

In this study, muscle-specific BDNF knockout animals were generated and compared with BDNF−/− knockouts. Our findings show that muscle-derived BDNF plays an important role in 1) regulating satellite cell proliferation and differentiation and 2) early regeneration after muscle injury.
Abstract
In adult skeletal muscle, brain-derived neurotrophic factor (BDNF) is expressed in myogenic progenitors known as satellite cells. To functionally address the role of BDNF in muscle satellite cells and regeneration in vivo, we generated a mouse in which BDNF is specifically depleted from skeletal muscle cells. For comparative purposes, and to determine the specific role of muscle-derived BDNF, we also examined muscles of the complete BDNF−/− mouse. In both models, expression of the satellite cell marker Pax7 was significantly decreased. Furthermore, proliferation and differentiation of primary myoblasts was abnormal, exhibiting delayed induction of several markers of differentiation as well as decreased myotube size. Treatment with exogenous BDNF protein was sufficient to rescue normal gene expression and myotube size. Because satellite cells are responsible for postnatal growth and repair of skeletal muscle, we next examined whether regenerative capacity was compromised. After injury, BDNF-depleted muscle showed delayed expression of several molecular markers of regeneration, as well as delayed appearance of newly regenerated fibers. Recovery of wild-type BDNF levels was sufficient to restore normal regeneration. Together, these findings suggest that BDNF plays an important role in regulating satellite cell function and regeneration in vivo, particularly during early stages.
INTRODUCTION
Neurotrophins are best known for their roles in regulating neuronal survival, plasticity, growth, and death (Hamburger and Yip, 1984; Oppenheim, 1991; Kalb, 2005; Reichardt, 2006). As such, they have been studied predominantly in the context of nervous system development and function. However, accumulating evidence suggests that neurotrophins play a more widespread role than originally thought. Accordingly, they are now the focus of study in numerous cell populations across multiple tissue systems. Of these populations, skeletal muscle is of particular interest, because it acts as an abundant source of neurotrophic support throughout development (Hamburger, 1934; Griesbeck et al., 1995; Kablar and Belliveau, 2005). In addition, skeletal muscles express several neurotrophin receptors, providing the basis for neurotrophin signaling within the muscle compartment (Tessarollo et al., 1993; Gonzalez et al., 1999; Ip et al., 2001; Sheard et al., 2002; reviewed; Pitts et al., 2006). Indeed, neurotrophin knockout mice often exhibit distinct defects in muscle development and function, and to date NT-4/5 has been implicated in muscle fiber type transformation, NT-3 in muscle spindle formation, and nerve growth factor (NGF) in dystrophic muscle pathology (Ernfors et al., 1994b; Capsoni et al., 2000; Carrasco and English, 2003).
The role of BDNF in skeletal muscle development and function has been more difficult to determine due in part to the early postnatal lethality of the BDNF knockout mouse (BDNF−/−; Ernfors et al., 1994a). However, expression profiling studies have shown that BDNF is differentially expressed in skeletal muscle under various physiological and pathological conditions (reviewed in Chevrel et al., 2006). During embryonic muscle development BDNF is highly expressed, with down-regulation early in postnatal life (Griesbeck et al., 1995). In adult skeletal muscle, BDNF expression is found in myogenic progenitors known as satellite cells (Mousavi and Jasmin, 2006). In healthy adult skeletal muscle, satellite cells are mitotically quiescent, sequestered between the basement membrane and sarcolemma of myofibers (Schultz et al., 1978; Lipton and Schultz, 1979). In response to injury, however, they become activated, triggering proliferation and differentiation to repair damaged fibers (Snow, 1978; Charge and Rudnicki, 2004). Muscle injury also results in up-regulation of BDNF expression at a time when satellite cell activation and proliferation occurs, suggesting that BDNF may play a role in mediating the satellite cell response to injury (Griesbeck et al., 1995; Omura et al., 2005).
To examine the functional role of muscle-derived BDNF in vivo, we generated a mouse in which BDNF is specifically depleted from skeletal muscle cells. This model circumvents the problem of early postnatal lethality and breeding issues presented by the complete BDNF knockout mouse (BDNF−/−), and allows examination of postnatal muscle development, maintenance, and regeneration in the absence of muscle-derived BDNF. To further support our findings, and to determine the specific role of muscle-derived BDNF, we compared these animals to neonatal BDNF−/− mice. Our results suggest that BDNF plays a role in regulating satellite cell function and that loss of muscle-derived BDNF results in abnormal myogenic differentiation and regeneration after injury.
MATERIALS AND METHODS
BDNF−/− and Muscle-specific BDNF Knockout Mice (BDNFMKO)
BDNF knockout mice (BDNF−/−) mice were purchased from The Jackson laboratory (Bar Harbor, ME; B6.129S4-BDNFtm1Jae/J). Homozygous BDNF−/− mice exhibit severe neuronal defects caused by degeneration of sensory ganglia, and they die within 2–3 wk of birth. In our facilities, however, heterozygous BDNF+/− mothers showed highly aggressive behavior, and they frequently cannibalized pups within hours of birth.
For generation of muscle-specific BDNF knockout colonies, mice carrying the LoxP-targeted BDNF allele (BDNFf/f) (BDNFtm2Jae/J; The Jackson Laboratory; Rios et al., 2001) were crossed with Myf5-Cre mice (kindly provided by M. Rudnicki, Sprott Center for Stem Cell Research, Ottawa, ON, Canada; Tallquist et al., 2000) to generate BDNFf/wt; Myf5-Cre progeny. These were backcrossed to BDNFf/f homozygotes to produce BDNFf/f; Myf5-Cre mice (BDNFMKO), in which skeletal muscle-BDNF is specifically knocked out (Figure 1A). To assess the efficiency of cAMP response element (Cre)-mediated BDNF excision, genomic DNA was polymerase chain reaction (PCR) genotyped using primers that flank the LoxP-targeted BDNF locus. BDNFwt/wt; Myf5-Cre mice were used as controls (CTL) in all experiments. Adult animals used in this study were aged 5–8 wk unless otherwise indicated. All animals were housed in the animal care facility at the University of Ottawa and were provided with unlimited access to food and water. Surgical procedures were approved by the University of Ottawa Animal Care and Use Committee.
Figure 1.

Generation of the muscle-specific BDNF knockout mouse. (A) Schematic representation of the breeding scheme used in generation of the muscle-specific BDNF knockout mouse. (B) Quantification of the relative levels of BDNF mRNA detected by qRT-PCR in hindlimb musculature at P7 and from adult EDL, GAS, TA, SOL, and DIA muscles of BDNFwt/wt; Myf5-Cre (CTL), and BDNFf/f; Myf5-Cre (MKO) animals (n = 6; *p < 0.01). (C) Relative levels of BDNF protein per milligram wet tissue mass in brain and GAS muscle preparations from adult CTL, HET and MKO animals (n = 3; *p < 0.05). (D) Relative levels of BDNF mRNA detected in CTL and MKO single fiber myoblast preparations cultured for 5 d in growth-promoting medium. (E) Relative levels of BDNF mRNA detected in hindlimb musculature of BDNF−/− (KO) compared with control littermates (CTL) at P1 (n = 3; *p < 0.001). Error bars represent SE.
Single-Fiber Satellite Cell Isolation
Flexor digitorum brevis (FDB) muscles were removed from 5- to 6-wk-old BDNFMKO and control mice and digested in a Petri dish containing type I collagenase (Sigma-Aldrich, St. Louis, MO) in DMEM (Invitrogen, Burlington, ON, Canada) for 1.5 h. Satellite cells were isolated as described previously (Rosenblatt et al., 1995). In brief, single fibers were dissociated by repeated titration in DMEM, and single intact fibers were cultured on Matrigel-coated culture plates containing basal medium (10% horse serum, 1% antibiotic in DMEM). Within 2 d of plating, satellite cells displaying myogenic potential detached from single fibers and basal medium was replaced by growth medium containing 20% fetal bovine serum, 10% horse serum, and 1% antibiotic in DMEM. Growth medium was changed daily for 5 d. Differentiation was induced by serum deprivation of cultures using 2% horse serum and 1% antibiotic in DMEM.
Cardiotoxin Injections
For regeneration experiments, 25 μl of 10−5 M cardiotoxin (Latoxan, Rosans, France) were injected into the tibialis anterior (TA) muscle of 5- to 6-wk-old BDNFMKO and control mice to induce muscle degeneration and regeneration as described previously (Condrea, 1974; Gramolini et al., 1999). One, 2, 5, and 7 d after injection, right TA muscles were excised and frozen in liquid nitrogen. For immunofluorescence experiments, left TA muscles were embedded in OCT compound (Thermo Fisher Scientific, Waltham, MA) and frozen in melting isopentane precooled with liquid nitrogen. All muscles were stored at −80°C until use.
RNA Extraction and Reverse Transcription (RT)-PCR
RNA was extracted from muscle and brain using TRIzol reagent (Invitrogen) as recommended by the manufacturer. TRIzol extracted RNA was treated for 1 h with DNAse I (Invitrogen) to eliminate possible DNA contamination. Reverse transcription was carried out using an RT reaction containing 5 mM MgCl2, 1× PCR buffer, 1 mM dNTP, 1 U/μl RNase inhibitor, 5 U/μl Moloney murine leukemia virus reverse transcriptase, and 2.5 μM random hexamers. Real-time quantitative RT-PCR was performed on an MX3005p real-time PCR system (Stratagene, La Jolla, CA) using QuantiTect SYBR Green PCR kit (QIAGEN, Valencia, CA). For these experiments, amplification of 18S ribosomal subunit, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Pax7, MyoD, myogenin, and embryonic myosin heavy chain (embMyHC) was performed in triplicate by using primers outlined in Supplemental Table 1. The ΔCT method was used to quantify expression levels based on normalization to 18S rRNA and/or GAPDH. Controls consisted of RT mixture in which total RNA was replaced by diethylpyrocarbonate (DEPC)-treated water, and/or RT mixture in which reverse transcriptase was replaced with DEPC-treated water. For endpoint PCR amplification (see Supplemental Data), cycle numbers varied depending on the primers used; however, all were optimized to be within the linear range of amplification as described in detail previously (Mousavi et al., 2004). For these experiments, the 28S ribosomal subunit was used for standardization.
Western Blotting
Total protein was extracted from muscle and brain samples using radioimmunoprecipitation assay extraction buffer (150 mM NaCl, 1% IPGAL, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0) supplemented with protease inhibitors (Complete; Roche, Indianapolis, IN). Protein concentration was determined using Bradford assay (Bio-Rad Laboratories, Hercules, CA). Samples (30 μg) were run on 12% SDS-polyacrylamide gel electrophoresis gels, and transferred to polyvinylidene difluoride membranes. Western blot analysis was performed as described previously (Mousavi and Jasmin, 2006). In brief, membranes were blocked with 5% nonfat milk in Tris-buffered saline Triton X-100, before incubation with either α-Pax7 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) or α-GAPDH (Advanced Immunochemical, Long Beach, CA) antibodies. Secondary detection was performed using horseradish peroxidase-conjugated secondary antibodies (Millipore Bioscience Research Reagents, Temecula, CA). Antibody complexes were detected using western lighting chemiluminescent reagent (PerkinElmer Life and Analytical Sciences, Boston, MA) and exposed to BioMAX MR film (Eastman Kodak, Rochester, NY). Quantification of band density was performed using ImageJ image analysis software (National Institutes of Health, Bethesda, MD).
Enzyme-linked Immunosorbent Assay (ELISA)
BDNF protein was quantified from muscle and brain samples using the Emax ImmunoAssay system following the protocol of the manufacturer (Promega, Madison, WI) as described previously (Mousavi and Jasmin, 2006). In brief, samples were homogenized in lysis buffer (2:1, vol/wt) containing 137 mM NaCl, 20 mM Tris-HCl, pH 8.0, 1% NP-40, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 0.5 mM sodium vanadate, and one protease inhibitor tablet (Roche). Samples were diluted with 4 volumes of Dulbecco's phosphate-buffered saline, sonicated, and centrifuged at 14,000 × g for 20 min. Sample absorbance was measured at 450 nm with an ELISA plate reader (SpectraMax M2; Molecular Devices, Sunnyvale, CA).
Muscle Histology
For all histological procedures, muscles were removed from animals, immediately frozen in OCT embedding medium (ThermoFisher Scientific, Pittsburgh, PA) and stored at −80°C until use. Muscles were cross-sectioned at 10 μm and placed on positively charged slides for histological examination. Bright field and fluorescent images were digitally acquired (Sony, Tokyo, Japan) by using an Axioscope 2 microscope (Carl Zeiss, Jena, Germany). Analysis was performed using Northern Eclipse Imaging software, and further image processing software was used to enhance color and clarity for publication (Photoshop CS; Adobe Systems, Mountain View, CA). All histological assessments were performed in a blinded manner such that the identity of the samples remained unknown until data analysis was complete.
For visualization of muscle structure, sections were stained with hematoxylin and eosin. The average number of newly regenerated myofibers was quantified as the average number of centrally nucleated fibers counted across five separate fields of view for at least three sections of each muscle.
Immunofluorescence
Immunofluorescence was performed using primary antibodies against BDNF (Promega), CD11b (Abcam, Cambridge, MA), and MF20 (Developmental Studies Hybridoma Bank, University of Iowa). Muscle sections were fixed with 2% paraformaldehyde for 10 min at room temperature, and blocked (1% bovine serum albumin and 0.1% Triton X in phosphate-buffered saline [PBS]) for an additional hour. For culture experiments, cells were fixed with 90% cold methanol and blocked for 1 h at room temperature. All samples were incubated in primary antibodies overnight at 4°C. The next day, sections were washed (3× PBS) and incubated in appropriate secondary antibodies for 1 h. Slides were mounted with VECTASHIELD (Vector Laboratories, Burlington, ON, Canada). Bromodeoxyuridine (BrdU) labeling was performed using a BrdU labeling and detection kit, as described in the manufacturer's protocol (Roche).
Statistical Analysis
Student's t test was used to determine the significant difference between two groups. The level of significance was set at p < 0.05.
RESULTS
Generation of the Muscle-specific BDNF Knockout Mouse
To examine the role of skeletal muscle-derived BDNF in postnatal muscle development, maintenance, and regeneration, we generated a mouse in which BDNF is specifically depleted from skeletal muscle cells. To this end, mice homozygous for the LoxP-targeted BDNF allele (BDNFf/f) were crossed with Myf5-Cre knockin mice, in which Cre recombinase expression is driven by the muscle-specific Myf5 promoter (Tallquist et al., 2000; as used previously; Huh et al., 2004). BDNFf/wt; Myf5-Cre progeny were then backcrossed to BDNFf/f mice to generate BDNFf/f; Myf5-Cre mice (BDNFMKO), in which muscle-BDNF is specifically depleted (Figure 1A).
BDNFMKO mice were viable and fertile, and they genotyped at expected Mendelian ratios. Cre-mediated excision of the BDNF gene was assessed in postnatal day (P)7 and adult hindlimb muscles using quantitative (q)RT-PCR and BDNF immunodetection assay (BDNF ELISA). At P7, BDNF transcript levels were decreased by ∼50% in BDNFMKO muscles compared with controls (Figure 1B; p < 0.01). Greater decreases in BDNF transcript (∼60%) were detected in extensor digitorum longus (EDL), gastrocnemius (GAS), TA, soleus (SOL), and diaphragm (DIA) muscles of adult (5- to 8-wk-old) BDNFMKO mice (Figure 1B; p < 0.01). Similarly, BDNF protein was decreased by 25% in heterozygous BDNFf/wt; Myf5-Cre mice and 50% in homozygous BDNFf/f; Myf5-Cre (BDNFMKO) mice (Figure 1C; p < 0.05). Importantly, BDNF levels were similar at both the transcript and protein level in brain samples isolated from adult BDNFMKO and control littermates, confirming the specificity of BDNF depletion to skeletal muscle (Figure 1, B and C). Furthermore, we detected no change in the transcript levels of other neurotrophins or their receptors, suggesting that expression of other neurotrophin family members is not altered to compensate for the loss of muscle-BDNF in the BDNFMKO mouse (Supplemental Figure 1; p > 0.05).
Based on the observation that BDNF levels were reduced by 50–60% in BDNFMKO muscle, it became important to determine the source of remaining BDNF expression in our model. Considering that the muscle compartment is made up of several cell types (Schwann, endothelial, and neuronal), and because several of these cell-types have been shown to express and secrete BDNF, we hypothesized that the remaining BDNF detected in BDNFMKO mice was derived from resident nonmuscle cells (Lomen-Hoerth and Shooter, 1995; Vega et al., 2003; Kermani and Hempstead, 2007; reviewed in Gordon, 2009). To support this hypothesis, and to ensure that BDNF expression was indeed eliminated from muscle cells, we isolated and cultured single myofiber explants (Rosenblatt et al., 1995). These preparations allow examination of a relatively pure sample of muscle fibers and their associated satellite cells. As expected, we found greater reductions (85%) in BDNF expression in cultures derived from BDNFMKO mice, whereas BDNF was highly expressed in proliferating control cultures (Figure 1D; p < 0.01). It is important to note that compared with BDNF−/− muscle, in which BDNF is abolished, detectable levels of BDNF remain in BDNFMKO muscles (Figure 1, D and E). This may be due to low levels of contaminating nonmuscle cells in our culture system (i.e., fibroblasts, neurons, and Schwann cells). Alternatively, remaining BDNF expression could be related to the existence of satellite cells that never express Myf5 (13 ± 4%) and thus continue to express BDNF in the BDNFMKO mouse (Kuang et al., 2007).
Hindlimb Muscle Morphology the Muscle-specific BDNF Knockout Mouse
The muscle-specific BDNF knockout mouse showed no overt phenotypic abnormalities and was similar in weight and size to control littermates from birth to 1 year. Histological examination of hematoxylin- and eosin-stained cross sections of SOL and EDL muscles from P7 and adult mice revealed no significant change in total myofiber size or number between BDNFMKO and control littermates (Figure 2; p > 0.05). Similar results were obtained upon examination of hindlimb muscles of BDNF−/− full knockout mice at postnatal day 1 (Supplemental Figure 2; p > 0.05). Together, these findings suggest that overall muscle fiber number and size are not adversely affected in the absence of BDNF.
Figure 2.

Overall muscle histology is not affected in the absence of muscle-BDNF. (A) Representative hematoxylin- and eosin-stained cross sections from control (CTL) and BDNFMKO (MKO) hindlimb sections at P7 (top) and adult muscle (bottom). (B) Quantification of the average cross-sectional area. (C) Fiber number from P7 and adult EDL and SOL muscles (n = 6). Error bars represent SE.
Expression of Muscle Regulatory Genes in the Muscle-specific BDNF Knockout Mouse
To determine whether depletion of muscle-derived BDNF resulted in changes in the expression of genes controlling muscle growth and differentiation, we examined transcript levels of the myogenic regulatory factors (MRFs) in EDL, TA, and DIA muscles of adult (5- to 8-wk-old) BDNFMKO and control mice. We observed no significant changes in transcript levels for positive regulators of myogenesis (MyoD and myogenin) in healthy adult skeletal muscle. Likewise, transcript levels of myostatin, a negative regulator of muscle growth showed no significant change compared with control littermates (Supplemental Figure 3; p > 0.05).
In contrast, transcript and protein levels for the satellite cell marker Pax7 were consistently decreased in adult BDNFMKO muscle compared with controls. Although there were no changes in Pax7 expression in neonatal BDNFMKO muscle, adult transcript levels were decreased by 15% (Figure 3A; p < 0.05), whereas Pax7 protein levels showed a 30% reduction compared with controls (Figure 3B). Similar changes in Pax7 expression were observed in satellite cell-derived myoblast cultures after single fiber isolation (Figure 3A; p < 0.05). For comparative purposes, we also examined Pax7 transcript levels in neonatal BDNF−/− hindlimb muscle. By P1, Pax7 transcript levels were decreased by 30%, suggesting a more pronounced deficit in Pax7 expression when BDNF is abolished (Figure 3C).
Figure 3.
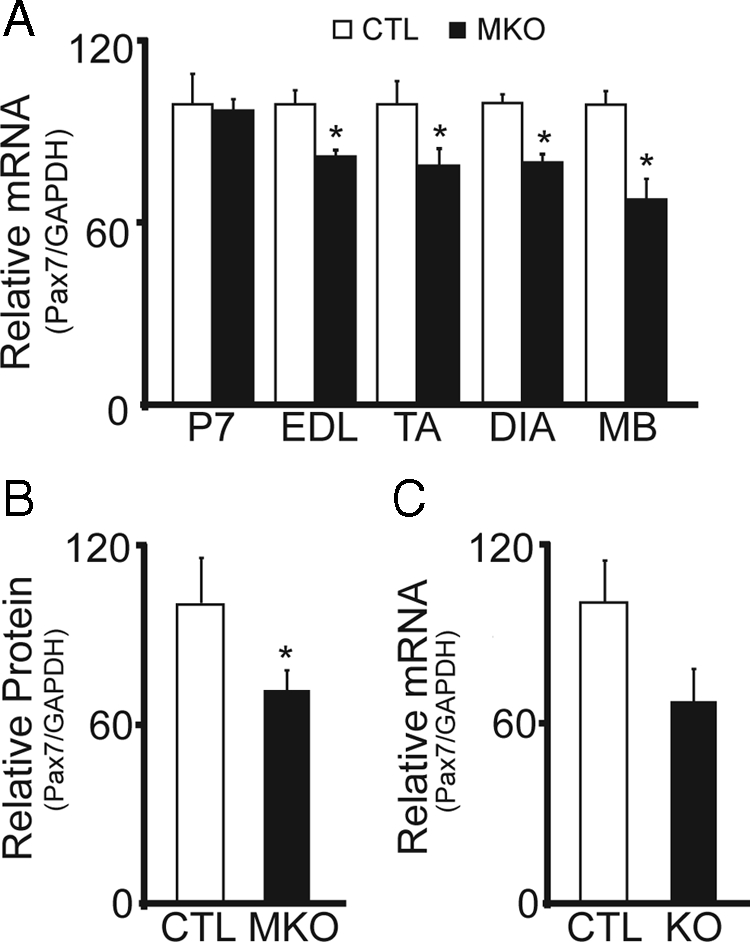
Decreased expression of the satellite cell marker, Pax7, in the absence of muscle-BDNF. (A) Quantification of the relative levels of Pax7 mRNA in P7, adult EDL, TA, and DIA muscles, and single fiber myoblast preparations (MB) from BDNFMKO (MKO) and control littermates (CTL). (B) Quantification of Pax7 protein levels standardized to GAPDH (n = 6; p < 0.05). (C) Relative levels of Pax7 mRNA in BDNF−/− (KO) compared with control littermates (CTL) at P1 (n = 3). Error bars represent SE.
Muscle-BDNF Is Required for Normal Satellite Cell Function
To determine whether loss of muscle-derived BDNF compromised the integrity of the satellite cell pool, we examined proliferation and differentiation of satellite cell-derived myoblasts after single fiber isolation. In BDNFMKO cultures, we found no significant difference in the number of satellite cell-derived myoblasts over 5-d growth, despite the observed decrease in Pax7 levels (Figure 4A). Instead, we found an increase in the number of BrdU-labeled nuclei associated with BDNFMKO fibers, suggesting that proliferation was enhanced in the absence of muscle-BDNF (Figure 4, A and B; p < 0.05).
Figure 4.

Abnormal satellite cell proliferation in single fiber myoblast preparations from BDNFMKO mice. (A) Representative BrdU labeling (green) of proliferating (day 0, top) and early differentiating (day 1, bottom) myoblasts isolated from BDNFMKO (MKO) and control (CTL) littermates. Images are counterstained with 4′,6-diamidino-2-phenylindole (DAPI) in blue. (B) Quantification of the ratio of BrdU-labeled nuclei relative to the total number of nuclei per field of view (n = 6, *p < 0.05). C) Relative induction of MyoD, myogenin, p21, embryonic (emb), and neonatal (neo) MyHC transcripts compared with proliferating cultures after 1-d exposure to differentiation promoting medium (n = 6; *p < 0.05). Error bars represent SE.
We next examined the rate of cell cycle withdrawal after 24-h exposure to differentiation promoting medium. Upon induction of differentiation, we observed a greater number of BrdU-labeled nuclei in BDNFMKO cultures compared with controls, suggesting that in the absence of BDNF, myoblasts remained in a proliferative state, whereas control cultures exited the cell cycle and began to differentiate (Figure 4, A and B). Furthermore, BDNFMKO cultures failed to induce control levels of several molecular markers of differentiation, including myogenin, p21, and embMyHC (Figure 4C). Importantly, however, MyoD expression was not changed, suggesting that myogenic determination of satellite cell-derived myoblasts was not affected (Figure 4C). Together, these results suggest that BDNF is required for early phases of myogenic differentiation and that in the absence of BDNF, myogenic lineage progression is delayed.
To examine the effects of BDNF on later stages of myogenic differentiation, we examined myotube formation in BDNFMKO cultures after 5-d differentiation. Compared with controls, BDNFMKO-derived myotubes contained fewer myonuclei and exhibited an increase in the number of unfused (mononuclear) myocytes expressing MyHC (Supplemental Figure 4). Consequently, the average size of MyHC-expressing myotubes was significantly decreased (Figure 5, A and B; p < 0.05), and MyHC transcript was expressed at significantly lower levels than controls (Figure 5C; p < 0.01).
Figure 5.

Decreased size and MyHC expression in BDNF-depleted myotubes is rescued by treatment with exogenous BDNF protein. (A) immunofluorescence labeling of total MyHC in control (CTL), BDNFMKO (MKO), and MKO cultures treated with 20 ng/ml recombinant BDNF protein (MKO+) after 5-d differentiation. Counterstaining with 4′,6-diamidino-2-phenylindole (DAPI) was performed. (B) Quantification of the average area of MyHC-positive myotubes. (C) Relative MyHC mRNA levels standardized to GAPDH after 5-d differentiation (n = 6; *, significant decrease compared with CTL; #, significant increase compared with MKO; p < 0.05). Error bars represent SE.
To determine whether differentiation defects observed in BDNFMKO cultures were the result of intrinsic changes to the satellite cell pool, or whether muscle-derived BDNF is itself required for normal myogenic differentiation, we examined whether daily treatment with recombinant BDNF protein could normalize differentiation of BDNFMKO cultures. Addition of exogenous BDNF protein was sufficient to rescue myotube size and MyHC expression in BDNFMKO cultures (Figure 5, A–C), demonstrating that BDNF itself is required for normal myogenic differentiation of satellite cell-derived myoblasts.
BDNF Expression during Cardiotoxin-induced Muscle Regeneration
After injury, muscle satellite cells become activated, proliferate, differentiate, and fuse with each other or with existing fibers to repair damaged muscle tissue. To determine whether muscle-BDNF is important for satellite cell function in vivo, we examined skeletal muscle regeneration in BDNFMKO mice. Unfortunately, these studies were not possible in the BDNF−/− mouse due to early postnatal lethality of complete BDNF knockout. To determine whether satellite cell function and muscle regeneration were compromised in the absence of muscle-BDNF, we performed time course studies on regenerating muscles after cardiotoxin (CTX) injury (Condrea, 1974; Gramolini et al., 1999). In control muscles, BDNF transcript levels were significantly up-regulated within 2 d after injury, followed by a gradual decrease over the 7-d time course (Figure 6A; p < 0.05). In contrast, BDNF levels remained significantly reduced throughout early time points in regenerating BDNFMKO muscle, reaching only 50% of uninjected control levels 0–2 d after injury (Figure 6A; p < 0.05). The disparity in BDNF levels was most apparent 2 d after injection, at which time control BDNF levels were increased by more than threefold compared with BDNFMKO.
Figure 6.

BDNF expression by CD11b-positive cells in regenerating skeletal muscle. (A) Quantification of the relative levels of BDNF transcript in regenerating BDNFMKO (MKO) and control (CTL) muscles at 0, 1, 2, 5, and 7 d postcardiotoxin injection (n = 6; *, decrease and #, increase compared with controls at day 0; p < 0.05). (B) immunofluorescence labeling of BDNF (green) and CD11b (red) in regenerating TA muscles of MKO and CTL animals 5 d after injury.
Interestingly, however, BDNF expression increased during later time points in BDNFMKO muscle, achieving control levels 5–7 d after injury. Although we were surprised to find such high levels of BDNF in regenerating BDNFMKO muscle, we hypothesized that BDNF was secreted by resident nonmuscle cells and/or infiltrating immune cells after injury (see below; Lomen-Hoerth and Shooter, 1995; Kerschensteiner et al., 1999; Vega et al., 2003). To examine the source of BDNF in regenerating BDNFMKO muscle, we performed immunolabeling experiments on TA sections 5 d after injury, when BDNF levels were at their peak. Because previous studies had shown that BDNF levels correlate with immune cell infiltration in several human neuropathies (Sobue et al., 1998), we examined BDNF localization with respect to the immune cell marker CD11b in regenerating muscle. BDNF staining colocalized with CD11b+ immune cells in both control and BDNFMKO sections (Figure 6B). Importantly, control cross sections showed BDNF+/CD11b+ and BDNF+/CD11b− labeling, whereas BDNFMKO sections showed exclusively BDNF+/CD11b+ labeling. The observation that BDNF labeled cells coexpressed CD11b in BDNFMKO tissue indicates that BDNF expression is purely immune cell derived in regenerating BDNFMKO muscle.
Regeneration Is Delayed in the Absence of Muscle-BDNF
Although BDNF transcript levels were eventually up-regulated in injured BDNFMKO muscle, expression levels did not reach control values until 5 d after injection (Figure 6A). Thus, BDNF levels were significantly reduced during the early time points in regenerating BDNFMKO muscle. To determine whether delayed expression of BDNF adversely affected the early regenerative response to injury, we examined several markers of myogenic differentiation in regenerating BDNFMKO muscle. In agreement with our culture studies, we found that induction of several molecular markers of regeneration, including Pax7, MyoD, myogenin, and embMyHC was impaired at day 5, when proliferating myoblasts begin to differentiate and fuse to repair damaged myofibers (Figure 7, A–D; p < 0.05). We also performed histological examination of regenerating muscle 5 and 7 d after injection. Newly formed fibers were quantified as the number of centrally nucleated fibers per field of view in areas of regeneration. Consistent with a decrease in regenerative markers, we observed 40% fewer regenerated fibers in BDNFMKO muscle 5 d after injury compared with control littermates (Figure 7, E and F; p < 0.05). Concomitantly, we observed an approximate 25% increase in the number of mononuclear cells within regenerating BDNFMKO compared with control littermates (Figure 7, E and G; p < 0.05). Together, these findings suggest that in the absence of muscle-derived BDNF, early regeneration is markedly delayed.
Figure 7.

Delayed regeneration in the absence of muscle-BDNF. Quantification of the relative levels of Pax7 (A), myogenin (B), MyoD (C), and embMyHC (D) transcript in regenerating BDNFMKO (MKO) and control (CTL) muscles. All values are relative to control day 0 transcript levels (n = 6; *p < 0.05). (E) Representative hematoxylin and eosin staining of regenerating TA muscles from CTL and MKO animals at days 5 (top) and 7 (bottom) after injury. (F) Quantification of the number of newly regenerated (centrally nucleated) fibers in CTL and MKO muscles at 5 and 7 d after injury. (G) Relative number of mononuclear cells within regenerating MKO and CTL muscles at 5 and 7 d after injury. All values are relative to control levels (n = 3; *p < 0.05). Error bars represent SE.
DISCUSSION
In the present study, we examined the role of BDNF in skeletal muscle using BDNF−/− mice and transgenic mice in which BDNF was specifically depleted from skeletal muscle cells. The muscle-specific BDNFMKO circumvents the problem of early postnatal lethality and breeding issues presented by BDNF−/− mice, and it allows examination of the specific role of muscle-derived BDNF in skeletal muscle development, maintenance, and regeneration. In characterizing the BDNFMKO mouse, we demonstrate that muscle-derived BDNF plays a role in 1) regulating proliferation and differentiation of satellite cells and 2) plays a role during muscle regeneration in vivo. Our present results demonstrate an important role for BDNF in regulating satellite cell function and muscle regeneration, particularly during early stages of myogenic differentiation.
In our initial characterization of the BDNFMKO mouse, we found that BDNF was depleted by 50–60% in whole muscles. However, BDNF was depleted by >85% in muscle fibers and associated satellite cells. These findings suggest that a portion of the BDNF detected in whole muscle preparations is derived from resident nonmuscle cells within the muscle compartment. This was further supported in our regeneration studies, where BDNF staining colocalized with CD11b+ immune cells. These findings are not entirely surprising, as the adult muscle compartment is composed of several cell types, including endothelial cells, neurons, Schwann cells, and immune cells, all of which have been shown to express and secrete BDNF (Kerschensteiner et al., 1999; Vega et al., 2003; Kermani and Hempstead, 2007; reviewed; Gordon, 2009). Thus, it becomes important to consider that low levels of secreted BDNF are available to BDNFMKO muscle cells, and because p75NTR and TrkB receptor levels are unchanged (Supplemental Figure 1B), some BDNF signaling may persist. As such, the phenotype of BDNFMKO muscle should be considered less severe than it would be in the absence of BDNF.
In the present study, we isolated single fibers and their associated satellite cells to examine myogenic properties in the absence of BDNF. In this system, it became obvious that muscle-derived BDNF is important for normal satellite cell function. Although Pax7 expression was reduced compared with controls, the number of satellite cell-derived myoblasts was similar, as were levels of the myogenic determination gene MyoD, suggesting that commitment of BDNFMKO satellite cells to the myogenic lineage was not affected. However, later steps in myogenic lineage progression, including efficient induction of “late” MRFs, p21, and myosin heavy chain (MyHC) require muscle-BDNF. Addition of exogenous BDNF protein was sufficient to rescue myotube size and normal MyHC levels, suggesting that BDNF itself is required for normal satellite cell function. Furthermore, these findings suggest that depletion of muscle-BDNF does not alter the intrinsic ability of muscle satellite cells to undergo normal differentiation.
Similar results were observed upon activation of the adult myogenic program in vivo. After injury, early regeneration was delayed in BDNFMKO muscles compared with controls, with delayed induction of molecular and histological signatures of repair. Interestingly, however, normal regeneration was restored within 7 d. Recovery of normal regeneration occurred as BDNF expression increased to control levels, probably due to secretion from infiltrating CD11b+ immune cells. As such, resident nonmuscle cell-derived BDNF appears to comprise a compensatory mechanism by which normal myogenic potential is restored to BDNFMKO tissue. In agreement with our culture studies, the delay in regeneration appears to be in response to a specific requirement for BDNF during myogenic differentiation, rather than an intrinsic defect within BDNFMKO satellite cells. Given that BDNF receptor levels are unchanged, secreted BDNF would be capable of normal signaling within the muscle compartment, allowing for recovery of normal regeneration. If this is the case, BDNF may play a similar role to other inflammatory cytokines (transforming growth factor-β and interleukin-6) in regulating muscle regeneration (Tidball, 2005). Furthermore, it becomes apparent that immune-cell derived BDNF is an important source of BDNF during muscle injury and repair. Similar findings have been reported in human peripheral neuropathies, in which BDNF expression was proportional to the extent of invasion by T cells and macrophages rather than to the specific disease pathology (Sobue et al., 1998).
Although further studies are required to elucidate the precise molecular mechanisms involved in BDNF-mediated progression of myogenic differentiation, evidence suggests that signaling through the low-affinity p75 neurotrophin receptor (p75NTR) plays an important role. P75NTR can mediate a number of downstream signaling cascades depending on the availability of ligands, coreceptors, and downstream signaling molecules. Several targets of p75NTR have been implicated in neuronal differentiation, and several studies have suggested that these targets may play a similar role in skeletal muscle (Seidl et al., 1998; Rende et al., 1999; Reddypalli et al., 2005). Of particular interest are the recent findings of Deponti et al., (2009), who show that p75NTR signaling is required for normal satellite cell function and muscle repair. In this study, inhibition of NGF-mediated p75NTR signaling resulted in increased expression of RhoA-GTP, which negatively affected myoblast fusion and cytoskeletal organization. These findings were recapitulated in vivo during muscle regeneration, where injection of an NGF-competing peptide, or myoblasts infected with constitutively active form of RhoA resulted in decreased regeneration after cardiotoxin injury (Deponti et al., 2009). However, this study did not detect a change in muscle-specific gene expression, suggesting that additional or alternate p75NTR-mediated signaling pathways are affected in the BDNFMKO mouse.
Previous studies in our laboratory showed that siRNA-mediated depletion of BDNF resulted in precocious differentiation of rat L6 myoblasts (Mousavi and Jasmin, 2006). Based on these findings, we had expected to see accelerated differentiation of BDNFMKO myoblasts in culture and regeneration. Instead, we found that in the absence of muscle-BDNF, both differentiation and regeneration were delayed. Thus, our more recent findings suggest that rather than playing an inhibitory role, BDNF is required for normal myogenic differentiation, specifically during early stages. There are many possible causes for the disparity observed between these studies. First, there are intrinsic differences in the properties of immortalized cells lines and primary cultures. Second, the developmental timing of BDNF depletion is different between the two studies. These differences, in conjunction with the different mechanisms used to deplete BDNF expression (siRNA transfection vs. transgenic knockout) could result in altered response of satellite cells to signals that promote growth, differentiation, or both.
Finally, it becomes important to consider that many features of embryonic muscle development are recapitulated during muscle regeneration, with similar changes in muscle gene expression, physiological properties, and functional characteristics. In BDNFMKO muscle, Pax7 expression is decreased, satellite cell differentiation is defective, and regeneration is delayed. Together, these findings clearly show that BDNF is required for normal satellite cell function and suggest that complete ablation of BDNF, if it did not result in early death due to severe neuronal deficits, would considerably exacerbate the observed muscle phenotype. Intriguingly, Pax7−/− mice lack satellite cells and fail to grow postnatally, resulting in death by ∼2 wk (Seale et al., 2000). Considering that satellite cell function is impaired in the BDNFMKO mouse, we could expect to see similar deficiencies in postnatal growth and survival. However, because resident nonmuscle cells can secrete BDNF, and because BDNF receptor expression is not affected (Supplemental Figure 1B), it seems likely that normal muscle development ensues, resulting in the subtle phenotype of the BDNFMKO mouse that we observed postnatally.
Supplementary Material
ACKNOWLEDGMENTS
We thank John Lunde and Amanda Shaver for technical assistance in various aspects of this study. We are also grateful to Dr. Kambiz Mousavi for thoughtful discussion and Drs. Phillip Soriano (Fred Hutchinson Cancer Research Center, Seattle, WA) and Michael Rudnicki (Sprott Center for Stem Cell Research, Ottawa, ON, Canada) for providing the Myf5-Cre mouse. This work was supported by research grants from the Canadian Institutes of Health Research and the Muscular Dystrophy Association (USA) (to B.J.J.). C. C. was supported by a Canadian Institutes of Health Research (CIHR) Canada Graduate Scholarship (CGS) master's award during the course of this work and is presently a recipient of a CIHR CGS doctoral award.
Abbreviations used:
- BDNF
brain-derived neurotrophic factor
- MRF
myogenic regulatory factor
- MyHC
myosin heavy chain.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-02-0154) on April 28, 2010.
REFERENCES
- Capsoni S., Ruberti F., Di Daniel E., Cattaneo A. Muscular dystrophy in adult and aged anti-NGF transgenic mice resembles an inclusion body myopathy. J. Neurosci. Res. 2000;59:553–560. doi: 10.1002/(SICI)1097-4547(20000215)59:4<553::AID-JNR11>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Carrasco D. I., English A. W. Neurotrophin 4/5 is required for the normal development of the slow muscle fiber phenotype in the rat soleus. J. Exp. Biol. 2003;206:2191–2200. doi: 10.1242/jeb.00412. [DOI] [PubMed] [Google Scholar]
- Charge S. B., Rudnicki M. A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- Chevrel G., Hohlfeld R., Sendtner M. The role of neurotrophins in muscle under physiological and pathophysiological conditions. Muscle Nerve. 2006;33:462–476. doi: 10.1002/mus.20444. [DOI] [PubMed] [Google Scholar]
- Condrea E. Membrane-active polypeptides from snake venom: cardiotoxins and haemocytotoxins. Experimentia. 1974;30:121–129. doi: 10.1007/BF01927688. [DOI] [PubMed] [Google Scholar]
- Deponti D., et al. The low affinity receptor for neurotrophins p75NTR plays a key role for satellite cell function in muscle repair acting via RhoA. Mol. Biol. Cell. 2009;20:3620–3627. doi: 10.1091/mbc.E09-01-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P., Lee K. F., Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994a;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Ernfors P., Lee K. F., Kucera J., Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994b;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- Gonzalez M., Ruggiero F. P., Chang Q. Disruption of Trk-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- Gordon T. The role of neurotrophic factors in nerve regeneration. Neurosurg. Focus. 2009;26:E3. doi: 10.3171/FOC.2009.26.2.E3. [DOI] [PubMed] [Google Scholar]
- Gramolini A. O., Karpati G., Jasmin B. J. Discordant expression of utrophin and its transcript in human and mouse skeletal muscles. J. Neuropathol. Exp. Neurol. 1999;58:235–244. doi: 10.1097/00005072-199903000-00003. [DOI] [PubMed] [Google Scholar]
- Griesbeck O., Parsadanian A. S., Sendtner M., Thoenen H. Expression of neurotrophins in skeletal muscle: quantitative comparison and significance of motorneuron survival and maintenance of function. J. Neurosci. Res. 1995;42:21–33. doi: 10.1002/jnr.490420104. [DOI] [PubMed] [Google Scholar]
- Hamburger V. The effects of wing bud extirpation on the development of the central nervous system in chick embryos. J. Exp. Zool. 1934;68:449–494. [Google Scholar]
- Hamburger V., Yip J. W. Reduction of experimentally induced neuronal death in spinal ganglia of chick embryo by nerve growth factor. J. Neurosci. 1984;4:767–774. doi: 10.1523/JNEUROSCI.04-03-00767.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh M. S., Parker M. H., Scime A., Parks R., Rudnicki M. A. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J. Cell Biol. 2004;166:865–876. doi: 10.1083/jcb.200403004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip F. C., Cheung J., Ip N. Y. The expression profiles of neurotrophins and their receptors in rat and chicken tissues during development. J. Neurosci. Lett. 2001;301:107–110. doi: 10.1016/s0304-3940(01)01603-2. [DOI] [PubMed] [Google Scholar]
- Jin X., et al. Opposite roles of MRF4 and MyoD in cell proliferation and myogenic differentiation. Biochem. Biophys. Res. Commun. 2007;364:476–482. doi: 10.1016/j.bbrc.2007.10.042. [DOI] [PubMed] [Google Scholar]
- Kablar B., Belliveau A. C. Presence of neurotrophic factors in skeletal muscle correlates with survival of spinal cord motor neurons. Dev. Dyn. 2005;234:659–669. doi: 10.1002/dvdy.20589. [DOI] [PubMed] [Google Scholar]
- Kalb R. The protean actions of neurotrophins and their receptors on the life and death of neurons. Trends Neurosci. 2005;28:5–11. doi: 10.1016/j.tins.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Kermani P., Hempstead B. Brain-derived neurotrophic factor: a newly described mediator of angiogenesis. Trends Cardiovasc. Med. 2007;17:140–143. doi: 10.1016/j.tcm.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M., et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J. Exp. Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S., Kuroda K., Le Grand F., Rudnicki M. A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton B. H., Schultz E. Developmental fate of skeletal muscle satellite cells. Science. 1979;205:1292–1294. doi: 10.1126/science.472747. [DOI] [PubMed] [Google Scholar]
- Lomen-Hoerth C., Shooter E. M. Widespread neurotrophin expression in the immune system and other nonneuronal rat tissues. J. Neurochem. 1995;64:1780–1789. doi: 10.1046/j.1471-4159.1995.64041780.x. [DOI] [PubMed] [Google Scholar]
- Mousavi K., Jasmin B. J. BDNF is expressed in skeletal muscle satellite cells and inhibits myogenic differentiation. J. Neurosci. 2006;26:5739–5749. doi: 10.1523/JNEUROSCI.5398-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi K., Parry D. J., Jasmin B. J. BDNF rescues myosin heavy chain IIB muscle fibers after neonatal nerve injury. Am. J. Physiol. Cell Physiol. 2004;287:C22–C29. doi: 10.1152/ajpcell.00583.2003. [DOI] [PubMed] [Google Scholar]
- Omura T., Sano M., Omura K., Hasegawa T., Doi M., Sawada T., Nagano A. Different expressions of BDNF, NT3 and NT4 in muscle and nerve after various types of peripheral nerve injuries. J. Peripher. Nerv. Syst. 2005;10:293–300. doi: 10.1111/j.1085-9489.2005.10307.x. [DOI] [PubMed] [Google Scholar]
- Oppenheim R. W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pitts E. V., Potluri S, Hess D. M., Balice-Gordon R. J. Neurotrophin and Trk-mediated signaling in the neuromuscular system. Int. Anesthesiol. Clin. 2006;44:21–76. doi: 10.1097/00004311-200604420-00004. [DOI] [PubMed] [Google Scholar]
- Reddypalli S., Roll K., Lee H. K., Lundell M., Barea-Rodriguez E., Wheeler E. F. p75NTR-mediated signaling promotes the survival of myoblasts and influences muscle strength. J. Cell. Physiol. 2005;204:819–829. doi: 10.1002/jcp.20330. [DOI] [PubMed] [Google Scholar]
- Rende M., Brizi E., Sorci G., Bianchi R., Provenzano C., Bruno R., Donato R. Regulation of the p75 neurotrophin receptor in a rat myogenic cell line (L6) Histochem. J. 1999;31:589–601. doi: 10.1023/a:1003851024732. [DOI] [PubMed] [Google Scholar]
- Reichardt L. F. Neurotrophin-regulated signaling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios M., Fan G., Fekete C., Kelly J., Bates B., Kuehn R., Lechan R. M., Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol. Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J. F., Lunt A. I., Parry D. J., Partridge T. A. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell. Dev. Biol. 1995;31:773–779. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- Schultz E., Gibson M. C., Champion T. Satellite cells are mitotically quiescent in mature mouse muscle: an EM and radioautographic study. J. Exp. Zool. 1978;206:451–456. doi: 10.1002/jez.1402060314. [DOI] [PubMed] [Google Scholar]
- Seale P., Sabourin L. A., Girgis-Gabardo A., Mansouri A., Gruss P., Rudnicki M. A. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Seidl K., Erck C., Buchberger A. Evidence for the participation of nerve growth factor and its low-affinity receptor in the regulation of the myogenic program. J. Cell. Physiol. 1998;176:10–21. doi: 10.1002/(SICI)1097-4652(199807)176:1<10::AID-JCP2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Sheard P. W., Musaad K., Duxson M. J. Distribution of neurotrophin receptors in the mouse neuromuscular system. Int. J. Dev. Biol. 2002;46:569–575. [PubMed] [Google Scholar]
- Snow M. H. An autoradiographic study of satellite cell differentiation into regenerating myotubes following transplantation of muscles in young rats. Cell Tissue Res. 1978;186:535–540. doi: 10.1007/BF00224941. [DOI] [PubMed] [Google Scholar]
- Sobue G., Yamamoto M., Doyu M., Yasuda T., Mitsuma T. Expression of mRNA for neurotrophins (NGF, BDNF, and NT-3) and their receptors (p75NGFR, Trk, TrkB and TrkC) in human peripheral neuropathies. Neurochem. Res. 1998;23:821–829. doi: 10.1023/a:1022434209787. [DOI] [PubMed] [Google Scholar]
- Tallquist M. D., Weismann K. E., Hellstrom M., Soriano P. Early myotome specification regulates PDGFA expression and axial skeleton development. Development. 2000;127:5059–5070. doi: 10.1242/dev.127.23.5059. [DOI] [PubMed] [Google Scholar]
- Tessarollo L., Tsoulfas P., Martin-Zanca D., Gilbert D. J., Jenkins N. A., Copeland N. G., Parada L. F. TrkC, a receptor for neurotrophin-3 is widely expressed in the developing nervous system and in non-neuronal tissues. Development. 1993;118:463–475. doi: 10.1242/dev.118.2.463. [DOI] [PubMed] [Google Scholar]
- Tidball J. G. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288:R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- Vega J. A., Garcia-Suarez O., Hannestad J., Perez-Perez M., Germana A. Neurotrophins and the immune system. J. Anat. 2003;203:1–19. doi: 10.1046/j.1469-7580.2003.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.