

dHDAC6 functions to suppress α-synuclein-induced neurodegeneration and locomotion defects in a Drosophila PD model through promoting α-synuclein-enriched inclusion formation while reducing the toxic oligomers.
Abstract
Parkinson's disease (PD) is associated with progressive degeneration of dopaminergic (DA) neurons. We report for the first time that the Drosophila histone deacetylase 6 (dHDAC6) plays a critical role in the protection of DA neurons and the formation of α-synuclein inclusions by using a Drosophila PD model constructed by ectopic expression of human α-synuclein. Depletion of dHDAC6 significantly enhances the effects caused by ectopic expression of α-synuclein, namely, loss of DA neurons, retinal degeneration, and locomotor dysfunction. Expression of α-synuclein in the DA neurons leads to fewer inclusions in the brains of dHDAC6 mutant flies than in wild-type flies. Conversely, overexpression of dHDAC6 is able to suppress the α-synuclein–induced DA neuron loss and retinal degeneration and promote inclusion formation. Furthermore, mutation of dHDAC6 reinforces the accumulation of oligomers that are suggested to be a toxic form of α-synuclein. We propose that α-synuclein inclusion formation in the presence of dHDAC6 protects DA neurons from being damaged by oligomers, which may uncover a common mechanism for synucleinopathies.
INTRODUCTION
As the second most common neurodegenerative disease, Parkinson's disease (PD) often impairs the sufferer's motor skills, speech, and other neuron-innervated functions (Jankovic, 2008). Neurodegenerative diseases are often featured as vulnerable neurons under misfolded protein stress (Taylor et al., 2002). The prominent pathological characteristics of PD are progressive loss of dopaminergic (DA) neurons and emergence of proteinaceous inclusions named Lewy bodies (LBs). The major component of LBs is a small neuronal protein α-synuclein (Lang and Lozano, 1998), which is the first-identified genetic cause of PD (Polymeropoulos et al., 1996, 1997). However, the link between α-synuclein and the neuron degeneration is not simply a matter of dosage of α-synuclein, although excess of this protein leads to higher cytotoxicity (Singleton et al., 2003). The conformation of α-synuclein has also been shown to play an important role in the process of α-synuclein–induced neuron degeneration (Outeiro et al., 2008). The oligomeric intermediates of α-synuclein aggregates are implicated to be the toxic species (Outeiro et al., 2008). Despite of the establishment of PD models in flies (Feany and Bender, 2000), mice (Masliah et al., 2000), and primates (Kirik et al., 2003), the detailed mechanism responsible for inclusion formation and α-synuclein neurotoxicity in vivo remains largely unclear.
Based on genetic studies of either inducing or suppressing PD pathological phenotypes, several genes have been identified to be involved in two types of PD pathogenesis. One type is autosomal recessive-juvenile Parkinson's (AR-JP) disease in which dopaminergic neuron loss is not associated with LB formation (Kitada et al., 1998). The other type is autosomal dominant familial PD mainly caused by missense mutation in α-synuclein (Polymeropoulos et al., 1997; Kruger et al., 1998). In the latter case, the development of α-synuclein–induced PD is considered to be dependent on the forms and the dosage of α-synuclein (Singleton et al., 2003). We set off to explore factors responsible for this complex event, and the relationship among the pathological hallmarks of Parkinson's disease.
The candidate factors relevant to the pathogenesis of PD should meet at least two criteria: 1) they must be modifiers (either suppressors or enhancers) of PD-like degenerative phenotypes; and 2) they must regulate α-synuclein aggregation, eventually its neurotoxicity. In this context, histone deacetylase 6 (HDAC6) possesses properties that place it on the top of the list. HDAC6 has been reported to rescue polyglutamine-mediated neurodegeneration in an autophagy-dependent manner (Pandey et al., 2007). It senses ubiquitinated aggregates and consequently activates the expression of major chaperones, such as heat-shock protein (Hsp)70 and Hsp25 (Boyault et al., 2007b). It directly facilitates aggresome formation via interacting with ubiquitinated proteins as a response to misfolded protein stress (Kawaguchi et al., 2003), and its concentration determines the fate of ubiquitinated proteins (Boyault et al., 2006). Here, we describe studies to investigate the physiological functions of HDAC6 in PD pathogenesis by using the well established Drosophila PD model (Feany and Bender, 2000). Based on analyses of the dHDAC6 null alleles generated with targeted gene knockout technique, our data reveal that dHDAC6 is a suppressor of α-synuclein–induced PD-like phenotypes in Drosophila. It promotes α-synuclein-enriched inclusion formation while reducing the oligomer form, a function that we suggest to be crucial for inhibiting the toxicity of α-synuclein. Our results provide a direct evaluation of the toxicity of different forms of α-synuclein during PD pathogenesis and highlight the role of dHDAC6 in handling the toxic α-synuclein forms by promoting them to become inclusions in vivo.
MATERIALS AND METHODS
Fly Stocks and Genetics
Flies were cultured under standard conditions at 25°C otherwise stated. Transgenic fly lines of UAS-α-synuclein, UAS-mCD8::GFP, GMR-GAL4, and UAS-Prosbeta21 were obtained from the Bloomington Stock Center (Bloomington, IN). TH-GAL4 line was a kind gift from Dr. Li Liu (The Institute of Biophysics, Chinese Academy of Sciences, Beijing, China). TH-GAL4, UAS-α-synuclein/TM6B, Tb flies were generated through recombining TH-GAL4 transgene and UAS-α-synuclein transgene which are both on the third chromosome. To generate the UAS-dHDAC6 construct, dHDAC6 coding sequence which encodes 1128 amino acids referring to HDAC6-RA on the Flybase was amplified from wild-type Drosophila cDNA library and cloned into the pUAST vector. The final transformation construct was confirmed by DNA sequencing. P-element mediated germline transformation was performed using standard procedures.
Generation of dHDAC6 Mutant Flies
The dHDAC6 genomic segment with intended modifications was cloned into the pTARG vector (Egli et al., 2006) to make the gene targeting construct. Mutations were introduced with polymerase chain reaction (PCR) by changing AAACATGGT to AGATCT (dHDAC6 start codon is boxed). Primers used in this process were 5′-CAACAGATCTGAGTACTGGCAGATTATTGCCG-3′ and 5′-TGACAGATCTGTGGGATTTCGAGTTGGC-3′ (the restriction site is underlined). Mutation of AAACATGGT to AGATCT generates a new restriction site (BglII) for identification of mutant DNA. Oligonucleotides used to introduce I-SceI recognition site were AATTTAGGGATAACAGGGTAAT and AATTATTACCCTGTTATCCCTA. The donor plasmid was transformed into Drosophila by using standard procedures. The dHDAC6 mutant flies were generated by “ends-in” targeting strategy (Rong and Golic, 2000). First, the donor transgenic flies were crossed to flies bearing heat-inducible FLP and I-SceI genes. The extrachromosomal target-homologous molecule carrying w+ marker was produced after FLP-mediated excision and I-SceI–mediated cutting. Homologous recombination of this targeting molecule with the endogenous dHDAC6 locus produced a tandem duplication which could be screened by w+ marker. Subsequently, for the reduction of the duplicates (wild-type and mutant forms of dHDAC6 locus) to a single copy allele, the homologous recombination was stimulated by an I-CreI–generated double-strand break between the duplicated sequences. In the reduction step, w− flies were selected and further tested for presence of the introduced mutation.
Western Blot and Immunoprecipitation
Adult fly heads with correct genotypes were collected, and proteins were extracted with radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 1 mM phenylmethylsulfonyl fluoride) and analyzed by SDS-10% polyacrylamide gel electrophoresis (PAGE). The antibodies used in this experiment are: rabbit anti-dHDAC6 (1:1000; a kind gift from Dr. Hao Li, Novartis Institutes for Biomedical Research, Cambridge, MA; Chen et al., 2008) and rabbit anti-actin (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA). For coimmunoprecipitations, proteins were extracted from 300 brains of w;TH-GAL4/UAS-α-synuclein and w;TH-GAL4/+ of 20-d-old flies and then incubated with agarose beads (Invitrogen, Carlsbad, CA) coupled with A11 antibody (Millipore Bioscience Research Reagents, Temecula, CA) or immunoglobulin G as a control for 8 h at 4°C. After five washes, precipitates were eluted and subjected to SDS-10% PAGE. Rabbit anti-dHDAC6 antibody (1:1000) and anti-α-synuclein antibody (clone 42, 1:1000, BD Biosciences, San Jose, CA) were used for Western blot.
Fractionation Assay
Fractionations of adult fly head lysates were carried out according to a protocol described previously (Chen and Feany, 2005). In brief, 350 heads of correct genotypes with right age were homogenized at 4°C in buffer A (50 mM Tris-HCl, pH 7.5, 1 mM EGTA, 1 mM dithiothreitol with protease inhibitors) and centrifuged at 100,000g for 30 min. The resulting pellets were sequentially extracted by homogenization in Triton X-100 (buffer A containing 1% Triton X-100, 10% sucrose, and 0.5 M sodium chloride), Sarkosyl (50 mM Tris-HCl, pH 7.5, 1% Sarkosyl, and 1 mM EGTA), and urea (50 mM Tris-HCl, pH 7.5, 8 M urea, and 1 mM EGTA) followed by centrifugation at 100,000g for 30 min. Sarkosyl-insoluble and urea-soluble pellets (P) and Tris-soluble supernatants (S) were separated by SDS-PAGE and analyzed by immunoblotting with anti-α-synuclein antibody (clone 42; 1:1000).
Climbing Assay
Climbing assay was performed essentially according to the published protocols (Feany and Bender, 2000; Coulom and Birman, 2004), with some modifications. Twenty adult males were placed into a vertical plastic tube (18 cm in length and 2 cm in diameter). Thirty seconds after being tapped to the bottom of the tube, the number of the flies climbed up over 15 cm was counted. The climbing scores represent the mean percentage of flies that reached the 15-cm line in the total number of flies that were tested. Five trials were performed in each experiment at 1-min intervals, and six experiments were carried out for each genotype. The results were presented as mean ± SEM of the scores obtained in six independent experiments.
Histological Examination of Adult Fly Retinas
For semithin sections, fly heads were fixed with 2.5% glutaraldehyde and postfixed in 1% osmium tetroxide. Then increasing gradient dehydration was performed using ethanol and propylene oxide. Finally, the fixed tissues were embedded in epoxy resin. Tangential retinal sections were performed at a thickness of 1 μm and stained with toluidine blue. Images were obtained with a light microscope (Nikon, Tokyo, Japan) using 60× objective lens.
Immunostaining and Confocal Imaging
Immunostaining of whole-mount adult fly brains were performed as described previously (Wang et al., 2008). In brief, adult fly brains with correct genotypes were dissected in phosphate-buffered saline (PBS) and then fixed with 4% paraformaldehyde for 1 h, followed by washing 3 × 15 min in 0.5% PBT (PBS with 0.5% Triton X-100). The brains were incubated with primary antibody and then secondary antibody, and finally were mounted in VECTASHIELD (Vector Laboratories, Burlingame, CA). The following primary antibodies were used: rabbit anti-green fluorescent protein (GFP) (1:1000; Invitrogen), mouse monoclonal anti-human α-synuclein (1:10; kindly provided by Dr. Qi-Hong Sun, Beijing Proteome Research Center and Beijing Institute of Radiation Medicine, Beijing, China), and rabbit A11 (1:100; Millipore Bioscience Research Reagents). The fluorescent secondary antibodies (1:100; Jackson ImmunoResearch Laboratories, West Grove, PA) were selected accordingly to visualize the protein expression patterns in the brain. Confocal images were obtained on an SP5 microscope (Leica, Nussloch, Germany).
Automated Quantification of Inclusions and Oligomers
The automated quantification was performed by analyzing the individual confocal Z-series with the image processing software ImageJ version 1.37 (National Institutes of Health, Bethesda, MD) as described previously (Botella et al., 2008). After background correction and color depth reduction to 8 bits, the detection threshold was set to no <20. Then, automated quantification was performed with the “analyze particles” function with a minimal size of 1 pixel, and the particles were scored automatically. For inclusion number, the automatic particle counts were analyzed. For inclusion size analysis, we defined large inclusions as >1 μm at diameter and assessed their number. For oligomer amount quantification, the pixel area of A11-positive signals was recorded from the dorsal regions of the brain as shown in Figure 7.
Figure 7.

Oligomers accumulate in dHDAC6KO fly brains with α-synuclein overexpression. Oligomers in the dorsal brain area of α-syn flies (A and C) and dHDAC6KO; α-syn flies (B and D) at 10 (A and B) and 20 d (C and D) were visualized in red by oligomer-specific antibody A11. (E) α-syn flies at 20 d with dHDAC6 overexpression showing A11 signals. (F) Wild-type control flies at 20 d probed with A11. Bar, 20 μm. (G) Quantitative graphs showing the total A11-positive signals (oligomers) in the dorsal region of the brain as shown in A–F. Data were analyzed by Student's t test and presented as mean ± SEM (n > 5) with *p < 0.05, **p < 0.01, and ***p < 0.001. Genotypes: α-syn flies are w; TH-GAL4/UAS-α-synuclein. dHDAC6KO; α-syn flies are w, dHDAC6KO; TH-GAL4/UAS-α-synuclein. Control flies are TH-GAL4/+.
RESULTS
Generation and Characterization of the dHDAC6 Mutants
HDAC6 has been shown to function in the clearance of misfolded protein in mammalian cell lines (Boyault et al., 2007a). We used loss-of-function study for dHDAC6, the Drosophila homologue of mammalian HDAC6, to investigate the role of dHDAC6 in Drosophila PD model. In this aim, we generated null allele of dHDAC6 (dHDAC6KO) by using targeted mutagenesis (Rong and Golic, 2000). A translation start codon mutation and a frame shift were introduced in the dHDAC6 open reading frame (Figure 1, A and B). Western blot result confirmed that no dHDAC6 protein was produced in homozygous dHDAC6KO flies (Figure 1C). The rescue experiments validated the specificity of the knockout mutation (Supplemental Figure S1; data not shown). The mutant flies were viable and fertile and exhibited no obvious morphological abnormalities under normal culture conditions. With oxidative or heat stress dHDAC6KO flies did not show any notable difference from wild-type animals (Supplemental Figure S2). These results demonstrate that dHDAC6 is dispensable for viability, fertility and resistance to heat and oxidative stress in Drosophila. The viability of the homozygous dHDAC6KO flies makes it genetically tractable to ablate dHDAC6 in α-synuclein-induced PD adult flies in the subsequent study.
Figure 1.

Characterization of the dHDAC6KO mutant. (A) Genomic organization of the Drosophila dHDAC6 locus and the mutant allele of dHDAC6. Asterisk indicates the mutation of the ATG start codon. Filled bars are the coding regions; open bars are the untranslated regulatory regions. (B) Part of the dHDAC6KO mutant genomic sequence that contains point mutation and frame shift at the dHDAC6 translation start codon. The boxed sequence is dHDAC6 start codon in the wild-type gene. (C) dHDAC6 protein is absent in the dHDAC6 mutant flies as shown by Western blot. Protein extracts from wild-type (WT) and dHDAC6 knockout (KO) flies were blotted with anti-HDAC6 and anti-actin antibodies. Actin was used as a loading control.
dHDAC6 Mutation Exacerbates Neurodegeneration in the Drosophila PD Model
The expression of human α-synuclein in the Drosophila nervous system stimulates the loss of tyrosine hydroxylase (TH)-positive dopaminergic neurons (Feany and Bender, 2000; Auluck et al., 2002). To determine whether dHDAC6 participates in this process, we used the GAL4/UAS system to express α-synuclein specifically in the DA neurons of dHDAC6KO (dHDAC6KO; α-syn) and wild-type (α-syn) flies. Because the expression pattern of the driver-TH-GAL4 has been shown to be specifically associated with the DA system (Friggi-Grelin et al., 2003; this study; and Supplemental Figure S3), we used TH-GAL4 to drive the expression of UAS-mCD8::GFP to visualize and quantitatively measure the DA neurons in the adult fly brain.
Four Drosophila strains of dHDAC6KO, α-syn, dHDAC6KO; α-syn, and the wild type as a control were examined for DA neurons. No significant difference in the number of DA neurons was detected in the 1-d-old flies of these strains (Figure 2, A and D) or in the 10- and 20-d-old wild-type and dHDAC6KO flies (Figure 2, B and C and E and F). Significant loss of DA neurons was observed not only in the dorsomedial (DM) but also in the posteriomedial (PM) and the dorsolateral (DL)1 clusters of the α-syn flies at the age of 20 d (Figure 2, C and F), whereas there was no distinguishable loss of DA neurons in that of 10-d-old α-syn flies (Figure 2, B and E). The abrogation of dHDAC6 enhanced the α-synuclein-induced DA neuron loss phenotype, especially in the DM and DL1 clusters (Figure 2, B and C and E and F). In the dHDAC6KO; α-syn flies, the neuron degeneration phenotype appeared at the age of 10 d, which is earlier than in α-syn flies, and the phenotype worsened with age. The total DA neuron number in DM, PM, and DL1 clusters of dHDAC6KO; α-syn flies decreased ∼29% at 10 d of age and ∼36% at 20 d compared with that in 1-d-old wild-type flies, whereas the total DA neuron number in the α-syn flies only declined ∼5% at 10 d of age and ∼11% at 20 d of age. Quantitative analysis of the total number of DA neurons in DM, PM, and DL1 clusters revealed that ectopic α-synuclein expression driven by TH-GAL4 resulted in progressive DA neuron loss, and the absence of dHDAC6 aggravated this α-synuclein-induced DA neuron loss phenotype (Figure 2G). Together, these results demonstrate that the loss of dHDAC6 enhances the degeneration of DA neurons caused by overexpression of α-synuclein.
Figure 2.

dHDAC6KO promotes α-synuclein–induced dopaminergic neuron loss. Confocal images of dopaminergic neurons in DM, PM, and DL1 regions immunostained with anti-GFP antibody on 1-d-old (A), 10-d-old (B), and 20-d-old (C) fly brains, respectively, are shown, with arrows for PM and arrowheads for DM clusters and circles for DL1 clusters. (D–F) Quantitative graphs are shown corresponding to confocal images of 1-d-old, 10-d-old, and 20-d-old fly brains, respectively. (G) Graphs showing total numbers of dopaminergic neurons in the DM, PM, and DL1 clusters of different genotypes as indicated at 1 d, 10 d, 20 d. Data were analyzed by Student's t test and presented as mean ± SEM (n = 20∼40) with * for p < 0.05, ** for p < 0.01, and *** for p < 0.001. Bar, 50 μm. Genotypes: control flies are w; UAS-mCD8::GFP/+; TH-GAL4/+. dHDAC6KO flies are w, dHDAC6KO; UAS-mCD8::GFP/+; TH-GAL4/+. α-syn flies are w; UAS-mCD8::GFP/+; TH-GAL4, UAS-α-synuclein/+. dHDAC6KO; α-syn flies are w, dHDAC6KO; UAS-mCD8::GFP/+; TH-GAL4, UAS-α-synuclein/+.
To gain more insight into the impact of dHDAC6 on α-synuclein–induced neurodegeneration, additional neural context was examined. We used GMR-GAL4 to express α-synuclein in the eyes. The photoreceptor cells, neuronal cells in the ommatidia, are prone to the effect of neurotoxicity (Feany and Bender, 2000). The GMR-GAL4/+ flies did not show any detectable morphological defects in the eyes. Retinal sections reveal that the inner structure of eyes of these flies was intact and normal, with seven photoreceptor cells properly arranged in a regular pattern (Figure 3, A and B). However, the α-syn flies between the age of 20 and 30 d exhibited neurodegenerative phenotype in the eyes, with less photoreceptor cells and retinal distortion (Figure 3). This age-dependant phenotype was worsened in dHDAC6 mutant flies. The 20-d-old dHDAC6KO; α-syn flies exhibited fewer photoreceptor cells in a severely disrupted internal eye structure as compared with α-syn flies (Figure 3, A and C). In dHDAC6KO flies, the eye morphology and inner retinal structure are comparable to GMR-GAL4/+ control flies (Figure 3, A and C). These observations further support that dHDAC6 plays a protective role in coping with the α-synuclein neurotoxicity.
Figure 3.

Loss of dHDAC6 exacerbates retinal degeneration in α-synuclein–expressing eyes and overexpression of dHDAC6 counteracts the α-synuclein neurotoxicity. Semithin sections of retina showing inner retinal structures of 1-d-old (A and B, top), 20-d-old (A, bottom), and 30-d-old (B, bottom) flies. Arrows indicate outer ring disintegration of the ommatidia. Circles indicate ommatidia with loss of photoreceptor cells. (C) Quantification of disrupted ommatidia in control, dHDAC6-LKO, α-syn and dHDAC6-LKO;α-syn flies at 1 and 20 d after eclosion. (D) Quantification of disrupted ommatidia in control, α-syn, and α-syn;dHDAC6 flies at 1 and 30 d. (E) Quantification of ommatidia with photoreceptor cell loss in 20-d-old dHDAC6KO;α-syn flies, 30-d-old α-syn flies, and α-syn;dHDAC6 flies. Note that the retinal pattern and seven photoreceptor cells were normal in GMR-GAL4 control and dHDAC6-LKO flies. No discernible photoreceptor loss was detected in 1-d-old and 20-d-old α-syn flies. Data were analyzed by Student's t test and presented as mean ± SEM (n > 5) with *p < 0.05, **p < 0.01, and ***p < 0.001. Genotypes: control flies are w; GMR-GAL4/+. dHDAC6-LKO flies are w, dHDAC6-LKO. α-syn flies are w; GMR-GAL4/+; UAS-α-synuclein/+. dHDAC6-LKO; α-syn flies are w, dHDAC6-LKO; GMR-GAL4/+; UAS-α-synuclein/+. α-syn; dHDAC6 flies are w; GMR-GAL4/+; UAS-α-synuclein/UAS-dHDAC6.
Next, we sought to examine whether increased dose of dHDAC6 can suppress α-synuclein neurotoxicity. To this end, a copy of UAS-dHDAC6 transgene was introduced into the α-syn flies and the analysis was carried out at 30 d after eclosion. Overexpression of dHDAC6 restored ommatidia integrity, especially the number of photoreceptor cells caused by α-synuclein overexpression, suggesting that dHDAC6 can alleviate the α-synuclein toxicity (Figure 3, B, D, and E). Furthermore, the role of dHDAC6 that overcomes the α-synuclein toxicity is manifested by the observation that overexpression of dHDAC6 rescued the DA neuron loss phenotype (Supplemental Figure S4). Collectively, both loss-of-function and gain-of-function data support the conclusion that dHDAC6 protects neurons from α-synuclein–derived toxicity.
dHDAC6 Mutation Promotes α-Synuclein–induced Locomotor Dysfunction
In the fly PD model, progressive DA neuron degeneration was reported to be accompanied with an age-dependent locomotor dysfunction (Feany and Bender, 2000). We used climbing assay (Coulom and Birman, 2004) to test the impacts of dHDAC6 on the locomotion ability of the modeled PD flies (dHDAC6KO; α-syn flies vs. α-syn flies) at 5-d intervals for a period of 20 d (Figure 4). Compared with the wild-type control, both of the transgenic lines had no detectable climbing defect in their early ages. At the age of 10 d, the climbing ability of dHDAC6KO; α-syn flies but not α-syn flies was significantly impaired (Figure 4), and progressively deteriorated in an age-dependent manner. In contrast, noticeable climbing defects in α-syn flies did not appear until the age of 20 d in our study. Furthermore, the climbing defects in 20-d-old dHDAC6KO; α-syn flies were also more pronounced than in α-syn flies. It also should be mentioned that there was no locomotion defect in dHDAC6KO flies at all five individual surveyed ages (Figure 4). Together, dHDAC6 mutation enhances the locomotor defects in PD modeled flies. Considering the other observations that 1) a sudden loss of DA neurons happens at ∼10th day after eclosion (Figure 2, B and E); and 2) a sharp decrease of survival rate occurs at the age of 10 d (Supplemental Figure S5) in dHDAC6KO; α-syn flies, we suggest that the enhancement of locomotor defects and life span shortening by dHDAC6 mutation is associated with DA neuron degeneration in PD modeled flies. We propose that 10 d after eclosion may be a “checkpoint” for the fly parkinsonism. These three lines of evidence (neurodegeneration, climbing ability, and longevity) demonstrate that dHDAC6 suppresses the α-synuclein-induced pathological progression in fly PD model.
Figure 4.

dHDAC6KO enhances the climbing defects caused by α-synuclein overexpression. Bars show the percentage of the flies that climbed to reach 15 cm in 30 s at different ages with the genotypes as indicated. Data were analyzed by Student's t test and presented as mean ± SEM (n = 120) with *p < 0.05, **p < 0.01, and ***p < 0.001. Genotypes: control flies are w; TH-GAL4/+. dHDAC6-LKO flies are w, dHDAC6-LKO; TH-GAL4/+. α-syn flies are w; TH-GAL4/UAS-α-synuclein. dHDAC6KO; α-syn flies are w, dHDAC6KO; TH-GAL4/UAS-α-synuclein.
dHDAC6 Is Essential for the Formation of α-Synuclein–positive Inclusions in the Drosophila PD Model
PD is usually associated with the formation of LBs (Lang and Lozano, 1998). LB-like inclusions containing α-synuclein have been found in flies that ectopically express α-synuclein (Feany and Bender, 2000). To evaluate the biological importance of dHDAC6 on LB-like inclusion formation, α-synuclein antibody was used to visualize the density of this property on whole-mount fly brains.
Aggregates detectable with 20× magnification objective were considered as LB-like inclusions (McLean et al., 2002). In 1-d-old α-syn fly brains, the immunostaining of α-synuclein showed diffuse pattern (data not shown), which is similar to the case of young fly brains reported previously (Feany and Bender, 2000). The α-synuclein-positive inclusions in the neuropil adjacent to the DM region of the brains were clearly observed in 10- and 20-d-old α-syn flies (Figure 5, A and C). The number and the size of inclusions in α-syn flies were gradually increased along with age (Figure 5, F and G). However, fewer LB-like inclusions were present in the same region of dHDAC6KO; α-syn flies at 20 d of age (Figure 5, D and F), and no such inclusion was detectable in 10-d-old flies (Figure 5, B and F). The α-syn, dHDAC6KO; α-syn and α-syn; UAS-dHDAC6 flies have comparable amounts of total α-synuclein, arguing against the possibility that the observed effects are a consequence of reduced α-synuclein expression level (Figure 6B). To confirm the immunostaining results, we took advantage of sarkosyl-insoluble, urea-soluble feature of inclusions (Chen and Feany, 2005), and performed fractionations of fly head lysates. Our results show a reduced amount of sarkosyl-insoluble and urea-soluble pellets in α-syn flies lacking dHDAC6 (Figure 6A). These data demonstrate that dHDAC6 plays a crucial role in the formation of LB-like inclusions in α-synuclein–expressing flies. It also indicates that the inclusions are cytoprotective in the process of neurodegeneration.
Figure 5.

The amount of LB-like inclusion decreases in dHDAC6KO flies with α-synuclein overexpression. LB-like inclusions in the DM region of α-syn (A and C) and dHDAC6KO; α-syn flies (B and D) at 10 d (A and B) and 20 d (C and D) were labeled by whole-mount immunostaining with anti-α-synuclein antibody. (E) Inclusions when UAS-dHDAC6 transgene is expressed in α-syn flies at 10 d old. Bar, 40 μm. (F and G) Quantification of the numbers of total inclusions (F) and large inclusions that are >1 μm (G) at 10 d old and 20 d old with the genotypes as indicated on the right. Data were analyzed by Student's t test and presented as mean ± SEM (n > 5) with *p < 0.05, **p < 0.01, and ***p < 0.001. Genotypes: flies in A and C are w; TH-GAL4/UAS-α-synuclein. Flies in B and D are w, dHDAC6KO; TH-GAL4/UAS-α-synuclein. Fly in E is w; UAS-dHDAC6/+; TH-GAL4, UAS-α-synuclein/+.
Figure 6.

dHDAC6 positively regulates the accumulation of insoluble α-synuclein. (A) Immunoblots detecting α-synuclein from the fractions (see Materials and Methods for details of fractionation) of Tris-soluble supernatant (S) and Sarkosyl-insoluble/urea-soluble pellets (P). The flies were 1 d old, 10 d old, and 20 d old, respectively. (B) Western blot of α-synuclein from the heads of 1-d-old flies that express α-synuclein under the control of TH-GAL4 in the backgrounds of wild-type, dHDAC6KO, and dHDAC6 overexpression, as indicated on the top. The membrane was first probed with α-synuclein antibody and then stripped before immunoblotted with anti-actin that serves as a loading control. Genotypes: α-syn flies are w; TH-GAL4/UAS-α-synuclein. dHDAC6KO; α-syn flies are w, dHDAC6KO; TH-GAL4/UAS-α-synuclein. α-syn; dHDAC6 flies are w; UAS-dHDAC6/+; TH-GAL4, UAS-α-synuclein/+.
To further assess the dHDAC6 function in the formation of LB-like inclusions, we generated transgenic flies that conditionally express dHDAC6 by using the GAL4/UAS system. When α-synuclein and dHDAC6 were coexpressed in DA neurons, the amount of inclusions was substantially elevated compared with the flies that expressed α-synuclein solely (Figure 5, A, E, and F). Overexpression of dHDAC6 also led to an increase in sarkosyl-insoluble and urea-soluble pellets in fractionation experiments (Figure 6A). This observation suggests that the dHDAC6 dosage directly controls the formation of inclusions, further supporting the notion that dHDAC6 facilitates inclusion formation to attenuate α-synuclein–induced toxicity.
dHDAC6 Suppresses Oligomer Accumulation through Physical Interaction in the Drosophila PD Model
In vivo and in vitro studies have shown that α-synuclein monomers can be sequentially assembled into several conformations, including oligomers, protofibrils, fibrils, and tightly packed form in LBs (Conway et al., 1998; Periquet et al., 2007). The oligomeric or protofibrillar forms of α-synuclein have been suggested to be more toxic than the mature fibril forms (Lashuel et al., 2002; Sharon et al., 2003). To further ascertain that dHDAC6 functions in protecting neurons from α-synuclein toxicity through reducing the toxic forms of α-synuclein, we examined oligomer accumulation in α-synuclein expressing flies in the presence and absence of dHDAC6. We probed oligomers with A11 antibody that recognizes a structure-specific epitope of amyloidogenic proteins and reacts with all the soluble oligomeric aggregates, including oligomers and protofibrils (Kayed et al., 2003). The specificity of A11 to oligomeric α-synuclein has been reported in the previous studies (Kayed et al., 2003; Kim et al., 2009; Yu et al., 2009) and shown in this study (Supplemental Figure S8). In wild-type flies at different ages, no oligomer in the dorsal region of the brain was detected by A11 antibody (Figure 7F). In both transgenic flies (α-syn and dHDAC6KO; α-syn) at 1 d old, no oligomer-specific deposit was found in the dorsal region of the brain either (data not shown). Oligomer-specific immunoreactivity was detected in 20-d-old α-syn flies (Figure 7C) but not of 10-d-old α-syn flies (Figure 7A). Compared with α-syn flies of the same age, the oligomer staining in dHDAC6KO; α-syn flies were considerably increased (Figure 7, B, D, and G). Conversely, overexpression of dHDAC6 led to detectable reduction of the oligomers (Figure 7, E and G). These results indicate that DA neuron loss is correlated to the increased level of α-synuclein oligomers (Figures 2 and 7), and the oligomers are thus very likely toxic to DA neurons. In addition, neither DA neuron loss (Figure 2, A and D) nor α-synuclein aggregates (data not shown) were observed in 1-d-old α-syn flies, suggesting that α-synuclein monomers do not induce DA neuron loss. These data indicate that dHDAC6 protects cells from α-synuclein toxicity by decreasing the amount of oligomers.
To determine whether dHDAC6 physically interacts with oligomers, we performed coimmunoprecipitation assay. Because the A11 antibody recognizes the structure that is vulnerable to SDS (Karpinar et al., 2009), we used this antibody for immunoprecipitation to determine whether dHDAC6 could be coimmunoprecipitated instead of performing the reverse immunoprecipitation. The extracts were prepared from the heads of 20-d-old α-syn flies and flies that do not express α-synuclein. The physical interaction between dHDAC6 and α-synuclein oligomer was detected in the coimmunoprecipitation experiment as shown in Figure 8A. Immunoblotting results with α-synuclein antibody validated the ability of A11 to immunoprecipitate oligomeric α-synuclein aggregates but not monomeric α-synuclein (Figure 8A and Supplemental Figure S8). No dHDAC6 was precipitated in the flies without α-synuclein (Figure 8B). These results further strengthen the view that dHDAC6 regulates the amount of α-synuclein oligomers by physically interacting with each other in vivo.
Figure 8.
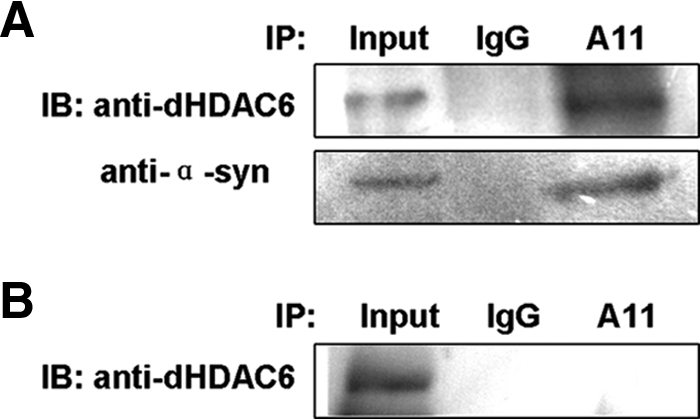
dHDAC6 physically interacts with oligomeric α-synuclein in Drosophila. Protein extracts for immunoprecipitation were prepared from the brains of 20-d-old flies: w; TH-GAL4/UAS-α-synuclein (A, with α-synuclein expression) and w; TH-GAL4/+ (B, without α-synuclein expression). The SDS-10% PAGE of the coimmunoprecipitation samples was immunoblotted with anti-dHDAC6 (A, top; and B) or anti-α-synuclein antibody (A, bottom). Input: 20% of the extracts that were used for immunoprecipitation.
DISCUSSION
In this study, we provide evidence for the first time that dHDAC6 protects DA neurons from α-synuclein toxicity by promoting inclusion formation and decreasing the amount of α-synuclein oligomers in the Drosophila PD model. We show that mutation of dHDAC6 notably affects the key features of PD in the α-synuclein modeled flies, namely selective loss of DA neurons, formation of α-synuclein-containing inclusions in the brain, decrease of locomotion ability, and shortening of life span. In addition, our genetic experiments show that dHDAC6 also functions to prevent the toxicity induced by overexpression of glutamine expanded human Spinocerebellar ataxia type 3 (SCA3) which is also known as Machado-Joseph disease (MJD) (Supplemental Figure S6). Furthermore, as shown in Supplemental Figure S1 mutation of dHDAC6 leads to a more severe rough eye phenotype characterizing impaired ubiquitin proteasome system (UPS) (Belote and Fortier, 2002), suggesting that dHDAC6 may play a compensatory role in the impaired UPS. Together, these lines of evidence indicate multiple roles of dHDAC6 in protein quality control machinery in vivo.
In vitro studies in our laboratory (Huang et al., 2006) and other laboratories (Cookson, 2005) show that α-synuclein exists as soluble and unfolded monomers under physiological conditions, but with a tendency to form fibrils under certain conditions through gradual polymerization to oligomers/protofibrils and finally to inclusions/fibrils. Although LBs are present in most of the clinically diagnosed PD patients, not every factor involved in this disorder has clear relationship with LB inclusion formation (Cookson et al., 2008). In this study, we demonstrated alteration of dHDAC6 dose remarkably affects the density of α-synuclein inclusions, suggesting that dHDAC6 is an integral factor in the process of inclusion formation in vivo. Outeiro et al. (2007) reported that Sirtuin 2 regulates the inclusion morphology in human H4 neuroglioma cells. Chen and Feany reported that blocking phosphorylation at Ser129 of α-synuclein substantially increased inclusion formation in Drosophila PD model (Chen and Feany, 2005). Mutation of Synphilin-1 was reported to be able to reduce the formation of α-synuclein inclusions in human neuroblastoma SH-SY5Y cells (Marx et al., 2003). It seems that the formation of inclusions is a complex issue hardly dominated by a single gene. We used genetic approaches to test for potential cooperators of dHDAC6 in this event and found that transgenic expression of Hsp70 can rescue, at least partially, the inclusion formation defect caused by abrogation of dHDAC6 (Supplemental Figure S7). This suggests that Hsp70 may be partially epistatic to dHDAC6, however, the possibility that they function in parallel in large inclusion body formation is not excluded by this experiment. Because the inclusions in the brains of the dHDAC-LKO; α-syn line are not abolished (Figure 5, B and D), we speculate that besides the dHDAC6-related pathway, other parallel pathway(s) also may be implicated in α-synuclein inclusion formation. Further experiments are required to elucidate the mechanism of formation and regulation of inclusions.
Oligomer is a conformational intermediate of α-synuclein aggregates. Our data have shown that abrogation of dHDAC6 results in increase of oligomers. Kawaguchi et al., (2003) demonstrate the association of HDAC6 with ubiquitinated misfolded proteins to form aggresomes in cell culture. Our coimmunoprecipitation assays show that HDAC6 associates with oligomeric α-synuclein, suggesting a role of dHDAC6 in the dynamics of α-synuclein oligomers. We propose that dHDAC6 binds to and promotes the toxic oligomers to change their conformations and form protective inclusions. However, the possibility that dHDAC6 may inhibit the oligomerization of α-synuclein cannot be excluded. A few factors have been identified to contribute to oligomerization with biochemical pathology methods. Polyunsaturated fatty acids increase oligomerization, whereas saturated fatty acids and rifampicin show opposite effects (Sharon et al., 2003; Xu et al., 2007). G protein-coupled receptor kinase (GRK)5 and Hsp70 have been characterized to be related to α-synuclein oligomerization. GRK5 promotes, whereas Hsp70 inhibits, oligomer formation (Arawaka et al., 2006; Outeiro et al., 2008). Given the functional relationship between HDAC6 and Hsp70 (Boyault et al., 2007b), a potential cooperative role of these two proteins in the oligomerization process may emerge.
The problem of which form/s of α-synuclein are toxic has been controversial. Inclusions used to be widely considered as a toxic component (Conway et al., 1998; Tu et al., 1998), whereas, increasing evidence suggests its role as a successful defense by lowering the concentration of toxic forms (Taylor et al., 2003; Arrasate et al., 2004). We monitored neuron cell mortality and retinal structure to assess the toxicity of different conformations of α-synuclein. Comparison of the neuron cell number, inclusion population and oligomer abundance between young flies and aged flies makes us to draw three sets of conclusions. First, monomeric α-synuclein is not toxic for neuron cells. Second, oligomers are toxic to the neuron cells. Finally, inclusions are cytoprotective form and protect neurons from α-synuclein toxicity. The three features, fewer neurons, fewer inclusions, and increased oligomers in aged dHDAC6KO; α-syn flies recapitulate the intrinsic essence of PD symptom progression. Our results provide the first insight into the in vivo evaluation of the toxicity of different α-synuclein conformation and build avenues for putative targets for dealing with α-synuclein toxicity.
Functional studies of the mechanism behind PD pathogenesis are required for therapeutic drug design. Our findings suggest that formation of inclusions or elimination of soluble α-synuclein oligomers, a process that involves dHDAC6, represents potential preventative targets. If this mechanism is conserved in humans, HDAC6 may offer a promising target for clinical therapy of PD patients through the up-regulation or enhancement of its activity.
Supplementary Material
ACKNOWLEDGMENTS
We thank Qi-Hong Sun, Hao Li, and Cayetano Gonzalez for the antibodies; John Belote, Mel Feany, Paul Taylor, Hao Li, Li Liu, Debapratim Kar Chowdhuri, and the Bloomington Fly Stock Center (Indiana University, Bloomington, IN) for fly stocks; and Jun Ma for critical reading of the manuscript. We are grateful to Xuehong Liang, Nan Wen and Shufeng Sun for technical assistance. Han Cheng was involved in the initiation of this study. We enjoyed stimulating discussions with Li Liu, Zhefeng Gong, and all the group members. This work was supported by the Chinese Ministry of Science and Technology (2009CB918702, 2005CB522804, 2006CB806508, and 2006CB910903), the China National Science Foundation (30623005, 90608029, and 30771217), and the Chinese Academy of Sciences (KSCX1-YW-R-70, KSCX2-YW-R-119).
Abbreviations used:
- DA
dopaminergic
- DM
dorsomedial
- DL
dorsolateral
- HDAC6
histone deacetylase 6
- LB
Lewy body
- PD
Parkinson's disease
- PM
posteriomedial.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-03-0200) on May 5, 2010.
REFERENCES
- Arawaka S., et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson's disease. J. Neurosci. 2006;26:9227–9238. doi: 10.1523/JNEUROSCI.0341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M., Mitra S., Schweitzer E. S., Segal M. R., Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Auluck P. K., Chan H. Y., Trojanowski J. Q., Lee V. M., Bonini N. M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Belote J. M., Fortier E. Targeted expression of dominant negative proteasome mutants in Drosophila melanogaster. Genesis. 2002;34:80–82. doi: 10.1002/gene.10131. [DOI] [PubMed] [Google Scholar]
- Botella J. A., Bayersdorfer F., Schneuwly S. Superoxide dismutase overexpression protects dopaminergic neurons in a Drosophila model of Parkinson's disease. Neurobiol. Dis. 2008;30:65–73. doi: 10.1016/j.nbd.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Boyault C., Gilquin B., Zhang Y., Rybin V., Garman E., Meyer-Klaucke W., Matthias P., Muller C. W., Khochbin S. HDAC6–p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C., Sadoul K., Pabion M., Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007a;26:5468–5476. doi: 10.1038/sj.onc.1210614. [DOI] [PubMed] [Google Scholar]
- Boyault C., et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007b;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Shi X., Padmanabhan R., Wang Q., Wu Z., Stevenson S. C., Hild M., Garza D., Li H. Identification of novel modulators of mitochondrial function by a genome-wide RNAi screen in Drosophila melanogaster. Genome Res. 2008;18:123–136. doi: 10.1101/gr.6940108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Feany M. B. alpha-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Conway K. A., Harper J. D., Lansbury P. T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- Cookson M. R. The biochemistry of Parkinson's disease. Annu. Rev. Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Cookson M. R., Hardy J., Lewis P. A. Genetic neuropathology of Parkinson's disease. Int. J. Clin. Exp. Pathol. 2008;1:217–231. [PMC free article] [PubMed] [Google Scholar]
- Coulom H., Birman S. Chronic exposure to rotenone models sporadic Parkinson's disease in Drosophila melanogaster. J. Neurosci. 2004;24:10993–10998. doi: 10.1523/JNEUROSCI.2993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli D., et al. A family knockout of all four Drosophila metallothioneins reveals a central role in copper homeostasis and detoxification. Mol. Cell. Biol. 2006;26:2286–2296. doi: 10.1128/MCB.26.6.2286-2296.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany M. B., Bender W. W. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Friggi-Grelin F., Coulom H., Meller M., Gomez D., Hirsh J., Birman S. Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J. Neurobiol. 2003;54:618–627. doi: 10.1002/neu.10185. [DOI] [PubMed] [Google Scholar]
- Huang C., Cheng H., Hao S., Zhou H., Zhang X., Gao J., Sun Q. H., Hu H., Wang C. C. Heat shock protein 70 inhibits alpha-synuclein fibril formation via interactions with diverse intermediates. J. Mol. Biol. 2006;364:323–336. doi: 10.1016/j.jmb.2006.08.062. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Parkinson's disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Karpinar D. P., et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 2009;28:3256–3268. doi: 10.1038/emboj.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y., Kovacs J. J., McLaurin A., Vance J. M., Ito A., Yao T. P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kim H. Y., et al. Structural properties of pore-forming oligomers of alpha-synuclein. J. Am. Chem. Soc. 2009;131:17482–17489. doi: 10.1021/ja9077599. [DOI] [PubMed] [Google Scholar]
- Kirik D., Annett L. E., Burger C., Muzyczka N., Mandel R. J., Bjorklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson's disease. Proc. Natl. Acad. Sci. USA. 2003;100:2884–2889. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kruger R., Kuhn W., Muller T., Woitalla D., Graeber M., Kosel S., Przuntek H., Epplen J. T., Schols L., Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Lang A. E., Lozano A. M. Parkinson's disease. Second of two parts. N. Engl. J. Med. 1998;339:1130–1143. doi: 10.1056/NEJM199810153391607. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A., Hartley D., Petre B. M., Walz T., Lansbury P. T., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- Marx F. P., et al. Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson's disease. Hum. Mol. Genet. 2003;12:1223–1231. doi: 10.1093/hmg/ddg134. [DOI] [PubMed] [Google Scholar]
- Masliah E., Rockenstein E., Veinbergs I., Mallory M., Hashimoto M., Takeda A., Sagara Y., Sisk A., Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- McLean P. J., Kawamata H., Shariff S., Hewett J., Sharma N., Ueda K., Breakefield X. O., Hyman B. T. TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J. Neurochem. 2002;83:846–854. doi: 10.1046/j.1471-4159.2002.01190.x. [DOI] [PubMed] [Google Scholar]
- Outeiro T. F., et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- Outeiro T. F., Putcha P., Tetzlaff J. E., Spoelgen R., Koker M., Carvalho F., Hyman B. T., McLean P. J. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS ONE. 2008;3:e1867. doi: 10.1371/journal.pone.0001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey U. B., et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Periquet M., Fulga T., Myllykangas L., Schlossmacher M. G., Feany M. B. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 2007;27:3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos M. H., et al. Mapping of a gene for Parkinson's disease to chromosome 4q21–q23. Science. 1996;274:1197–1199. doi: 10.1126/science.274.5290.1197. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos M. H., et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Rong Y. S., Golic K. G. Gene targeting by homologous recombination in Drosophila. Science. 2000;288:2013–2018. doi: 10.1126/science.288.5473.2013. [DOI] [PubMed] [Google Scholar]
- Sharon R., Bar-Joseph I., Frosch M. P., Walsh D. M., Hamilton J. A., Selkoe D. J. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Singleton A. B., et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Taylor J. P., Hardy J., Fischbeck K. H. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Taylor J. P., Tanaka F., Robitschek J., Sandoval C. M., Taye A., Markovic-Plese S., Fischbeck K. H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003;12:749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- Tu P. H., Galvin J. E., Baba M., Giasson B., Tomita T., Leight S., Nakajo S., Iwatsubo T., Trojanowski J. Q., Lee V. M. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann. Neurol. 1998;44:415–422. doi: 10.1002/ana.410440324. [DOI] [PubMed] [Google Scholar]
- Wang Z., Pan Y., Li W., Jiang H., Chatzimanolis L., Chang J., Gong Z., Liu L. Visual pattern memory requires foraging function in the central complex of Drosophila. Learn. Mem. 2008;15:133–142. doi: 10.1101/lm.873008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Wei C., Xu C., Bennett M. C., Zhang G., Li F., Tao E. Rifampicin protects PC12 cells against MPP+-induced apoptosis and inhibits the expression of an alpha-Synuclein multimer. Brain Res. 2007;1139:220–225. doi: 10.1016/j.brainres.2006.12.074. [DOI] [PubMed] [Google Scholar]
- Yu W. H., Dorado B., Figueroa H. Y., Wang L., Planel E., Cookson M. R., Clark L. N., Duff K. E. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am. J. Pathol. 2009;175:736–747. doi: 10.2353/ajpath.2009.080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.