminals. A, right: capacitance jump as a function of the step depolarization protocol. Black bars (leftmost within protocol) represent terminals loaded with 0.5 mM EGTA; white bars (rightmost within protocol) represent terminals loaded with 10 mM EGTA. Comparing terminals within protocols shows that for all protocols except −28-mV P1 step depolarizations, the capacitance jump is significantly larger for 0.5 mM EGTA-loaded terminals. B, left: charge introduced during a step depolarization to −30 mV compared with that introduced during a step depolarization to −30 mV that was immediately preceded by a 5-ms prepulse to +180 mV, to introduce a calcium tail current (−30-mV tail). B, right: capacitance change due to a simple 250-ms step depolarization to −30 mV (black trace) compared with the capacitance change elicited by a 250-ms step depolarization to −30 mV immediately preceded by a 5-ms prepulse to +180 mV (red trace). C, left: current records from representative terminals stimulated with a step depolarization from −60 to −30 mV for 250 ms (black trace) or stimulated with a step depolarization from −60 to −30 mV for 250 ms immediately preceded by a 5-ms depolarization to +180 mV (red trace). These currents gave rise to capacitance changes of −1.4 fF (terminal from black trace) and 4.6 fF (terminal from red trace). C, right: capacitance jump comparing simple 250-ms step depolarizations to step depolarizations with 5-ms prepulse to +180 mV. Black bars are from terminals loaded with 10 mM and depolarized to −30 mV. Red bars are from terminals loaded with 15 mM EGTA and 7.5 mM Ca2+, to give a measured internal [Ca2+] of about 300 nM (see methods).
The number of open calcium channels changes significantly between −25 and −35 mV, whereas the conductance through individual channels is less sensitive to membrane potential, due to the very positive reversal potential of calcium. Thus one explanation for the greater effects of EGTA on release at more negative potentials is that EGTA could effectively restrict calcium near individual channels, but is less effective in blocking release if calcium simultaneously summates through the opening of multiple, spatially localized channels. To test this idea, we observed whether the brief opening of calcium channels during a tail current at −30 mV was sufficient to overcome the block of exocytosis by 10 mM EGTA. Under these conditions the single-channel conductance will be equivalent to that when holding the membrane potential at −30 mV, but transiently the number of channels open simultaneously will be greater. We gave a 5-ms prepulse to +180 mV to open voltage-gated calcium channels without allowing calcium ions to flow into the cell, followed immediately by a 250-ms step depolarization to −30 mV. The rapid step to −30 mV is faster than channels are capable of closing and thus generates a “tail current” in which most channels are open transiently and close with a time course that reflects the timing of channel closure at −30 mV. This prepulse increased the total charged carried through calcium channels by only 21% (mean 2.63 pC vs. mean 2.17 pC) beyond the total calcium entry during a 250-ms step to −30 mV, yet is sufficient to elicit a substantial capacitance jump in the presence of elevated EGTA, where there was none for the 250-ms step to 0 mV. The results of this experiment are presented in Fig. 5B. Figure 5B, left shows a comparison of the charge entering terminals stepped to −30 mV for 250 ms versus terminals first given the 5-ms prepulse to +180 mV before being repolarized to −30 mV for 250 ms. Figure 5B, right shows the average capacitance from all terminals normalized to the capacitance at the start of data recording. A clear capacitance jump can be seen for terminals given the 5-ms prepulse to +180 mV (shown in red; 11 ± 4 fF, n = 7). Figure 5C, left shows uncorrected current signals from representative terminals. Figure 5C, right provides a comparison of terminals loaded with 10 mM EGTA (shown in black; data also shown in Fig. 5B, right) or loaded with 15 mM EGTA and 7.5 mM Ca2+ to buffer calcium at near-physiological basal calcium concentrations (n = 7, shown in red; see methods). There was no significant difference in capacitance responses between the two conditions.
An alternative conclusion from this experiment is that opening most of the calcium channels by the prepulse allows enough Ca2+ entry to transiently overwhelm locally free EGTA and drive exocytosis before bound Ca2+-EGTA diffuses away and is replaced by free EGTA. However, the prepulse does not change the single-channel current during the −30-mV depolarization. Therefore if EGTA can suppress release near single or few channels, as shown by the simple 250-ms depolarization to−30 mV, simply increasing the number of open channels by giving a prepulse should allow release only if those new open channels are close spatially to other open channels allowing calcium summation.
Measuring release rate while varying calcium influx using membrane potential
The above-cited results suggest that under heavy buffering conditions, release rates are influenced more by changes in the number of calcium channels open at any given time than the amount of time channels are open. This implies that simultaneously open calcium channels may act cooperatively to set the release rate at different membrane potentials. To estimate the apparent degree of cooperativity of calcium channels, we measured the rates of exocytosis for several membrane potentials between −35 and 0 mV and plotted the rates as a function of calcium current. To measure release rates in response to −25 and 0 mV, we fit exponentials to our measurements of capacitance at different durations. At lower membrane potentials, where the small size of the capacitance jumps precluded a full exploration of the time course of release over a period where replenishment is negligible, we estimated release rate by assuming a linear fit from the origin to the amount of release observed in response to a 250-ms depolarization. The relationship between ICa and release rate is shown on a linear plot in Fig. 6 A and a log–log plot in Fig. 6B. As expected, the relationship between calcium entry and exocytic rate is highly nonlinear, with a calcium dependence that increases roughly with the second power of ICa in 0.5 mM EGTA and third power of ICa in 10 mM EGTA. By comparison, the relationship between calcium and neurotransmitter release was found to vary with the fourth power of calcium (Heidelberger et al. 1994).
FIG. 6.

Rates of exocytosis due to different calcium currents. A: linear plot of exocytic rate compared with calcium current in response to step depolarizations to different membrane potentials for terminals loaded with 0.5 mM EGTA (black squares; data from Figs. 1 and 3) or 10 mM EGTA (open circles; data from Figs. 1 and 4). See text for a description of how the exocytic rate was calculated. B: log–log plot of the data in A. The vertically hatched line represents a least-squares best-fit linear line to data from terminals loaded with 0.5 mM EGTA implemented in OriginLab (see methods). The solid line represents a best-fit linear line to data from terminals loaded with 10 mM EGTA.
Paired-pulse depression and refilling of the IRP
If vesicles within the IRP had different sensitivities to membrane potential, as may be expected if vesicles reside at different distances from calcium channels (Burrone and Lagnado 2000), our earlier results may reflect these heterogeneities in the IRP. To address whether the IRP acted as a single pool at each membrane potential, we looked at the properties of paired-pulse depression to estimate the pool size at each membrane potential.
Since our experimental conditions are slightly different from those of previous publications, we first revisited the properties of depression and its recovery under high and low EGTA concentrations following 20-ms depolarizations to 0 mV. To do so, bipolar cells were given pairs of 20-ms depolarizations and the magnitude of the second response was measured relative to the first. As Fig. 7 shows, we found that [EGTA] had no effect on rate of recovery of the complete IRP depleted at 0 mV. The left side of Fig. 7D shows data from all intervals tested for terminals loaded with either 0.5 or 10 mM EGTA. The right side of Fig. 7D shows the short intervals between depolarizations on an expanded timescale. The ordinate is the magnitude of the capacitance increase from the second depolarization divided by the increase from the first depolarization. The rate of IRP replenishment is the same whether terminals are under high (10 mM EGTA) or low (0.5 mM EGTA) buffering conditions. Our results essentially replicate those in previous work using whole cell patch clamp with strong depolarizations and 0.5 or 5 mM EGTA (Mennerick and Matthews 1996). Our data could be well fit by a single-exponential function with time constant τ = 1.1 or 1.4 s for terminals loaded with 0.5 or 10 mM EGTA, respectively. Fitting our data with two-exponential functions provided time constants of 0.15 and 4.9 s for terminals loaded with 0.5 mM EGTA and time constants of 1.2 and 1.2 s for terminals loaded with 10 mM EGTA. Two-exponential functions did not provide a better fit as determined by adjusted R2 (see methods), although this may reflect the noisiness in our data set rather than any inherent feature of the process itself, and thus a biexponential recovery could be masked under our experimental conditions. Indeed, previous experiments by other researchers have determined that two-exponential functions provide the best fit to recorded data not only in dissociated goldfish bipolar terminals (Gomis et al. 1999; Hull et al. 2006), but also in slice preparations (Palmer et al. 2003).
FIG. 7.

Simple paired-pulse depression of bipolar neurons. A, left: pulse protocol used in this experiment. Terminals were voltage clamped at −60 mV and given two 20-ms-long depolarizations to 0 mV with variable time between the depolarizations. The number of 0.5 mM EGTA loaded cells for each interpulse interval: 50 ms (n = 6), 100 ms (n = 8), 200 ms (n = 8), 400 ms (n = 6), 800 ms (n = 6), 1 s (n = 7), 2 s (n = 6), 5 s (n = 7), 20 s (n = 8), and 30 s (n = 9). The number of 10 mM EGTA-loaded cells for each interpulse interval: 50 ms (n = 9), 100 ms (n = 7), 200 ms (n = 10), 400 ms (n = 8), 800 ms (n = 7), 1 s (n = 7), 2 s (n = 7), 5 s (n = 6), 20 s (n = 9), and 30 s (n = 9). A, right: average peak current measured for both 0.5 mM and 10 mM EGTA-loaded terminals. The black (leftmost) bars represent current from the 1st depolarization, whereas the white (rightmost) bars represent current from the 2nd depolarization. B, left: raw current signal from a bipolar cell terminal loaded with 0.5 mM EGTA and depolarized twice with 200 ms between stimuli. B, middle: raw capacitance signal from the same terminal as at left. B, right: raw capacitance signal from a different terminal that had 2 s between stimuli. C, left: raw current signal from a bipolar cell terminal loaded with 10 mM EGTA and depolarized twice with 200 ms between stimuli. C, middle: raw capacitance signal from the same terminal as at left. C, right: raw capacitance signal from a different terminal that had 2 s between stimuli. D, left: composite data from all terminals. Filled circles represent terminals loaded with 0.5 mM EGTA, whereas open triangles are terminals loaded with 10 mM EGTA. Right: expanded abscissa from data at left to show short interpulse intervals more clearly.
Figure 8 explores paired-pulse depression at more physiologically relevant potentials (−30 to −25 mV). Figure 8, A and B shows raw capacitance traces from the same terminals used in Figs. 3 and 4, where we stimulated a second time, after a 50-ms interval, to the same membrane potential for 250 ms to measure recovery of the IRP. Figure 8A shows terminals loaded with 0.5 mM EGTA and depolarized (from left to right) to −30, −28, and −25 mV. Figure 8B shows terminals loaded with 10 mM EGTA and depolarized to the same membrane potentials as Fig. 8A. Figure 8C (left and right) shows the composite data for capacitance jumps due to the first depolarization compared with the capacitance jumps due to the second. Figure 8C, left shows the first (black) and second (white) capacitance jumps for terminals loaded with 0.5 mM EGTA; Fig. 8C, right shows capacitance jumps for terminals loaded with 10 mM EGTA. No capacitance jumps comparing the first and second were significantly different for terminals loaded with either 0.5 or 10 mM EGTA.
FIG. 8.

Simple paired-pulse depression of bipolar neurons stimulated to physiological membrane potentials. A, left: raw capacitance of bipolar neuron terminal voltage clamped at −60 mV, loaded with 0.5 mM EGTA and stimulated with two 250-ms depolarizations to −30 mV with a 50-ms interval between depolarizations. A, middle: different bipolar terminal stimulated the same as at left, only to a membrane potential of −28 mV. A, right: different bipolar terminal stimulated the same as at left, only to a membrane potential of −25 mV. B, left: raw capacitance of bipolar neuron terminal voltage clamped at −60 mV, loaded with 10 mM EGTA, and stimulated with two 250-ms depolarizations to −30 mV with a 50-ms interval between depolarizations. B, middle: different bipolar terminal stimulated the same as at left, only to a membrane potential of −28 mV. B, right: different bipolar terminal stimulated the same as at left, only to a membrane potential of −25 mV. C, left: composite capacitance jump data for all terminals loaded with 0.5 mM EGTA. The black bar (leftmost within depolarization protocol) is the capacitance jump from the 1st 250-ms depolarization, whereas the white bar (rightmost within protocol) represents the capacitance jump from the 2nd 250-ms depolarization started 50 ms after the cessation of the 1st stimulation. The 1st and 2nd depolarizations were not significantly different (P < 0.05) for any protocols. C, right: composite capacitance jump data for all terminals loaded with 10 mM EGTA. The black bar (leftmost within depolarization protocol) is the capacitance jump from the 1st 250-ms depolarization, whereas the white bar (rightmost within protocol) represents the capacitance jump from the 2nd 250-ms depolarization started 50 ms after the cessation of the 1st stimulation. The 1st and 2nd depolarizations were not significantly different (P < 0.05) for any protocols.
Our results show considerably less depression during pairs of stimuli at physiological potentials than we see in response to maximal stimuli. Is this a result of less vesicle depletion at physiological membrane potentials? To address this question, we estimated the expected capacitance response due to depletion, using Eq. 3, for each membrane potential. Using 70 and 28 fF for the pool size of terminals loaded with 0.5 and 10 mM EGTA, respectively, we calculated expected capacitance jumps of: 7.31, 13.1, and 15.1 fF for terminals loaded with 0.5 mM EGTA and depolarized to −30, −28, and −25 mV, respectively. Equation 3 assumes no recovery in the 400 to 800 ms between depolarizations. If we assume a recovery of about 25%, as we measured for intervals of 400–800 ms at 0 mV, we predict capacitance jumps of 7.6, 14.2, and 23.1 fF for depolarizations to −30, −28, and −25 mV, respectively. By comparison, we measured 5.1, 17.9, and 29 fF for the second 250-ms depolarization applied 50 ms after the first—not significantly different from our prediction.
We found similar results with terminals loaded with 10 mM EGTA. Our expected capacitance increase due to a second depolarization to −28 or −25 mV was 5.18 or 6.95 fF, respectively, if we assume no pool refilling; or 5.6 or 9.0 fF, if we assume 25% refilling. Our measured capacitance jumps for the second 250-ms depolarization was 5.1 and 10.8 fF for depolarizations to −28 or −25 mV, respectively, similar to the predictions. Together these results support the notion that the smaller degree of depression at physiological potentials is a consequence of less neurotransmitter being released and supports the notion that the IRP acts as a single homogeneous pool even at more negative membrane potentials.
Model of Ca2+ channel density at the synaptic ribbon of bipolar neurons
To account for our findings, we tested whether a simple model assuming a homogeneous pool of vesicles equidistant from multiple calcium channels could account for key features of our data. In particular, we wanted to see whether a “microdomain” model could account for 1) the differential effects of EGTA at different membrane potentials; 2) the roughly second-order dependence of release on number of open channels; and 3) the apparent increase in cooperativity with elevated EGTA. To do so, we built on the calcium dependence of exocytosis measured by Heidelberger et al. (1994) and the steady-state approximation of the spatial profile of calcium near an open calcium channel derived by Naraghi and Neher (1997), to estimate the effects of buffers and changing open channel number on exocytosis. This formulation resembles one used by Lagnado and colleagues for goldfish bipolar neurons (Burrone et al. 2002), except that it is adapted for multiple calcium channels. The relevant equations are the following
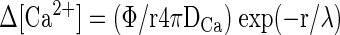 |
(5) |
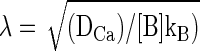 |
(6) |
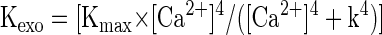 |
(7) |
where Φ is the Ca2+ flux through open calcium channels at any given time, DCa is the diffusion coefficient of free calcium, [B] is the free concentration of Ca2+ buffer, kB is the rate constant of Ca2+ buffer (EGTA in our experiments), r is the distance from the channel pore, Kexo is the exocytic rate, Kmax is the maximal rate (3,000 s−1), and k = 200 μM (Heidelberger et al. 1994). We set the values of Kmax (3,000 s−1) and k (200 μM) based on results from Heidelberger et al. (1994). The maximum open probability (Popen) of the channels was set to 0.5, similar to estimates of maximal Popen of L-type calcium channels in mouse hair cells (Brandt et al. 2005). The conductance of a single channel was set to 0.25 pA; the single L-type channel conductance measured in chick ciliary ganglion neurons (Church and Stanley 1996); and the same as assumed for goldfish bipolar cells in a previous study (von Gersdorff et al. 1998), the number of open channels determined assuming a Poisson process, and the release rate at any particular channel open probability determined as the weighted sum of the expected release rate for the number of channels open over the period of depolarization. Thus for a two-channel process with an open probability of 0.5, we would expect that 25% of the time both channels are open and the release rate is determined by the conductance through two calcium channels: 50% of the time one channel is open determines the conductance and 25% of the time no channels are open. The above-cited model determines the release rate expected as a function of distance from an open calcium channel and, to simplify the model, the vesicle is assumed to be equidistant from each open calcium channel.
We used the model to predict the degree of nonlinearity in the rate of release with membrane potential and the effects of increasing EGTA at each membrane potential. Membrane potential was emulated by varying the open probability (Popen) from 0.0135 to 0.5 (or about 37-fold), similar to the range in calcium current observed in our experiments. This neglects the effects of membrane potential on conductance, which we assume to be small since the reversal potential of Ca2+ is much more positive than 0 mV. Figure 9A plots the release rate (for 0.5 EGTA) as a function of open probability for 2-, 5-, 10-, and 20-channel arrays at distances where the release rate at Popen = 0.0135 was similar to our observed exocytic rate of 33 s−1 at −30 mV. For two channels, a distance of 9 nm gave us the closest rate, whereas 5, 10, and 20 channels gave closest rates at 12, 16, and 21 nm, respectively. As expected, release showed a greater degree of nonlinearity with increasing channel number. As Fig. 9B shows, using two channels per vesicle, a log–log plot of release rate against open probability has a slope of 1.38, indicating that release varies nearly linearly with membrane potential and results in a severe underestimate of release rate (72 s−1 from the model vs. 345 s−1 measured experimentally) at maximal open probability (Popen = 0.5). It should be noted that our measurements of rate at higher membrane potentials likely represent a lower limit, since activation kinetics for calcium channels are rate limiting for release at these potentials (Mennerick and Matthews 1996). As more channels contribute, the apparent cooperativity increases. For 5, 10, and 20 channels, the slope was 1.84, 2.15, and 2.38, respectively, similar to what we measured experimentally (1.9).
FIG. 9.

Exocytic rate compared with calcium channel open probability calculated using a multiple-channel model. A: linear plot of vesicle release rate compared with channel open probability (Popen) as calculated by the model, assuming release sites are equidistant from 2 (blue triangles), 5 (green triangles), 10 (red circles), or 20 (black squares) calcium channels, assuming 0.5 mM EGTA. The model calculates the calcium concentration as a function of distance from each number of channels. The traces in A are chosen to have a separation (to the nearest nm) between the channels and release sites that gave a release rate (0.37 s−1) closest to our capacitance measurements at −30 mV at lowest open probability (0.014). B: log–log plot of data in A. Red lines indicate best-fit lines through the data. The slopes of the lines were the following: 1.38 for 2 channels, 1.83 for 5 channels, 2.15 for 10 channels, and 2.38 for 20 channels. C: same as in A, except model calculates release rates in the presence of 10 mM EGTA. The same channel-release site separation distances are used as in A. D: log–log plot of data in C. Lines are linear best-fit curves through the data. The slopes were the following: 1.39 for 2 channels, 1.87 for 5 channels, 2.21 for 10 channels, and 2.54 for 20 channels. Symbols in B through D are the same as in A.
Our results also showed a differential sensitivity to EGTA at different membrane potentials and we adapted the same model to look at the effects of 10 mM EGTA for different Popen values (see Fig. 9, C and D). Similar to the expectations for cooperativity, a model in which many channels contribute to release better fit our results than a model in which few channels were needed. This is because in order to drive release from single channels at the high rates we observe, the vesicle must be very near the channel where EGTA has little effect, whereas with multiple channels the vesicle must be close, but not so close that EGTA has no effect on calcium concentrations. Moreover, since the slope of the relationship between calcium and release rate flattens at high calcium concentrations (e.g., when more channels are open), EGTA has less effect on release rate when more channels are open. Figure 9C shows the predicted relationship between open probability and release rate for the model for the 2-channel (blue triangles), 5-channel (green triangles), 10-channel (red circles), and 20-channel (black squares) cases. EGTA has essentially the same effect at both low and high Popen values for two channels. If we take the distance of 9 nm from the vesicle to the calcium channels, which gave us a best fit for the release rate at low open probability for two channels, then we get a 39% reduction in release rate with a 0.5 Popen and a 41% reduction with a Popen of 0.013, or a 5% greater effect (i.e., 41%/39%) with fewer open channels. By contrast, for 20 channels 21 nm away from the release site, 10 mM EGTA reduces release from 0.41 to 0.12 s−1 at Popen = 0.013 and from 1,745 to 935 at Popen = 0.5, an almost 50% greater effect on relative release rate at low Popen than high Popen. The results for 5- and 10-channel arrays give intermediate results, with a 14 and 35% greater suppression of release at lower Popen. It should be noted that the rates of exocytosis we measure at higher membrane potential are limited by the activation rate of the calcium current (Mennerick and Matthews 1996), which could mask any effects of EGTA on the rate of release at higher membrane potentials.
Since both in the model and in the data we found greater effects of EGTA on release rates with fewer open channels, one may also expect that EGTA affects the apparent cooperativity between channels. To measure this effect in the model, we also plotted the calculated release rate against Popen on a log–log plot in Fig. 9D. By comparing the slopes of the curves in Fig. 9D with those in Fig. 9B, one can look at the effect of EGTA on the apparent degree of cooperativity of release on channel open number. The slopes in 10 EGTA are 1.39 (1.38 in 0.5 EGTA), 1.87 (1.84), 2.21 (2.15), and 2.54 (2.38) for 2-, 5-, 10-, and 20-channel arrays, respectively. Thus the model predicts greater effects of EGTA on the apparent cooperativity of release as channel number increases, although the predictions of the model even with 20 channels are more subtle than the observed results (cooperativity of 3 vs. 2 for 10 mM vs. 0.5 mM EGTA, respectively; see Fig. 6).
DISCUSSION
We have investigated the kinetics and magnitude of exocytosis, paired-pulse depression, and recovery of the immediately releasable pool of vesicles (IRP) with different concentrations of the exogenously applied Ca2+ buffer EGTA, in pursuit of characterizing bipolar neuron properties in response to physiologically relevant membrane depolarizations. We show here that: 1) the pool of vesicles capable of rapid exocytosis at 0 mV in low buffering conditions is larger than the cohort of vesicles that are contained at the base of the synaptic ribbon. 2) Release at physiological potentials draws from a subset of these releasable vesicles for depolarizations ≤250 ms. 3) Elevated EGTA concentrations more strongly reduce release at more negative membrane potentials. 4) The relationship between membrane potential and release rate is highly nonlinear. Our results refine previous studies using stronger stimuli, which showed that exocytosis of membrane equivalent to the expected number of vesicles located on the lowest row of synaptic vesicles is accomplished rapidly (<15 ms). We propose that under physiological conditions, exocytosis is accomplished predominantly at the same spatial location as synaptic ribbons, the bipolar neuron is capable of transient and sustained exocytosis, and that individual vesicles are sensitive to the coordinated opening of multiple calcium channels to achieve high exocytic rates.
Maximal Ca2+ current activation and density of synaptic ribbons
A number of previous studies have demonstrated that when bipolar terminals are stimulated to voltages that allow for maximal Ca2+ entry, exocytosis has rapid kinetics for depolarizations <15 ms and is rate limited by the opening speed of Ca2+ channels. After this initial burst, exocytosis increases at a slower rate (Gomis et al. 1999; Mennerick and Matthews 1996). In this work, we have confirmed these results while discovering that under buffering conditions of 0.5 mM EGTA there is little additional exocytosis between 20 and 60 ms after initiation of the depolarization, similar to results looking at glutamate release measured using catfish horizontal cells to report neurotransmitter release (von Gersdorff et al. 1998). The magnitude of the capacitance response for these depolarization times is about 70 fF and represents 2,470–2,750 vesicles. By contrast, under buffering conditions of 10 mM EGTA, we were able to abolish any exocytosis in excess of about 30 fF, until step depolarizations were nearly 500 ms in duration. The close to 30 fF represents approximately the same magnitude as that seen in previous studies designed to probe the size and potentially discrete nature of the immediately releasable pool (IRP) (Burrone et al. 2002; Gomis et al. 1999; Mennerick and Matthews 1996). In our work, similar to Burrone et al. (2002), the magnitude of the capacitance jump for >10-ms depolarizations is significantly smaller when 10 mM EGTA is introduced (P < 0.05). In the study by Burrone et al. (2002), the authors also uncovered a pool of about 23 fF, which was insensitive to ≤70 mM EGTA, suggesting that vesicles located within 30 nm of Ca2+ channels would be unaffected by EGTA due to its relatively slow Kon. Our experiments provide a measure of synaptic ribbon number within cells of the same preparation and in relation to synaptic terminal size and allow us to provide reasonable estimates of the number of vesicles that make up both the EGTA- sensitive and -insensitive pools. From our data it is possible to anticipate about 41 ribbons in our typical bipolar neuron terminal. If exocytosis occurs solely at synaptic ribbons during these brief stimuli, 28 fF at around 41 ribbons corresponds to about 0.7 fF/ribbon, which is equivalent to approximately 27 vesicles/ribbon. This value is very similar to the anticipated size of the lowest row of vesicles on synaptic ribbons (von Gersdorff et al. 1996).
By contrast, under low buffering conditions, we found a capacitance increase that saturated within 20 ms that was about threefold larger than the number of vesicles thought to reside at the bottom of the ribbon, a larger pool than that previously found in most studies, but approximately the same as that found in terminal recorded in slice preparations (Palmer et al. 2003). To account for this extra release, we consider three possible sources for the additional release: 1) additional vesicles from locations distal to the plasma membrane on the ribbon; 2) clusters of vesicles nearby, but not directly tethered to the base of the ribbon (Lenzi et al. 1999); and 3) extra-ribbon release of vesicles or large endosomal-like structures (Coggins et al. 2007). Since total internal reflection fluorescence microscopy imaging studies have shown little contribution of extra-ribbon release of vesicles or endosomes within 30 ms of the initiation of a depolarization (Coggins et al. 2007; Midorikawa et al. 2007; Zenisek et al. 2002) we suggest that extra-ribbon release is not a major contributor to the EGTA-sensitive portion of the IRP. Instead, we favor the idea that the extra capacitance increase elicited under low buffering conditions is either due to fusion of vesicles in more distal locations on the synaptic ribbon or due to docked vesicles just adjacent to the ribbon.
Membrane distal release sites on the ribbon would be consistent with a model of release due to compound fusion: the exocytosis of multiple vesicles with one another prior to or after exocytosis (Edmonds et al. 2004; Heidelberger et al. 2002; LoGiudice et al. 2008; Matthews and Sterling 2008). Although our results cannot distinguish compound exocytosis from other mechanisms, the idea of compound exocytosis is consistent with numerous recent studies describing large quantal events from ribbon synapses, thought to arise simultaneously from the release of multiple quanta of neurotransmitter (Glowatzki and Fuchs 2002; Goutman and Glowatzki 2007; Singer et al. 2004). Although multivesicular release does not require compound exocytosis, it provides an attractive mechanism by which multiple quanta could be released simultaneously. Consistent with this idea, tubular structures resembling multiple vesicles fused together have been found near synaptic ribbons in goldfish bipolar cells following strong stimulation (Matthews and Sterling 2008). Moreover, compound exocytosis is also consistent with studies measuring rates of exocytosis in response to the rapid release of calcium from photolyzable calcium buffers (Heidelberger 1998; Heidelberger et al. 1994). In these studies, Heidelberger and colleagues demonstrated that a pool of vesicles, approximately the size of the number of vesicles populating the ribbon, could be released in <1 ms. Through a series of studies looking at the ability to deplete the pool of vesicles releasable by photolysis by instead using electrical step depolarizations, they demonstrated that the same vesicles released by photolysis and step depolarizations populated both pools. Together, these results could all be explained by a mechanism in which synaptic vesicles fused together prior to fusing with the plasma membrane.
One must also consider synaptic vesicles on the periphery of the ribbon as a potential source for the additional vesicles we find in the IRP. Electron tomographic reconstructions of the frog saccular hair cells, another ribbon synapse, indicate that many apparently docked vesicles reside just adjacent to the synaptic ribbon, but not attached to it. Although near-ribbon regions of enhanced vesicle docking have not been described in bipolar cells, electron microscopic images often show vesicles proximal to the membrane even outside of ribbon locations (e.g., Coggins et al. 2007). Given that extra-ribbon release has been described to occur at regions distal to the ribbon during long depolarizations (e.g., Midorikawa et al. 2007; Zenisek 2008), it seems plausible that vesicles more proximal to the ribbon, but not directly bound to it, may be released during even brief depolarizations to 0 mV.
Exocytosis at physiological membrane potentials
The maximal currents elicited by membrane depolarizations to −10 to 0 mV are unlikely to occur in vivo, as demonstrated by studies designed to test the voltage response of bipolar neurons to spot and annular light (Joselevitch and Kamermans 2007; Kaneko 1970; Lasansky 1992; Werblin and Dowling 1969). Thus the wealth of data on exocytosis under maximal stimulation might not fully capture response properties of bipolar neurons and we sought to add data to previous work testing representative membrane depolarizations (Burrone and Lagnado 2000; von Gersdorff and Matthews 1997).
In our studies, long depolarizations (250 ms) elicited small capacitance increases for terminals loaded with 0.5 mM EGTA and stimulated by step depolarizations between −35 and −28 mV. These capacitance jumps were fractions of the IRP measured by using 15-ms depolarizations to 0 mV (Fig. 1). Shorter-duration depolarizations showed progressively smaller capacitance increases (data not shown) in a qualitatively similar manner as depolarizations to 0 mV (Gomis et al. 1999; Mennerick and Matthews 1996).
It is important to note that the charge carried by Ca2+ ions was not the determining factor for the small capacitance increases observed. The amount of charge carried through channels when terminals were depolarized to −30 or −28 mV for 250 ms was greater than the charge entering during 15-ms depolarizations to 0 mV, on average, yet the capacitance increase was significantly smaller (Figs. 1 and 3). Even more dramatically, transient calcium entry on repolarization to −30 mV was capable of driving neurotransmitter release in 10 mM EGTA, whereas 250 ms at that potential resulted in no release (Fig. 5). Thus having multiple channels open simultaneously, even briefly, is more effective in driving release than having fewer channels open for longer. This is expected from the nonlinear relationship between calcium and exocytosis (Dodge Jr and Rahamimoff 1967; Heidelberger et al. 1994), but only under conditions where calcium through multiple channels sum together at the release site, a result also hinted at by Ca2+ current calculations in goldfish bipolar neurons (Palmer et al. 2003). Thus we favor a “microdomain” model, where each release site senses calcium in the vicinity of multiple channels rather than a “nanodomain” model in which release is usually driven by a “nanodomain” of elevated calcium concentrations just proximal to single open calcium channels. This idea is further supported by our capacitance measurements with elevated EGTA concentrations. We find that exocytosis is effectively blocked by 10 mM EGTA at low membrane potentials (Fig. 4), indicating that vesicles reside at distances sufficiently far from calcium channels to allow for EGTA to significantly buffer the entering calcium. Last, the nanodomain model predicts near-linear changes in release rates with calcium entry when calcium levels are changed by membrane potential (Brandt et al. 2005). We instead find a highly nonlinear relationship (Fig. 8).
To account for our results, we used a simple microdomain model that calculates the expected release rates as a function of distance from an array of channels equidistant from the release site. Using reasonable estimates for the parameters, we find that the summation of calcium through many (5–20) channels fits our data much better than when release is driven by few channels. Specifically, the “many-channel” model predicts: 1) the nonlinearity in release we observe with depolarization; 2) the increase in the degree of apparent cooperativity with elevated EGTA; and 3) the stronger effects of EGTA with fewer channels open. By comparison, a model with only two channels predicts a near-linear change in release rate with membrane potential with no effect of elevated EGTA.
It should be noted, however, despite the successes of the model, it likely does not represent a realistic representation of the active zone architecture. For example, in the absence of information about the exact location or numbers of channels near release sites, we chose to put channels at equal distances from the vesicle, which is unlikely to be true. It seems more likely that vesicles may reside near different numbers and different distances from calcium channels, as suggested by the differential sensitivity of some vesicles to EGTA (e.g., Figs. 4 and 5 and Burrone and Lagnado 2000). The single-channel conductance of calcium channels in the goldfish bipolar cell has not yet been measured and may be different from what we assumed here. Moreover, endogenous buffers and local diffusion barriers may act to change the profile in the spread of calcium near channels. Indeed, our “many-channel” model, although predictive of certain aspects of release, does not predict our results with quantitative precision. Specifically, the effects of EGTA on apparent cooperativity and release rate at lower channel open probability is more modest in the model than we measured.
Of note, our results are in stark contrast to those in the mouse inner hair cells, where changes in calcium channel number cause near-linear changes in release rate (Brandt et al. 2005). This may reflect differences in the physiology of hair cells, which encode sound frequency in part by releasing neurotransmitter in phase with changes of membrane potential. In these cells, release driven by nanodomains is thought to be critical to maintain phase-locking across different sound intensities.
Our results should not be interpreted to mean that single channels are incapable of driving release from goldfish bipolar cells. The capacitance technique lacks the sensitivity to accurately measure release rates at the most negative physiological membrane potentials and it remains possible that an undetected linear relationship between membrane potential and release may exist at these potentials. Instead, our results indicate that synaptic vesicles can and do sense calcium across many channels and this gives rise to a nonlinear relationship between membrane potential and release at sufficiently positive potentials. Across the range of physiological potentials, the sampling of calcium from many channels would be expected to reduce the noise associated with the stochastic opening of individual calcium channels, thus allowing for more stereotyped responses to a given depolarizing stimulus (Doering et al. 2005). Similarly, response variability is further reduced by the ribbon having many vesicles poised to respond to their own subset of channel openings. Thus one potentially important role for the arrangement of the bipolar cell ribbon-type active zone may be to reduce the stochastic variability inherent in vesicle release and channel opening by signal averaging across multiple channels and multiple vesicles (Barg et al. 2001).
Paired-pulse depression at physiological membrane potentials
Previous measurements of neurotransmitter release from bipolar cell synapses (e.g., von Gersdorff and Matthews 1994a) have indicated that maximal stimulation of bipolar cells results in profound depression of neurotransmitter release in response to even very brief stimuli (Mennerick and Matthews 1996). Such a profound degree of depression might render bipolar neurons unresponsive to the second of a pair of light flashes within its receptive field; therefore we tested whether depression is also exhibited in response to more modest depolarizations within the physiological range. We find that responses to pairs of depolarizations still exhibit depression of the second response at most membrane potentials, but that the amount of depression is considerably less pronounced at these potentials. In addition, bipolar cells are still capable of release of a rapid transient component even after a prolonged depolarization to −28 mV (Figs. 3 and 4). Our results fit a model in which the IRP for each buffering condition is a homogeneous pool, which is released as a single kinetic component.
Summary
Our data suggest a different model of the spatial organization of channels and vesicles than that proposed in inner hair cells, where researchers have concluded that only a few channels drive exocytosis of any particular vesicle (Brandt et al. 2005). Instead we find that many more Ca2+ channels contribute to release probability of each vesicle, consistent with overlapping Ca2+ microdomains contributing to each release site, previously predicted based on the size of the calcium current in these cells (von Gersdorff et al. 1998). This arrangement will have the beneficial effect of reducing stochastic noise in the synaptic response and thus may be important for maintaining the exquisite sensitivity and reliability of the visual system.
Acknowledgments
This work was supported by National Eye Institute Grant EY-014990.
REFERENCES
- Adler et al. 1991.Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci 11: 1496–1507, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine et al. 1991.Augustine GJ, Adler EM, Charlton MP. The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann NY Acad Sci 635: 365–381, 1991. [DOI] [PubMed] [Google Scholar]
- Axelrod 2001.Axelrod D Total internal reflection fluorescence microscopy in cell biology. Traffic 2: 764–774, 2001. [DOI] [PubMed] [Google Scholar]
- Barg et al. 2001.Barg S, Ma X, Eliasson L, Galvanovskis J, Gopel SO, Obermuller S, Platzer J, Renstrom E, Trus M, Atlas D, Striessnig J, Rorsman P. Fast exocytosis with few Ca(2+) channels in insulin-secreting mouse pancreatic B cells. Biophys J 81: 3308–3323, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt et al. 2005.Brandt A, Khimich D, Moser T. Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J Neurosci 25: 11577–11585, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone and Lagnado 2000.Burrone J, Lagnado L. Synaptic depression and the kinetics of exocytosis in retinal bipolar cells. J Neurosci 20: 568–578, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone et al. 2002.Burrone J, Neves G, Gomis A, Cooke A, Lagnado L. Endogenous calcium buffers regulate fast exocytosis in the synaptic terminal of retinal bipolar cells. Neuron 33: 101–112, 2002. [DOI] [PubMed] [Google Scholar]
- Church and Stanley 1996.Church PJ, Stanley EF. Single L-type calcium channel conductance with physiological levels of calcium in chick ciliary ganglion neurons. J Physiol 496: 59–68, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins et al. 2007.Coggins MR, Grabner CP, Almers W, Zenisek D. Stimulated exocytosis of endosomes in goldfish retinal bipolar neurons. J Physiol 584: 853–865, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge Jr and Rahamimoff 1967.Dodge FA Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol 193: 419–432, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering et al. 2005.Doering CJ, Hamid J, Simms B, McRory JE, Zamponi GW. Cav1.4 encodes a calcium channel with low open probability and unitary conductance. Biophys J 89: 3042–3048, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds et al. 2004.Edmonds BW, Gregory FD, Schweizer FE. Evidence that fast exocytosis can be predominantly mediated by vesicles not docked at active zones in frog saccular hair cells. J Physiol 560: 439–450, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet et al. 2000.Gentet LJ, Stuart GJ, Clements JD. Direct measurement of specific membrane capacitance in neurons. Biophys J 79: 314–320, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki and Fuchs 2002.Glowatzki E, Fuchs PA. Transmitter release at the hair cell ribbon synapse. Nat Neurosci 5: 147–154, 2002. [DOI] [PubMed] [Google Scholar]
- Gomis et al. 1999.Gomis A, Burrone J, Lagnado L. Two actions of calcium regulate the supply of releasable vesicles at the ribbon synapse of retinal bipolar cells. J Neurosci 19: 6309–6317, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutman and Glowatzki 2007.Goutman JD, Glowatzki E. Time course and calcium dependence of transmitter release at a single ribbon synapse. Proc Natl Acad Sci USA 104: 16341–16346, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger 1998.Heidelberger R Adenosine triphosphate and the late steps in calcium-dependent exocytosis at a ribbon synapse. J Gen Physiol 111: 225–241, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger et al. 1994.Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature 371: 513–515, 1994. [DOI] [PubMed] [Google Scholar]
- Heidelberger and Matthews 1992.Heidelberger R, Matthews G. Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. J Physiol 447: 235–256, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger et al. 2002.Heidelberger R, Sterling P, Matthews G. Roles of ATP in depletion and replenishment of the releasable pool of synaptic vesicles. J Neurophysiol 88: 98–106, 2002. [DOI] [PubMed] [Google Scholar]
- Hudspeth and Lewis 1988.Hudspeth AJ, Lewis RS. Kinetic analysis of voltage- and ion-dependent conductances in saccular hair cells of the bull-frog, Rana catesbeiana. J Physiol 400: 237–274, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull et al. 2006.Hull C, Studholme K, Yazulla S, von Gersdorff H. Diurnal changes in exocytosis and the number of synaptic ribbons at active zones of an on-type bipolar cell terminal. J Neurophysiol 96: 2025–2033, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa and Hudspeth 1996.Issa NP, Hudspeth AJ. Characterization of fluo-3 labelling of dense bodies at the hair cell's presynaptic active zone. J Neurocytol 25: 257–266, 1996. [DOI] [PubMed] [Google Scholar]
- Joselevitch and Kamermans 2007.Joselevitch C, Kamermans M. Interaction between rod and cone inputs in mixed-input bipolar cells in goldfish retina. J Neurosci Res 85: 1579–1591, 2007. [DOI] [PubMed] [Google Scholar]
- Kaneko 1970.Kaneko A Physiological and morphological identification of horizontal, bipolar and amacrine cells in goldfish retina. J Physiol 207: 623–633, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko 1973.Kaneko A Receptive field organization of bipolar and amacrine cells in the goldfish retina. J Physiol 235: 133–153, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagnado et al. 1996.Lagnado L, Gomis A, Job C. Continuous vesicle cycling in the synaptic terminal of retinal bipolar cells. Neuron 17: 957–967, 1996. [DOI] [PubMed] [Google Scholar]
- Lasansky 1992.Lasansky A Properties of depolarizing bipolar cell responses to central illumination in salamander retinal slices. Brain Res 576: 181–196, 1992. [DOI] [PubMed] [Google Scholar]
- Lenzi et al. 1999.Lenzi D, Runyeon JW, Crum J, Ellisman MH, Roberts WM. Synaptic vesicle populations in saccular hair cells reconstructed by electron tomography. J Neurosci 19: 119–132, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau and Neher 1988.Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflügers Arch 411: 137–146, 1988. [DOI] [PubMed] [Google Scholar]
- LoGiudice et al. 2008.LoGiudice L, Sterling P, Matthews G. Mobility and turnover of vesicles at the synaptic ribbon. J Neurosci 28: 3150–3158, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews and Sterling 2008.Matthews G, Sterling P. Evidence that vesicles undergo compound fusion on the synaptic ribbon. J Neurosci 28: 5403–5411, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick and Matthews 1996.Mennerick S, Matthews G. Ultrafast exocytosis elicited by calcium current in synaptic terminals of retinal bipolar neurons. Neuron 17: 1241–1249, 1996. [DOI] [PubMed] [Google Scholar]
- Midorikawa et al. 2007.Midorikawa M, Tsukamoto Y, Berglund K, Ishii M, Tachibana M. Different roles of ribbon-associated and ribbon-free active zones in retinal bipolar cells. Nat Neurosci 10: 1268–1276, 2007. [DOI] [PubMed] [Google Scholar]
- Mintz et al. 1995.Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron 15: 675–688, 1995. [DOI] [PubMed] [Google Scholar]
- Mitra et al. 2004.Mitra K, Ubarretxena-Belandia I, Taguchi T, Warren G, Engelman DM. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc Natl Acad Sci USA 101: 4083–4088, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi and Neher 1997.Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci 17: 6961–6973, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves and Lagnado 1999.Neves G, Lagnado L. The kinetics of exocytosis and endocytosis in the synaptic terminal of goldfish retinal bipolar cells. J Physiol 515: 181–202, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada et al. 1995.Okada T, Horiguchi H, Tachibana M. Ca(2+)-dependent Cl− current at the presynaptic terminals of goldfish retinal bipolar cells. Neurosci Res 23: 297–303, 1995. [DOI] [PubMed] [Google Scholar]
- Palmer et al. 2003.Palmer MJ, Hull C, Vigh J, von Gersdorff H. Synaptic cleft acidification and modulation of short-term depression by exocytosed protons in retinal bipolar cells. J Neurosci 23: 11332–11341, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts 1993.Roberts WM Spatial calcium buffering in saccular hair cells. Nature 363: 74–76, 1993. [DOI] [PubMed] [Google Scholar]
- Sakaba et al. 1997.Sakaba T, Tachibana M, Matsui K, Minami N. Two components of transmitter release in retinal bipolar cells: exocytosis and mobilization of synaptic vesicles. Neurosci Res 27: 357–370, 1997. [DOI] [PubMed] [Google Scholar]
- Singer and Diamond 2003.Singer JH, Diamond JS. Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci 23: 10923–10933, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer et al. 2004.Singer JH, Lassova L, Vardi N, Diamond JS. Coordinated multivesicular release at a mammalian ribbon synapse. Nat Neurosci 7: 826–833, 2004. [DOI] [PubMed] [Google Scholar]
- Spiwoks-Becker et al. 2004.Spiwoks-Becker I, Glas M, Lasarzik I, Vollrath L. Mouse photoreceptor synaptic ribbons lose and regain material in response to illumination changes. Eur J Neurosci 19: 1559–1571, 2004. [DOI] [PubMed] [Google Scholar]
- Tachibana and Okada 1991.Tachibana M, Okada T. Release of endogenous excitatory amino acids from ON-type bipolar cells isolated from the goldfish retina. J Neurosci 11: 2199–2208, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana et al. 1993a.Tachibana M, Okada T, Arimura T, Kobayashi K. Dihydropyridine-sensitive calcium current mediates neurotransmitter release from retinal bipolar cells. Ann NY Acad Sci 707: 359–361, 1993a. [DOI] [PubMed] [Google Scholar]
- Tachibana et al. 1993b.Tachibana M, Okada T, Arimura T, Kobayashi K, Piccolino M. Dihydropyridine-sensitive calcium current mediates neurotransmitter release from bipolar cells of the goldfish retina. J Neurosci 13: 2898–2909, 1993b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson et al. 2004.Thoreson WB, Rabl K, Townes-Anderson E, Heidelberger R. A highly Ca2+-sensitive pool of vesicles contributes to linearity at the rod photoreceptor ribbon synapse. Neuron 42: 595–605, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollrath and Spiwoks-Becker 1996.Vollrath L, Spiwoks-Becker I. Plasticity of retinal ribbon synapses. Microsc Res Tech 35: 472–487, 1996. [DOI] [PubMed] [Google Scholar]
- von Gersdorff and Matthews 1994a.von Gersdorff H, Matthews G. Dynamics of synaptic vesicle fusion and membrane retrieval in synaptic terminals. Nature 367: 735–739, 1994a. [DOI] [PubMed] [Google Scholar]
- von Gersdorff and Matthews 1994b.von Gersdorff H, Matthews G. Inhibition of endocytosis by elevated internal calcium in a synaptic terminal. Nature 370: 652–655, 1994b. [DOI] [PubMed] [Google Scholar]
- von Gersdorff and Matthews 1996.von Gersdorff H, Matthews G. Calcium-dependent inactivation of calcium current in synaptic terminals of retinal bipolar neurons. J Neurosci 16: 115–122, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff and Matthews 1997.von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. J Neurosci 17: 1919–1927, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff et al. 1998.von Gersdorff H, Sakaba T, Berglund K, Tachibana M. Submillisecond kinetics of glutamate release from a sensory synapse. Neuron 21: 1177–1188, 1998. [DOI] [PubMed] [Google Scholar]
- von Gersdorff et al. 1996.von Gersdorff H, Vardi E, Matthews G, Sterling P. Evidence that vesicles on the synaptic ribbon of retinal bipolar neurons can be rapidly released. Neuron 16: 1221–1227, 1996. [DOI] [PubMed] [Google Scholar]
- Werblin and Dowling 1969.Werblin FS, Dowling JE. Organization of the retina of the mudpuppy, Necturus maculosus. II. Intracellular recording. J Neurophysiol 32: 339–355, 1969. [DOI] [PubMed] [Google Scholar]
- Zenisek 2008.Zenisek D Vesicle association and exocytosis at ribbon and extraribbon sites in retinal bipolar cell presynaptic terminals. Proc Natl Acad Sci USA 105: 4922–4927, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek et al. 2003.Zenisek D, Davila V, Wan L, Almers W. Imaging calcium entry sites and ribbon structures in two presynaptic cells. J Neurosci 23: 2538–2548, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek et al. 2004.Zenisek D, Horst NK, Merrifield C, Sterling P, Matthews G. Visualizing synaptic ribbons in the living cell. J Neurosci 24: 9752–9759, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek et al. 2000.Zenisek D, Steyer JA, Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature 406: 849–854, 2000. [DOI] [PubMed] [Google Scholar]
- Zenisek et al. 2002.Zenisek D, Steyer JA, Feldman ME, Almers W. A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron 35: 1085–1097, 2002. [DOI] [PubMed] [Google Scholar]
- Zucker and Fogelson 1986.Zucker RS, Fogelson AL. Relationship between transmitter release and presynaptic calcium influx when calcium enters through discrete channels. Proc Natl Acad Sci USA 83: 3032–3036, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]