

Protein kinase D (PKD) is a critical regulator of Golgi structure and function. Biochemical evidence is presented that demonstrates the oxysterol-binding protein OSBP as a novel PKD substrate. Phosphorylation inhibits OSBP Golgi localization, impairs CERT Golgi localization, and promotes Golgi fragmentation.
Abstract
Protein kinase D (PKD) plays a critical role at the trans-Golgi network by regulating the fission of transport carriers destined for the plasma membrane. Two known Golgi-localized PKD substrates, PI4-kinase IIIβ and the ceramide transfer protein CERT, mediate PKD signaling to influence vesicle trafficking to the plasma membrane and sphingomyelin synthesis, respectively. PKD is recruited and activated at the Golgi through interaction with diacylglycerol, a pool of which is generated as a by-product of sphingomyelin synthesis from ceramide. Here we identify a novel substrate of PKD at the Golgi, the oxysterol-binding protein OSBP. Using a substrate-directed phospho-specific antibody that recognizes the optimal PKD consensus motif, we show that PKD phosphorylates OSBP at Ser240 in vitro and in cells. We further show that OSBP phosphorylation occurs at the Golgi. Phosphorylation of OSBP by PKD does not modulate dimerization, sterol binding, or affinity for PI(4)P. Instead, phosphorylation attenuates OSBP Golgi localization in response to 25-hydroxycholesterol and cholesterol depletion, impairs CERT Golgi localization, and promotes Golgi fragmentation.
INTRODUCTION
The Golgi apparatus controls secretory traffic through generation of vesicles that transport lipid and protein cargo destined for the plasma membrane (Warren and Malhotra, 1998; Emr et al., 2009). Despite the constant flux of lipids such as cholesterol, ceramide, and phosphoinositides, the Golgi maintains a unique lipid composition. Several lipid-modifying and -binding proteins function as regulators of Golgi lipid homeostasis. The Golgi is enriched in phosphatidylinositol 4-phosphate [PI(4)P], achieved by the phosphorylation of phosphatidylinositol by PI4-kinase IIIβ (PI4KIIIβ; Hausser et al., 2005, 2006; Weixel et al., 2005). Nir2, a PI-phosphatidylcholine transfer protein, is essential for Golgi vesicle transport through regulation of diacylglycerol (DAG; Vihtelic et al., 1993; Litvak et al., 2005). In addition, phosphatidylinositol-four-P adaptor proteins bind PI(4)P at the Golgi and regulate the formation of transport vesicles (Godi et al., 2004). Similarly, the ceramide transfer protein (CERT) transports ceramide from the endoplasmic reticulum (ER) to the Golgi complex, a process that is critical for sphingomyelin (SM) synthesis and production of DAG (Perry and Ridgway, 2005). A distinct Golgi-localized protein, the oxysterol-binding protein (OSBP), acts as a global sterol sensor or transfer protein for cholesterol and 25-hydroxycholesterol (25-OH). Importantly, both PI4KIIIβ and CERT are substrates of protein kinase D (PKD; Hausser et al., 2005; Fugmann et al., 2007), a serine/threonine kinase that also localizes to the Golgi (Van Lint et al., 2002).
PKD comprises a family of protein kinases in the calcium–calmodulin kinase family of the kinome. Three distinct isoforms exist in mammals: PKD1/PKCμ, PKD2, and PKD3/PKCν. The PKD regulatory domain comprises two cysteine-rich domains (C1a and C1b) that bind DAG and a pleckstrin homology (PH) domain that functions to autoinhibit the kinase (Van Lint et al., 2002; Rykx et al., 2003). The catalytic domain is highly conserved in all PKD orthologues, and a PDZ (postsynaptic-density-95, Drosophila-discs-large, zona-occludens 1) binding motif is found at the carboxy terminus of PKD1 and PKD2, but not PKD3. In addition to Golgi function, pools of PKDs have been observed at the plasma membrane, cytoplasm, mitochondria, and nucleus where they regulate a variety of cellular processes including cellular proliferation and survival, motility, transcriptional regulation, and immune cell responses (Van Lint et al., 2002; Rykx et al., 2003).
Studies on the function of PKD at the Golgi have revealed that localization at the trans-Golgi network (TGN) is mediated through DAG binding conferred by the C1a domain (Maeda et al., 2001; Baron and Malhotra, 2002). At the TGN, PKD is also activated by novel protein kinase C (PKC) isoforms (Diaz Anel and Malhotra, 2005) and control the fission of transport carriers destined for the plasma membrane (Liljedahl et al., 2001; Yeaman et al., 2004). At the Golgi, PKD transduces signals through phosphorylation and activation of PI4KIIIβ, leading to increased PI(4)P production (Hausser et al., 2005). CERT, a protein that regulates the transport of ceramide from the ER to the Golgi, is also phosphorylated by PKD thereby decreasing its affinity for PI(4)P (Hanada et al., 2003; Toth et al., 2006). In turn this decreases CERT activity and consequently reduces SM synthesis (Fugmann et al., 2007). Because both CERT and PI4KIIIβ are lipid-binding and -modifying proteins that are PKD substrates, we sought to identify additional substrates that might contribute to PKD function at the Golgi. Here we report the identification of a new PKD substrate, OSBP, that is phosphorylated and regulated at the Golgi.
OSBP is a member of the oxysterol-binding protein family that includes 11 other OSBP-related proteins (ORPs). OSBP and ORPs are characterized by a carboxyl-terminal sterol-binding domain that binds sterols such as 25-OH and cholesterol (Lehto and Olkkonen, 2003). OSBP localizes to the cytoplasm or with vesicle-associated membrane protein–associated protein (VAP) in the ER (Wyles et al., 2002; Loewen et al., 2003; Loewen and Levine, 2005). On binding 25-OH, OSBP translocates to the Golgi (Ridgway et al., 1992; Mohammadi et al., 2001). This translocation also occurs when cholesterol and sphingomyelin are depleted. OSBP localizes to the Golgi by binding PI(4)P through PH domain interaction (Levine and Munro, 1998, 2002). OSBP regulates the sterol-dependent activation and translocation of CERT, thereby integrating cholesterol and SM metabolism (Perry and Ridgway, 2006). OSBP does not directly regulate cholesterol homeostasis by binding 25-OH, but instead senses or transfers sterols as a mechanism to integrate cellular lipid metabolism (Wang et al., 2005; Ngo and Ridgway, 2009). Here, we provide evidence that PKD phosphorylates OSBP, thereby inhibiting Golgi localization in response to 25-OH and cholesterol depletion.
MATERIALS AND METHODS
DNA constructs, Reagents, and Antibodies
The anti-PKD (C20) and anti-c-Myc (9E10) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-PKD pSer 744/748 antibody was from Cell Signaling (Danvers, MA); anti-OSBP from Proteintech (Chicago, IL); anti-β-actin from Sigma (St. Louis, MO); anti-TGN46 from AbD Serotec (Raleigh, NC); anti-Giantin from Abcam (Cambridge, MA); anti-GM130 from Novus Biologicals (Littleton, CO); and anti-PI4KIIIβ from BD Transduction (San Jose, CA). The anti-vesicular stomatitis virus glycoprotein (VSVG; 8G5F11) antibody was a generous gift from Dr. Doug Lyles (Wake Forest University). The anti-HA antibody was purified in-house from the 12CA5 hybridoma. The anti-PKD pMOTIF antibody has been described (Doppler et al., 2005). Lipoprotein-deficient bovine calf serum and human low-density lipoprotein (LDL) were from Biomedical Technologies (Stoughton, MA). All expression plasmids for PKD1 are based on an amino-terminal HA-tagged PKD1 in pcDNA3 and have been described (Storz and Toker, 2003). The rabbit OSBP (rOSBP) in pcDNA3.1 (pCDNA3.1-rOSBP-myc-His) was a generous gift from Dr. R. G. W. Anderson (UT Southwestern). The tsO45-VSVG-GFP has been described (Hirschberg et al., 2000). The S242A, S242D, and S271A mutations were introduced using the following oligonucleotide primer pairs: for S242A: 5′-GGCACAGCCCTGCAGCGTTCCCTCGCCGA GCTGGAGTCCCTGAAGTTGCC-3′ and 5′-GGCAACTTCAGGGACTCCAGCTCGGCGAGGGAACGCTGCAGGGCTGTGCC-3′; 5′-GGCACAGCCCTGCAGCGTTCCCTCGACGAGCTGGAGTCCCTGAAGTTGCC-for S242D: 3′ and 5′-GGCAACTTCAGGGACTCCAGCTCGTCGAGGGAACGCTGCAGGGCTGTGCC-3′; and for S271A: and 5′-GCCACACTCTTCAGAATAACCGCCAATGCCATGATCAATGCTTGC-3′ and 5′-GCAAGCATTGATC ATGGCATTGGCGGTTATTCTGAAGAGTGTGGC-3′. Mutagenesis was carried out using the QuikChange strategy (Stratagene, La Jolla, CA), and all constructs were verified by sequencing. To silence the expression of OSBP, the following primer pair was cloned into the pLKO.1 vector (Moffat et al., 2006): 5′-CCGGGGCAAACACTCCTGGCAATGTCTCGAGACATTGCCAGGAGTGTTTGCCTTTTTG-3′ and 5′-AATTCAAAAAGGCAAACACTCCTGGCAATGTCTCGAGACATTGCCAGGAGTGTTTGCC-3′. Second-generation lentiviral packaging plasmids, pCMV-dR8.2 dvpr and pCMV-VSVG, were obtained from Addgene (Cambridge, MA).
Cell Culture
Human embryonic kidney (HEK) 293T cells and Hela cells were maintained in high-glucose DMEM supplemented with 10% fetal bovine serum. LnCap cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. Cells were transiently transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) or polyethylenimine (PEI) gene transfer reagent (Boussif et al., 1995) in its 25-kDa linear form (Polysciences, Warrington, PA). Typically, 6 μg of DNA was mixed with 18 μg of linear 25-kDa PEI in a total volume of 400 μl of GIBCO reduced serum OPTI-MEM medium (GIBCO, Rockford, MD). This mix was incubated at room temperature for 15 min and then was added to 6-cm plates of subconfluent Hela cell monolayers that had been washed twice with 1× PBS or HEK293T cells in suspension. Treatment of cells with phorbol 12,13-dibutyrate (PdBu; Sigma) was at 1 μM for 30 min after overnight serum starvation.
Lentiviral Infection
pLKO.1 plasmid constructs were cotransfected in HEK293T cells with packaging vectors pCMV-Δ8.2 and pCMV-VSVG using PEI. Two days after transfection, lentiviral particles were harvested, passed through a 0.45-μm filter, and used to infect target cells. Target cells at ∼70% confluency were infected for 24 h followed by selection in 2 μg/ml (HEK293T) or 4 μg/ml (LnCap) puromycin for 24 h (HEK293T) or 48 h (LnCap) before stimulation and cell lysis.
Immunoblotting and Immunoprecipitation
Cells were serum-starved for 24 h and stimulated and/or harvested 48 h after transfection. The cells were lysed in lysis buffer (50 mM Tris/HCl, pH 7.5, 0.5% NP-40, 150 mM NaCl, 1 mM EDTA, pH 7.5, 1 mM Na2HPO4, 20 mM NaF, 1 mM DTT, 50 nM calyculin, 1 mM PMSF) plus protease inhibitor cocktail (Sigma). Either the lysates were used for immunoblot analysis, or proteins were immunoprecipitated by a 2- or 4-h incubation with 1 μg anti-myc or anti-OSBP, respectively, followed by a 2-h incubation with protein G agarose (Amersham Biosciences, Piscataway, NJ). Immune complexes were washed three times with NETN (20 mM Tris/HCl, pH 8.0, 100 mM NaCl, 0.5% NP-40, 1 mM EDTA, pH 8.0) and resolved by SDS-PAGE or were subjected to kinase assays.
In Vitro Kinase Assay
Substrates were expressed in HEK293T cells and immunoprecipitated with myc antibody. Immune complexes were incubated with purified hemagglutinin (HA)-tagged PKD1 from baculovirus-infected Sf9 insect cells (Storz et al., 2003) in kinase buffer (25 mM Tris-HCl, pH 7.5, 5 mM β-glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, and 10 mM MgCl2) in the presence of 250 μM cold ATP for 1 h at 30°C. Samples were resolved by SDS-PAGE, blotted onto membrane, and detected with the indicated antibodies.
Immunofluorescence Microscopy
Cells were washed twice with PBS and then fixed in PBS containing 2% formaldehyde for 10 min. Cells were permeabilized with 0.5% Triton X-100 for 1 min and blocked with 1% BSA for 20 min. Fixing and permeabilizing solutions were made in buffer containing 100 mM KCl, 100 mM HEPES, pH 6.8, 200 mM sucrose, and 1 mM MgCl2. Blocking solution was made in buffer containing 20 mM Tris-HCl, pH 7.5 and 150 mM NaCl. Cells were then incubated with primary antibody diluted in blocking buffer for 1 h followed by three washes in PBS and incubation with secondary antibodies diluted in blocking buffer for 1 h. After washing three times with PBS, coverslips were mounted in ProLong Gold (Invitrogen) and analyzed on a fluorescence microscope (Eclipse TE2000, Nikon, Melville, NY) using a PL Fluor 100×/1.30 NA oil objective lens. Images were processed with Photoshop (Adobe Systems, San Jose, CA).
VSVG Transport Assay
After lentiviral infection, cells were transfected with myc-tagged OSBP plasmids and cultured at 37°C. After 24 h, cells were transfected with tsO45-VSVG-green fluorescent protein (GFP) and cultured at 40°C for 20 h. Cycloheximide, at 100 μg/ml, was added before a 15-min incubation at 32°C. This was followed by a 2-h incubation at 20°C and a final incubation at 32°C for the indicated times. Cells were fixed with 2% formaldehyde and blocked as described above. Surface VSVG was labeled for 1 h with anti-VSVG mAb 8G5F11, which is specific for the extracellular domain of VSVG. After washings with PBS, cells were incubated with secondary Cy3-conjugated anti-mouse IgG for 1 h.
Mass Spectrometry
Coomassie-stained SDS-PAGE gel bands containing myc-OSBP isolated from HEK293T cells with different treatments were excised and subjected to reduction with DTT and alkylation with iodoacetamide in-gel digestion with trypsin overnight at pH 8.3. Reversed-phase microcapillary liquid chromatography tandem mass spectrometry (LC-MS/MS) using a Easy-nLC nanoHPLC (Proxeon Biosystems, Odense, Denmark) using a self-packed 75μm-id × 15-cm C18 column connected to a LTQ-Orbitrap XL mass spectrometer (Thermo Scientific, Waltham, MA) in data-dependent acquisition and positive ion mode at 300 nl/min. MS/MS spectra collected via collision-induced dissociation in the ion trap were searched against the concatenated target and decoy (reversed) Swiss-Prot protein databases using Sequest (proteomics browser software, Thermo Scientific) with differential modifications for Ser/Thr/Tyr phosphorylation (+79.97) and the sample processing artifacts Met oxidation (+15.99) and Cys alkylation (+57.02) and deamidation of Asn and Gln (+0.984). Phosphorylated and unphosphorylated peptide sequences were identified if they initially passed the following Sequest scoring thresholds: 1+ ions, Xcorr ≥ 2.0 Sf ≥ 0.4, p ≥ 5; 2+ ions, Xcorr ≥ 2.0, Sf ≥ 0.4, p ≥ 5; and 3+ ions, Xcorr ≥ 2.60, Sf ≥ 0.4, p ≥ 5 against the target protein database. Passing MS/MS spectra were manually inspected to be sure that all b- and y-fragment ions aligned with the assigned sequence and modification sites. Determination of the exact sites of phosphorylation was aided using FuzzyIons and GraphMod software (proteomics browser software, ThermoFischer Scientific). For relative quantification of phosphorylation peptide signal levels, an isotope-free (label-free) method was used by first integrating the total ion counts (TIC) for each MS/MS sequencing event during a targeted ion MS/MS (TIMM) experiment or a data-dependant acquisition (Asara et al., 2008; Dibble et al., 2009; Zheng et al., 2009). For each targeted phosphorylation site (Ser242 and Ser271), a ratio of phosphorylated peptide signal (TIC of phosphorylated form) to the total peptide signal (TIC of phosphorylated form + TIC of nonphosphorylated form) from the OSBP, OSBP + SS/EE PKD, and OSBP + SS/EE PKD + PKD shRNA samples were calculated according to the following equation:
The ratios of the Ser242 site (no phosphorylation was observed for Ser271) from OSBP + EE PKD and OSBP + EE PKD + PKD shRNA samples were then compared with the same phosphopeptide ratios from the OSBP sample according to the following equation:
Although a direct comparison of phosphopeptide signals between different experiments is not accurate because of different total protein levels and sample environments, a comparison of the ratio of the phosphorylated to nonphosphorylated peptide forms is an accurate measure of signal-level change because of the total peptide signal (modified and unmodified). The above calculations were performed using automated in-house developed software, Protein Modification Quantifier v1.0 (Beth Israel Deaconess Medical Center, Boston, MA).
RESULTS
OSBP Reactivity with PKD pMOTIF Requires PKD Activity
We recently developed a novel PKD phosphorylation-state specific substrate–directed antibody that recognizes the optimal phosphorylation motif of PKD, LXR(Q/K/E/M)(M/L/K/E/Q/A)S*XXXX (where X denotes any amino acid; Doppler et al., 2005). The PKD pMOTIF antibody has been used to identify several PKD substrates including two novel Golgi-localized PKD targets, PI4KIIIβ (Hausser et al., 2005) and CERT (Fugmann et al., 2007). We reasoned that additional Golgi-localized lipid-binding proteins that are also PKD substrates must exist, and screened phosphopeptides recently were identified by functional phospho-proteomics (Villen et al., 2007). Using this approach, we identified a phosphopeptide in human OSBP that conforms to the optimal PKD consensus motif at Ser240 (Figure 1A).
Figure 1.

OSBP is recognized by the PKD substrate antibody. (A) Domain structure of OSBP revealing the alignment of PKD phosphorylation sites Ser 240 and Ser 269 from different species. (B and C) HEK293T cells transfected with myc-OSBP stimulated with (B) 1 μM PdBu or (C) 50 ng/μL PDGF for 10 or 30 min. OSBP was immunoprecipitated with anti-myc and immunoblotted with anti-pMOTIF or anti-myc. Control immunoblotting of total lysate was with anti-actin and anti-pSS738/742. (D) HEK293T cells were transfected with myc-OSBP and HA-PKD.SS/EE or PKD.KW. OSBP was immunoprecipitated with anti-myc and immunoblotted with anti-pMOTIF or anti-myc. Total lysates were immunoblotted with anti-actin and anti-HA. (E) HEK293T cells transfected with myc-OSBP were treated with or without 1 μM Gö6976 for 1 h and then stimulated with 1 μM PdBu for 30 min. OSBP was immunoprecipitated with anti-myc and immunoblotted with anti-pMOTIF or anti-myc. Total lysates were immunoblotted with anti-pSS738/742 and anti-actin. Results are representative of at least three independent experiments.
To evaluate whether OSBP is a PKD substrate, we first determined whether OSBP is recognized by the pMOTIF antibody. Cells transfected with Myc-tagged OSBP were stimulated with PdBu, a potent PKD activator, and analyzed by immunoblotting. OSBP immunoreactivity with PKD pMOTIF coincides with PKD phosphorylation at Ser738/Ser742, a surrogate for PKD activation (Figure 1B). Similar kinetics of phosphorylation are also observed with platelet-derived growth factor (PDGF), a physiological agonist of PKD (Figure 1C). Immunoreactivity of OSBP with pMOTIF antibody is dependent on PKD catalytic activity because a constitutively active PKD allele (PKD.SS/EE) induces OSBP phosphorylation, whereas a kinase-inactive allele (PKD.KW) does not (Figure 1D). Moreover, immunoreactivity is completely blocked in cells pretreated with Gö6976, a potent PKD and PKC inhibitor, compared with control PdBu-stimulated cells (Figure 1E). Taken together, these data show that PKD kinase activity is required for OSBP recognition by the pMOTIF antibody.
PKD Phosphorylates OSBP at Ser242
The PKD pMOTIF antibody recognizes the phosphorylated consensus motif of other protein kinases such as AMP-activated kinase (AMPK), mitogen-activated protein kinase–activated protein kinase 2 (MAPKAP-K2 or MK2), and calcium–calmodulin-dependent kinases (CamK). To determine whether PKD is the relevant kinase for OSBP phosphorylation, PKD short hairpin RNA (shRNA) was used. PdBu stimulation increases endogenous OSBP phosphorylation as predicted; however, silencing PKD1 expression using lentiviral shRNA reduces OSBP recognition by pMOTIF (Figure 2A). To corroborate these results, a rescue experiment was performed in which OSBP was expressed with constitutively active PKD.SS/EE or an shRNA resistant PKD.SS/EE* allele in cells depleted of PKD1 and PKD2. In the presence of PKD, OSBP is phosphorylated by PKD.SS/EE and PKD.SS/EE* to similar levels (Figure 2B, lanes 3 and 4). Silencing of PKD.SS/EE by PKD1/2 shRNA decreases OSBP phosphorylation (Figure 2B, lane 7). PKD.SS/EE* is refractory to silencing by PKD1/2 shRNA, and this effectively rescues OSBP phosphorylation (Figure 2B, lane 8). Finally, silencing of PKD1 or PKD2 alone or in combination decreases OSBP phosphorylation to similar levels, indicating a lack of isoform specificity in OSBP phosphorylation (Figure 2C). This is in agreement with other Golgi-localized PKD substrates such as PI4KIIIβ and CERT that are also efficiently phosphorylated by PKD1 or PKD2 (Hausser et al., 2005; Fugmann et al., 2007).
Figure 2.
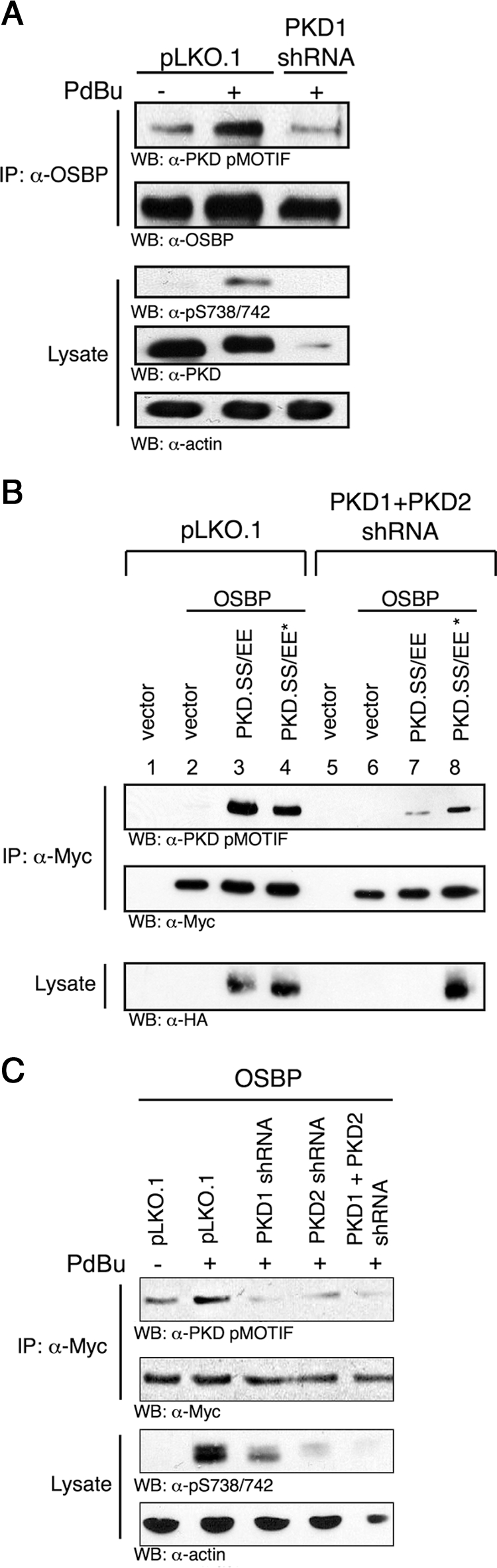
PKD phosphorylates OSBP. (A) LnCap cells were lentivirally infected with PKD1 shRNA, selected with puromycin, serum-starved, and stimulated with 1 μM PdBu for 30 min. Endogenous OSBP was immunoprecipitated with anti-OSBP and immunoblotted with anti-pMOTIF or anti-OSBP. Total lysates were immunoblotted with anti-pSS738/742, anti-PKD (C20), and anti-actin. (B) HEK293T cells were lentivirally infected with PKD1 and PKD2 shRNAs and then transfected with myc-OSBP and HA-PKD.SS/EE or PKD.SS/EE* (nonsilenceable PKD). OSBP was immunoprecipitated with anti-myc and immunoblotted with anti-pMOTIF and anti-myc. Total lysates were immunoblotted with anti-HA. (C) HEK293T cells were lentivirally infected with PKD1, PKD2, or PKD1, and PKD2 shRNAs then were transfected with myc-OSBP. Cells were serum-starved and stimulated with 1 μM PdBu for 30 min. OSBP was immunoprecipitated with anti-myc and immunoblotted with anti-pMOTIF. Total lysates were immunoblotted with anti-myc or anti-pSS738/742. Results are representative of at least three independent experiments.
Functional phospho-proteomics have revealed OSBP phosphorylation in vivo at Ser240 that conforms to a PKD consensus phosphorylation motif (Villen et al., 2007). This motif is evolutionarily conserved in all mammals and to Danio rerio. Examination of the amino acid sequence of OSBP reveals an additional putative PKD motif corresponding to human Ser269 that is conserved to Drosophila melanogaster (Figure 1A). Both sites conform to a minimal PKD consensus motif with a leucine at −5 and an arginine at −3 relative to the phosphoacceptor (Doppler et al., 2005). A rabbit (Oryctolagus cuniculus) OSBP cDNA was used in these studies, whereby the corresponding PKD phosphorylation sites are Ser242 and Ser271 (Figure 1A). To assess the relative contribution of these sites to OSBP phosphorylation by PKD, wild-type, and Ser-to-Ala OSBP alleles were coexpressed with constitutively active PKD (PKD.SS/EE) and immunoblotted with the pMOTIF antibody. Although wild-type OSBP is efficiently phosphorylated by activated PKD, phosphorylation of OSBP.S242A is completely eliminated (Figure 3A). In contrast, OSBP.S271A phosphorylation is unchanged from wild-type OSBP. Similarly, both wild-type and S271A OSBP are directly phosphorylated by purified PKD in an in vitro protein kinase assay, compared with mutant S242A (Figure 3B). These data demonstrate that PKD directly phosphorylates OSBP at Ser242. To corroborate these data and test for Ser242 and Ser271 phosphorylation using a distinct physiological approach, LC-MS/MS was used. Cells infected with shRNA control or PKD1/PKD2 shRNA were cotransfected with tagged OSBP and constitutively active PKD (PKD.SS/EE). OSBP was immunopurified, digested with trypsin and subjected to LC-MS/MS analysis to assess the phosphorylation status at Ser242 and Ser271. The relative quantities of the peptides containing phospho-Ser242 and phospho-Ser271 to nonphospho-peptides were analyzed using ratios of the TIC over the LC elution peaks (Asara et al., 2008; Dibble et al., 2009; Zheng et al., 2009). This analysis revealed that expression of constitutively active PKD induces a 4.5-fold increase in phosphorylation of OSBP at Ser242 (Figure 3, C and D). In the presence of PKD1/PKD2 shRNA, phosphorylation of Ser242 increases by <1.0 fold (Figure 3D). The relative expression levels of OSBP and PKD are shown in Figure 3E. Using this analysis, phosphorylation at Ser271 was undetectable (data not shown). Taken together, these data demonstrate that OSBP is a direct substrate of PKD in vitro and in vivo and that phosphorylation occurs exclusively at Ser242 (Ser240 in humans).
Figure 3.

PKD phosphorylates OSBP at Ser242. (A) HEK293T cells were transfected with HA-PKD.SS/EE and myc-WT.OSBP, OSBP.S242A, or OSBP.S271A. OSBP was immunoprecipitated with anti-myc and immunoblotted with anti-pMOTIF. Total lysates were immunoblotted with anti-myc and anti-HA. (B) HEK293T cells expressing myc-WT.OSBP, OSBP.S242A, or OSBP.S271A were serum-starved, and OSBP was immunoprecipitated with anti-myc. Immune complexes were incubated in kinase buffer containing ATP in the absence or presence of recombinant PKD1. PKD phosphorylation was analyzed by immunoblotting with anti-pMOTIF and control anti-myc. (C) The MS/MS spectrum of the phosphorylated tryptic peptide SLpSELESLK acquired via collision induced dissociation with an Orbitrap XL mass spectrometer. Notice that the mass of the y31+ ion does not contain a phosphate group while a prominent y71+ shows the addition of 79.97 Da (phosphate), suggesting the phosphate group is present on Ser3 in the above peptide sequence. Similarly, the b-ion series is consistent with the phosphate group at Ser3. (D) Quantification of peptide SLpSELESLK using a TIC quantification method. Quantification of peptide from HEK293T cells coexpressing OSBP and constitutively active PKD (SS/EE PKD) or OSBP and SS/EE PKD with PKD1/2 shRNA is relative to cells expressing only OSBP. (E) Expression of OSBP and knockdown of SS/EE PKD were confirmed by immunoblotting with anti-myc and anti-HA. Results are representative of at least three independent experiments.
PKD Phosphorylates OSBP at the Golgi
We next determined if OSBP phosphorylation by PKD occurs at the Golgi, because a pool of PKD localizes at the Golgi where it phosphorylates PI4KIIIβ and stimulates PI(4)P production, and in turn bind OSBP through its PH domain. An OSBP PI(4)P-binding mutant was generated with mutation of Arg109 and Arg110 to Glu, as these are residues in the PH domain that coordinate binding of the phosphate group of PI(4)P. OSBP was depleted from cells using shRNA and either wild-type OSBP or PI(4)P-binding mutant (OSBP.RR109/110EE) that are refractory to silencing were reintroduced. Although wild-type OSBP translocates from a perinuclear compartment to the Golgi upon 25-OH stimulation, OSBP.RR109/110EE has a more cytoplasmic staining pattern with minimal localization to the Golgi, with or without 25-OH treatment, as revealed by costaining with the TGN marker TGN46 (Figure 4A). In a parallel experiment, phosphorylation of the PI(4)P-binding mutant was determined in cells expressing constitutively active PKD (PKD.SS/EE) and either wild-type OSBP or OSBP.RR109/110EE. Immunoblotting with PKD pMOTIF reveals significantly reduced OSBP phosphorylation with the PI(4)P-binding mutant compared with wild-type OSBP (Figure 4B). The residual phosphorylation of OSBP.RR109/110EE can be explained by residual localization of the PI(4)P-binding mutant at the TGN (Figure 4A) under these conditions. Overall, these data indicate that PKD phosphorylation of OSBP occurs at the Golgi.
Figure 4.

PKD phosphorylates OSBP at the Golgi. (A) Hela cells infected with OSBP shRNA were transfected with myc-OSBP or OSBP.RR109/110EE. Cells were serum-starved and treated with 2.5 μg/ml 25-OH for 2 h, fixed, and immunostained with anti-myc or anti-TGN46. (B) HEK293T cells were cotransfected with HA-PKD.SS/EE and myc-WT.OSBP or OSBP.RE109/110EE. OSBP was immunoprecipitated with anti-myc and immunoblotted with anti-pMOTIF. Total lysates were immunoblotted with anti-actin and anti-HA. Results are representative of at least three independent experiments.
Ser242 Phosphorylation Affects OSBP trans-Golgi Network Localization
We next investigated the functional consequences of OSBP phosphorylation by PKD. We first evaluated OSBP homodimerization, as OSBP has been shown to exist as homodimers and Ser242 resides in a leucine zipper (Ridgway et al., 1992). Phospho-mimetic OSBP.S242D and OSBP.S242A were used in a glutaraldehyde cross-linking experiment, revealing that neither OSBP.S242D nor OSBP.S242A affect OSBP homodimerization compared with control (Supplemental Figure S1). This is consistent with deletion of the leucine zipper having a negligible effect on OSBP homodimerization (Ridgway et al., 1992). We next determined the effect of OSBP phosphorylation on sterol binding. In vitro sterol-binding assays revealed no significant effects on the maximal binding capacity of OSBP.S242A or OSBP.S242D for 25-OH (Supplemental Figure S2). This is consistent with the distal location of Ser242 from the sterol-binding domain of OSBP. We also evaluated the consequence of OSBP phosphorylation on PI(4)P binding, because OSBP localizes to the Golgi through binding of PI(4)P through its PH domain. However, in vitro lipid-binding assays revealed that neither OSBP.S242A or OSBP.S242D are impaired in PI(4)P binding compared with wild-type OSBP (Supplemental Figure S3).
We next focused on regulation of OSBP Golgi localization in response to PKD signaling. OSBP translocation to the Golgi was examined using indirect immunofluorescence microscopy in cells depleted of endogenous OSBP and expressing wild-type OSBP or mutant OSBP.S242A or OSBP.S242D that are refractory to silencing. To assess Golgi localization, the Golgi markers GM130, Giantin, and TGN46 were used to mark the cis-, cis-/medial-, and trans-Golgi, respectively. 25-OH treatment stimulates translocation of OSBP and OSBP.S242A to the Golgi, evident by colocalization with GM130, Giantin, and TGN46 (Figure 5, A–C). Similarly, 25-OH stimulates translocation of OSBP.S242D to the cis- and medial-Golgi (Figure 5, A and B). Moreover, under these conditions there is a minor pool of OSBP that does not colocalize with any one Golgi marker, consistent with the notion that OSBP is a medial- and trans-Golgi–associated protein. In contrast, OSBP.S242D does not localize to the TGN upon 25-OH stimulation, with OSBP and TGN46 staining distinct cellular compartments with no detectable colocalization (Figure 5C). These data indicate that phosphorylation of Ser242 specifically inhibits 25-OH-dependent OSBP localization to the TGN, whereas OSBP localization to the cis- and medial Golgi is not altered. This is consistent with PKD activity at the TGN and its established role in regulating the fission of budding vesicles (Liljedahl et al., 2001). To further investigate this functional mechanism of OSBP localization, we evaluated localization in response to cholesterol depletion. OSBP translocates to the Golgi in response to altered cholesterol trafficking and depletion of plasma membrane cholesterol (Ridgway et al., 1992; Storey et al., 1998). Experimentally, OSBP constitutively localizes to the Golgi in response to cholesterol depletion by treatment with cyclodextrin (Ridgway et al., 1992) or treatment with delipidated serum (Ridgway et al., 1992; Storey et al., 1998). Repletion of cellular cholesterol with LDL leads to the dissociation of OSBP from the Golgi (Storey et al., 1998). Cells depleted of endogenous OSBP with shRNA and reexpressing wild-type OSBP, OSBP.S242A, or OSBP.S242D that are refractory to silencing were grown in lipoprotein-deficient serum (LPDS) for 24 h and then stimulated with LDL. As previously reported, OSBP staining revealed constitutive localization to a concentrated perinuclear region that colocalizes with TGN46 and previously confirmed as the Golgi (Mohammadi et al., 2001; Figure 6A, WT.OSBP, −LDL, merge). Treatment of cells with LDL promotes dissociation from the Golgi, evident by diffuse cytoplasmic staining (Figure 6A, WT.OSBP, +LDL, merge). Although OSBP.S242A also localizes to the Golgi in the absence of cholesterol, it fails to dissociate in response to LDL treatment (Figure 6A, S242A, +LDL, merge). Interestingly, cells expressing OSBP.S242D reveal no detectable Golgi localization in either condition (Figure 6A, S242D, −/+LDL). Taken together, these data demonstrate that phosphorylation of Ser242 is required for LDL-mediated release of OSBP from the Golgi. Furthermore, Ser242 phosphorylation inhibits the Golgi localization of OSBP under cholesterol-depleted conditions.
Figure 5.

Ser242 phosphorylation inhibits 25-OH–induced TGN localization. HeLa cells infected with OSBP shRNA were transiently transfected with myc-WT OSBP, S242A, or S242D. Cells were serum-starved and treated with 2.5 μg/ml 25-OH for 2 h. Cells were then fixed and immunostained with anti-myc and (A) anti-GM130, (B) anti-Giantin, or (C) anti-TGN46. Results are representative of at least three independent experiments.
Figure 6.

Ser242 phosphorylation affects cholesterol-regulated Golgi localization. (A) HeLa cells infected with OSBP shRNA were transiently transfected with myc-WT OSBP, S242A, or S242D. Cells were grown in 5% lipoprotein-deficient serum (LPDS) for 24 h and treated with 50 μg human LDL/ml for 18 h. Cells were then fixed and immunostained with anti-myc or anti-TGN46. Results are representative of at least three independent experiments. (B) Quantitation of Golgi fragmentation was determined by counting the number of cells with dispersed TGN46 staining (to total number of cells with fragmented and nonfragmented Golgi). The percentage of cells with Golgi fragmentation was determined for OSBP-depleted cells expressing the indicated mutants −/+ LDL. Statistical evaluations were made between the following: WT and S242A, +LDL; WT and S242D, −LDL; and WT and S242D, +LDL. The data shown are mean values ± SD of four independent experiments, with n = 100 for each condition. *p < 0.05; **p < 0.01; ***p < 0.001.
Constitutive Ser242 Phosphorylation Promotes Golgi Fragmentation
We noticed that under conditions where OSBP displays a diffuse cytoplasmic localization (Figure 6A, WT.OSBP +LDL, S242D −/+LDL), TGN46 staining is dispersed, suggesting that inhibition of OSBP Golgi localization may promote fragmentation of the Golgi. Quantitation of cells with dispersed TGN46 staining confirms that increased Golgi fragmentation in cells expressing OSBP.S242D compared with wild-type is significant before (p < 0.001) and after (p < 0.01) LDL treatment (Figure 6B). Furthermore, the decrease in fragmentation in cells expressing OSBP.S242A after LDL treatment is also significant (p < 0.05), indicating that constitutive localization of OSBP at the Golgi prevents fragmentation. This is consistent with previous reports that have demonstrated complete vesiculation of the Golgi apparatus upon PKD activation (Bossard et al., 2007). These results indicate that nonfunctional OSBP that is impaired in Golgi localization also promotes fragmentation. This finding may explain the significant inhibition of 25-OH–activated SM synthesis upon cellular depletion of OSBP (Perry and Ridgway, 2006). Consistent with this, Golgi fragmentation is also induced upon depletion of the OSBP family member OSBP-related protein 9 (ORP9L; Ngo and Ridgway, 2009).
Ser242 Phosphorylation Affects CERT Golgi Localization
We next investigated whether alterations in OSBP localization affect CERT localization, as 25-OH–induced OSBP Golgi translocation is required for CERT localization and activation (Perry and Ridgway, 2006). 25-OH treatment stimulates translocation of OSBP and OSBP.S242A from a perinuclear/cytoplasmic compartment to the Golgi, whereas OSBP.S242D fails to translocate (Figure 7). In all conditions, a significant pool of CERT colocalizes with OSBP. As OSBP.S242D fails to localize to the TGN with 25-OH treatment (Figure 5), we conclude that CERT also fails to translocate to the TGN in cells expressing OSBP.S242D. Taken together, these data demonstrate that inhibition of 25-OH–induced OSBP Golgi localization by PKD-mediated Ser242 phosphorylation leads to the impairment of CERT translocation to the Golgi.
Figure 7.

Ser242 phosphorylation affects CERT Golgi localization. CHO-K1 cells stably expressing CERT-GFP were transiently transfected with myc-WT OSBP, S242A, or S242D. Cells were treated with 2.5 μg/ml 25-OH for 2 h. Cells were fixed and immunostained with anti-myc. Results are representative of at least three independent experiments.
OSBP Is Required for TGN-to-Cell Surface Protein Transport
Because PKD is required for secretory transport (Bossard et al., 2007), we next determined whether OSBP is also required for delivery of cargo destined for exocytosis. To investigate this, cells depleted of endogenous OSBP with shRNA were transfected with myc-tagged WT.OSBP, S242A, and S242D plasmids refractory to silencing. Cells were then transfected with the tsO45 strain of VSVG-GFP with a thermosensitive mutation causing it to unfold in the ER at the nonpermissive temperature of 40°C. After 24 h, cells were shifted to 32°C in the presence of cycloheximide to allow protein folding and then to 20°C to accumulate tsO45-VSVG in the Golgi. On shifting cells to the permissive temperature of 32°C, tsO45-VSVG is transported to the cell surface. VSVG transported to the cell surface is detected by immunofluorescence with an exofacial antibody (8G5F11) that recognizes the extracellular portion of VSVG. In control cells, VSVG is transported to the cell surface 60 min after temperature shift, whereas there is a severe delay in OSBP-depleted cells that is most evident at 120 min (Figure 8). The delay is rescued with WT.OSBP and OSBP.S242A, but not with OSBP.S242D. Furthermore, the level of intracellular VSVG-GFP localized to the Golgi decreases over time in cells in which the secretory pathway is intact, but remains in the Golgi when the secretory pathway is disrupted (Supplemental Figure S4). The defect in VSVG transport is likely a result of Golgi fragmentation that occurs upon OSBP depletion (Supplemental Figure S5). These data demonstrate that OSBP is required for proper Golgi structure and trafficking of cargo from the Golgi to the plasma membrane and that constitutive phosphorylation of OSBP impairs this process.
Figure 8.

Depletion of OSBP impairs VSVG transport. HeLa cells infected with OSBP shRNA were transiently transfected with myc-WT.OSBP, S242A, or S242D. After 24 h, cells were transfected with tsO45-VSVG-GFP. Cells were fixed and immunostained with anti-VSVG (8G5F11) to detect surface VSVG after the indicated times (min). Results are representative of at least three independent experiments.
DISCUSSION
In this study, we identify OSBP as a novel substrate of PKD at the Golgi and provide the first demonstration of phosphorylation-mediated regulation of OSBP function. The mechanism by which PKD regulates Golgi structure and function is defined by its substrates. PKD phosphorylation of PI4KIIIβ stimulates its kinase activity leading to increased PI(4)P production (Hausser et al., 2005, 2006). In addition to maintaining local PI(4)P levels at the TGN, PKD regulates Golgi lipid homeostasis through phosphorylation of another Golgi-localized substrate, CERT. PKD-mediated phosphorylation decreases the affinity of the CERT PH domain for PI(4)P, thereby inhibiting transport of ceramide, a SM precursor, from the ER to the Golgi (Fugmann et al., 2007). The inhibition of OSBP Golgi localization by phosphorylation impairs the 25-OH–induced Golgi localization of CERT (Figure 7). This is consistent with previous studies demonstrating the requirement of 25-OH–induced OSBP Golgi localization for CERT activity and SM synthesis (Perry and Ridgway, 2006). This suggests that PKD-mediated inhibition of OSBP Golgi association may affect CERT activity and SM synthesis, thus acting as a distinct mechanism of PKD signaling that influences SM metabolism. We propose that by phosphorylating OSBP and inhibiting its association with the Golgi, PKD regulates the ability of CERT to localize to the Golgi. In turn, PKD-mediated phosphorylation of CERT causes its release from the Golgi. The net effect is rapid termination of SM synthesis. SM and cholesterol are coregulated metabolically and associate physically in membrane microdomains involved in cargo sorting and signaling. An excess of these lipids would perturb the rigidity of and signaling from endomembranes. In line with this notion, disruption of lipid rafts by cholesterol depletion with methyl-β-cyclodextrin activates PKD (Cabrera-Poch et al., 2004). Termination of SM synthesis through substrate phosphorylation of OSBP and CERT may explain coordinate down-regulation of SM synthesis that is observed upon cholesterol depletion.
PKD phosphorylates OSBP on Ser242 (Figure 3) located in the putative leucine zipper region (Figure 1A). This region fits the criteria for α-helical sequences that mediate dimerization of certain DNA-binding proteins (Landschulz et al., 1988), with the exception that it lacks an adjacent basic domain that binds DNA. Although not required, the leucine zipper does contribute to OSBP homodimerization. Deletion of this domain results in dimerization; however, it is significantly reduced compared with wild-type OSBP, suggesting that the leucine zipper may function to stabilize homodimerization (Ridgway et al., 1992), although it is dispensable for initial dimer formation. Consistent with this, Ser242 mutations in the leucine zipper do not affect OSBP homodimerization (Supplemental Figure 1). This does not preclude the possibility that the leucine zipper facilitates heterodimerization with other proteins. Studies have shown that phosphorylation in the leucine zipper stabilizes multimeric complexes (Loriaux et al., 1993; Szilak et al., 1997b) and also disrupts α-helices (Szilak et al., 1997a). The importance of phosphorylation in regulating leucine zipper-mediated protein–protein interactions suggests that PKD phosphorylation of OSBP may regulate OSBP interaction with other proteins.
PKD phosphorylation of OSBP regulates TGN localization in response to 25-OH treatment or cholesterol depletion (Figures 5C and 6A). As OSBP phosphorylation occurs at the Golgi (Figure 4), this localization defect is likely due to the inability of OSBP to associate with the TGN upon phosphorylation. As the affinity for PI(4)P is unaffected in vitro (Supplemental Figure 3), it is likely that other factors at the TGN whose interactions with OSBP, possibly through the leucine zipper domain, are regulated by PKD phosphorylation. Phosphorylation may promote an interaction with a ternary binding protein, which serves to mask the PH domain of OSBP, thereby inhibiting PI(4)P binding. Interaction with a ternary protein may also inhibit ARF1 interaction. In addition to PI(4)P, studies have shown that the ARF1 GTPase is critical for OSBP Golgi localization (Levine and Munro, 2002). At the Golgi, the PH domains of OSBP (OSBP-PH) and FAPP (FAPP-PH) bind activated ARF1 (GTP-ARF1). This facilitates the stabilization of ARF1 at Golgi membranes by interfering with the interaction with ARF-GAP, effectively inhibiting recruitment to the Golgi and reducing GAP activity (Godi et al., 2004). Brefeldin A disrupts the Golgi by interfering with the ARF1 and ARF-GEF interaction, resulting in ARF1 inactivation (Cherfils and Melancon, 2005). Consistent with this, we demonstrate that a phospho-mimetic OSBP mutant (OSBP.S242D) is impaired in Golgi localization, and this is concomitant with increased Golgi fragmentation (Figure 6). We also show that OSBP depletion induces Golgi fragmentation (Supplemental Figure S5). It is worth noting that depletion of ORP9L also leads to Golgi fragmentation (Ngo and Ridgway, 2009). In further support of this model, OSBP that is constitutively localized to the Golgi protects from Golgi fragmentation (Figure 6). The increased Golgi fragmentation observed with OSBP.S242D expression is consistent with a model in which the OSBP that is phosphorylated by PKD at S242 is nonfunctional. This is also reminiscent of the functional role of PKD activation that leads to Golgi vesiculation (Bossard et al., 2007). We also show that OSBP is required for trafficking of cargo from the TGN to the plasma membrane and that constitutive phosphorylation of Ser242 impairs the secretory pathway. As complete vesiculation of Golgi membranes inhibits TGN to plasma membrane transport (Takizawa et al., 1993), our results are consistent with a model in which hyperactivation of PKD leads to constitutive phosphorylation of OSBP and Golgi fragmentation, which in turn impairs transport of cargo.
OSBP also serves as a cholesterol-sensitive scaffolding protein for two extracellular signal–regulated kinase (ERK) phosphatases, PP2A and HePTP, to regulate ERK1/2 activity (Wang et al., 2005). Depletion of cellular cholesterol with methyl-β-cyclodextrin triggers disassembly of the oligomer and leads to an increase in the level of pERK1/2. It has been reported that 25-OH treatment leads to increased ERK1/2 activation (Ares et al., 2000). This is most likely due to oligomer disassembly and inhibition of pERK1/2 dephosphorylation, as exposure to 25-OH results in disassembly of the OSBP/PP2A/HePTP oligomer (Wang et al., 2005). As both cholesterol depletion and 25-OH induce OSBP Golgi localization, we speculate that PKD-mediated phosphorylation and inhibition of OSBP Golgi localization may also attenuate ERK1/2 activation.
PKD is a critical regulator of Golgi structure and function through substrate phosphorylation of PI4KIIIβ, CERT, and OSBP. Our results are consistent with a model in which PKD phosphorylation of PI4KIIIβ leads to increased PI(4)P production at the TGN, allowing CERT and OSBP to localize to the Golgi through their PH domains (Figure 9). Phosphorylation of CERT attenuates SM and DAG production (Kumagai et al., 2007). Similarly, phosphorylation of OSBP causes its displacement from the Golgi. As OSBP Golgi localization regulates CERT activity, we propose that PKD phosphorylation of OSBP regulates localized production of SM and DAG by sterol sensing or a direct transfer activity that controls sterol levels in the Golgi (Ngo and Ridgway, 2009). Thus, PKD phosphorylation of CERT and OSBP may serve to tightly integrate SM and cholesterol levels at the Golgi, which in turn has a profound effect on lipid raft assembly and maintenance of lipid homeostasis in the cell.
Figure 9.

Model of PKD regulation of Golgi lipid homeostasis. (1) PKD phosphorylation of PI4KIIIβ leads to 14-3-3 binding and stabilization of its lipid kinase activity, resulting in increased PI(4)P production at the TGN. (2) CERT and OSBP localize to the TGN via binding of their PH domains to PI(4)P. (3) PKD phosphorylation of CERT decreases its affinity for PI(4)P, releasing it from the TGN and attenuating SM and DAG production. (4) PKD phosphorylation of OSBP inhibits localization to the TGN. This impairs CERT Golgi localization and activation, and therefore SM and DAG production.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Richard G. W. Anderson for providing the OSBP cDNA and also thank Drs. Lewis C. Cantley, Andrius Kazlauskas, and John Blenis for guidance, and all members of the Toker laboratory for discussions and advice. This work was supported in part by National Institutes of Health (NIH) Grant CA075134 (A.T.); Canadian Institutes of Health Research Grants MOP-15284 (N.R.), 5P01CA120964-03, and 3P30CA006516-45S6 (J.A.); the Burroughs Wellcome Fund and NIH Director's New Innovator Award (S.F.); and the American Cancer Society Postdoctoral Scholar Award (M.N.).
Abbreviations used:
- CERT
ceramide transfer protein
- DAG
diacylglycerol
- LDL
low-density lipoprotein
- PI(4)P
phosphatidylinositol 4-phosphate
- 25-OH
25-hydroxycholesterol
- OSBP
oxysterol-binding protein
- PdBu
phorbol 12,13-dibutyrate
- PH
pleckstrin homology
- PI4KIIIβ
phosphatidylinositol 4-kinase IIIβ
- PKD
protein kinase D
- SM
sphingomyelin
- TGN
trans-Golgi network.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-02-0090) on May 5, 2010.
REFERENCES
- Ares M. P., Porn-Ares M. I., Moses S., Thyberg J., Juntti-Berggren L., Berggren P., Hultgardh-Nilsson A., Kallin B., Nilsson J. 7beta-hydroxycholesterol induces Ca(2+) oscillations, MAP kinase activation and apoptosis in human aortic smooth muscle cells. Atherosclerosis. 2000;153:23–35. doi: 10.1016/s0021-9150(00)00380-4. [DOI] [PubMed] [Google Scholar]
- Asara J. M., Christofk H. R., Freimark L. M., Cantley L. C. A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics. 2008;8:994–999. doi: 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
- Baron C. L., Malhotra V. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science. 2002;295:325–328. doi: 10.1126/science.1066759. [DOI] [PubMed] [Google Scholar]
- Bossard C., Bresson D., Polishchuk R. S., Malhotra V. Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J. Cell Biol. 2007;179:1123–1131. doi: 10.1083/jcb.200703166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussif O., Lezoualc'h F., Zanta M. A., Mergny M. D., Scherman D., Demeneix B., Behr J. P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Poch N., Sanchez-Ruiloba L., Rodriguez-Martinez M., Iglesias T. Lipid raft disruption triggers protein kinase C and Src-dependent protein kinase D activation and Kidins220 phosphorylation in neuronal cells. J. Biol. Chem. 2004;279:28592–28602. doi: 10.1074/jbc.M312242200. [DOI] [PubMed] [Google Scholar]
- Cherfils J., Melancon P. On the action of Brefeldin A on Sec7-stimulated membrane-recruitment and GDP/GTP exchange of Arf proteins. Biochem. Soc. Trans. 2005;33:635–638. doi: 10.1042/BST0330635. [DOI] [PubMed] [Google Scholar]
- Diaz Anel A. M., Malhotra V. PKCeta is required for beta1gamma2/beta3gamma2- and PKD-mediated transport to the cell surface and the organization of the Golgi apparatus. J. Cell Biol. 2005;169:83–91. doi: 10.1083/jcb.200412089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble C. C., Asara J. M., Manning B. D. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler H., Storz P., Li J., Comb M. J., Toker A. A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J. Biol. Chem. 2005;280:15013–15019. doi: 10.1074/jbc.C400575200. [DOI] [PubMed] [Google Scholar]
- Emr S., et al. Journeys through the Golgi—taking stock in a new era. J. Cell Biol. 2009;187:449–453. doi: 10.1083/jcb.200909011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugmann T., Hausser A., Schoffler P., Schmid S., Pfizenmaier K., Olayioye M. A. Regulation of secretory transport by protein kinase D-mediated phosphorylation of the ceramide transfer protein. J. Cell Biol. 2007;178:15–22. doi: 10.1083/jcb.200612017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godi A., Di Campli A., Konstantakopoulos A., Di Tullio G., Alessi D. R., Kular G. S., Daniele T., Marra P., Lucocq J. M., De Matteis M. A. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat. Cell Biol. 2004;6:393–404. doi: 10.1038/ncb1119. [DOI] [PubMed] [Google Scholar]
- Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., Nishijima M. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- Hausser A., Link G., Hoene M., Russo C., Selchow O., Pfizenmaier K. Phospho-specific binding of 14–3-3 proteins to phosphatidylinositol 4-kinase III beta protects from dephosphorylation and stabilizes lipid kinase activity. J. Cell Sci. 2006;119:3613–3621. doi: 10.1242/jcs.03104. [DOI] [PubMed] [Google Scholar]
- Hausser A., Storz P., Martens S., Link G., Toker A., Pfizenmaier K. Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIbeta at the Golgi complex. Nat. Cell Biol. 2005;7:880–886. doi: 10.1038/ncb1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg K., Phair R. D., Lippincott-Schwartz J. Kinetic analysis of intracellular trafficking in single living cells with vesicular stomatitis virus protein G-green fluorescent protein hybrids. Methods Enzymol. 2000;327:69–89. doi: 10.1016/s0076-6879(00)27268-6. [DOI] [PubMed] [Google Scholar]
- Kumagai K., Kawano M., Shinkai-Ouchi F., Nishijima M., Hanada K. Interorganelle trafficking of ceramide is regulated by phosphorylation-dependent cooperativity between the PH and START domains of CERT. J. Biol. Chem. 2007;282:17758–17766. doi: 10.1074/jbc.M702291200. [DOI] [PubMed] [Google Scholar]
- Landschulz W. H., Johnson P. F., McKnight S. L. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- Lehto M., Olkkonen V. M. The OSBP-related proteins: a novel protein family involved in vesicle transport, cellular lipid metabolism, and cell signalling. Biochim. Biophys. Acta. 2003;1631:1–11. doi: 10.1016/s1388-1981(02)00364-5. [DOI] [PubMed] [Google Scholar]
- Levine T. P., Munro S. The pleckstrin homology domain of oxysterol-binding protein recognises a determinant specific to Golgi membranes. Curr. Biol. 1998;8:729–739. doi: 10.1016/s0960-9822(98)70296-9. [DOI] [PubMed] [Google Scholar]
- Levine T. P., Munro S. Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and -independent components. Curr. Biol. 2002;12:695–704. doi: 10.1016/s0960-9822(02)00779-0. [DOI] [PubMed] [Google Scholar]
- Liljedahl M., Maeda Y., Colanzi A., Ayala I., Van Lint J., Malhotra V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell. 2001;104:409–420. doi: 10.1016/s0092-8674(01)00228-8. [DOI] [PubMed] [Google Scholar]
- Litvak V., Dahan N., Ramachandran S., Sabanay H., Lev S. Maintenance of the diacylglycerol level in the Golgi apparatus by the Nir2 protein is critical for Golgi secretory function. Nat. Cell Biol. 2005;7:225–234. doi: 10.1038/ncb1221. [DOI] [PubMed] [Google Scholar]
- Loewen C. J., Levine T. P. A highly conserved binding site in vesicle-associated membrane protein-associated protein (VAP) for the FFAT motif of lipid-binding proteins. J. Biol. Chem. 2005;280:14097–14104. doi: 10.1074/jbc.M500147200. [DOI] [PubMed] [Google Scholar]
- Loewen C. J., Roy A., Levine T. P. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 2003;22:2025–2035. doi: 10.1093/emboj/cdg201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loriaux M. M., Rehfuss R. P., Brennan R. G., Goodman R. H. Engineered leucine zippers show that hemiphosphorylated CREB complexes are transcriptionally active. Proc. Natl. Acad. Sci. USA. 1993;90:9046–9050. doi: 10.1073/pnas.90.19.9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y., Beznoussenko G. V., Van Lint J., Mironov A. A., Malhotra V. Recruitment of protein kinase D to the trans-Golgi network via the first cysteine-rich domain. EMBO J. 2001;20:5982–5990. doi: 10.1093/emboj/20.21.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J., et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Mohammadi A., Perry R. J., Storey M. K., Cook H. W., Byers D. M., Ridgway N. D. Golgi localization and phosphorylation of oxysterol binding protein in Niemann-Pick C and U18666A-treated cells. J. Lipid. Res. 2001;42:1062–1071. [PubMed] [Google Scholar]
- Ngo M., Ridgway N. D. Oxysterol binding protein-related Protein 9 (ORP9) is a cholesterol transfer protein that regulates Golgi structure and function. Mol. Biol. Cell. 2009;20:1388–1399. doi: 10.1091/mbc.E08-09-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R. J., Ridgway N. D. Molecular mechanisms and regulation of ceramide transport. Biochim. Biophys. Acta. 2005;1734:220–234. doi: 10.1016/j.bbalip.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Perry R. J., Ridgway N. D. Oxysterol-binding protein and vesicle-associated membrane protein-associated protein are required for sterol-dependent activation of the ceramide transport protein. Mol. Biol. Cell. 2006;17:2604–2616. doi: 10.1091/mbc.E06-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway N. D., Dawson P. A., Ho Y. K., Brown M. S., Goldstein J. L. Translocation of oxysterol binding protein to Golgi apparatus triggered by ligand binding. J. Cell Biol. 1992;116:307–319. doi: 10.1083/jcb.116.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rykx A., De Kimpe L., Mikhalap S., Vantus T., Seufferlein T., Vandenheede J. R., Van Lint J. Protein kinase D: a family affair. FEBS Lett. 2003;546:81–86. doi: 10.1016/s0014-5793(03)00487-3. [DOI] [PubMed] [Google Scholar]
- Storey M. K., Byers D. M., Cook H. W., Ridgway N. D. Cholesterol regulates oxysterol binding protein (OSBP) phosphorylation and Golgi localization in Chinese hamster ovary cells: correlation with stimulation of sphingomyelin synthesis by 25-hydroxycholesterol. Biochem. J. 1998;336(Pt 1):247–256. doi: 10.1042/bj3360247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P., Doppler H., Johannes F. J., Toker A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J. Biol. Chem. 2003;278:17969–17976. doi: 10.1074/jbc.M213224200. [DOI] [PubMed] [Google Scholar]
- Storz P., Toker A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J. 2003;22:109–120. doi: 10.1093/emboj/cdg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilak L., Moitra J., Krylov D., Vinson C. Phosphorylation destabilizes alpha-helices. Nat. Struct. Biol. 1997a;4:112–114. doi: 10.1038/nsb0297-112. [DOI] [PubMed] [Google Scholar]
- Szilak L., Moitra J., Vinson C. Design of a leucine zipper coiled coil stabilized 1.4 kcal mol-1 by phosphorylation of a serine in the e position. Protein Sci. 1997b;6:1273–1283. doi: 10.1002/pro.5560060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa P. A., Yucel J. K., Veit B., Faulkner D. J., Deerinck T., Soto G., Ellisman M., Malhotra V. Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell. 1993;73:1079–1090. doi: 10.1016/0092-8674(93)90638-7. [DOI] [PubMed] [Google Scholar]
- Toth B., Balla A., Ma H., Knight Z. A., Shokat K. M., Balla T. Phosphatidylinositol 4-kinase IIIbeta regulates the transport of ceramide between the endoplasmic reticulum and Golgi. J. Biol. Chem. 2006;281:36369–36377. doi: 10.1074/jbc.M604935200. [DOI] [PubMed] [Google Scholar]
- Van Lint J., Rykx A., Maeda Y., Vantus T., Sturany S., Malhotra V., Vandenheede J. R., Seufferlein T. Protein Kinase D: an intracellular traffic regulator on the move. Trends Cell Biol. 2002;12:193–200. doi: 10.1016/s0962-8924(02)02262-6. [DOI] [PubMed] [Google Scholar]
- Vihtelic T. S., Goebl M., Milligan S., O'Tousa J. E., Hyde D. R. Localization of Drosophila retinal degeneration B, a membrane-associated phosphatidylinositol transfer protein. J. Cell Biol. 1993;122:1013–1022. doi: 10.1083/jcb.122.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villen J., Beausoleil S. A., Gerber S. A., Gygi S. P. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. USA. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. Y., Weng J., Anderson R. G. OSBP is a cholesterol-regulated scaffolding protein in control of ERK 1/2 activation. Science. 2005;307:1472–1476. doi: 10.1126/science.1107710. [DOI] [PubMed] [Google Scholar]
- Warren G., Malhotra V. The organisation of the Golgi apparatus. Curr. Opin. Cell Biol. 1998;10:493–498. doi: 10.1016/s0955-0674(98)80064-1. [DOI] [PubMed] [Google Scholar]
- Weixel K. M., Blumental-Perry A., Watkins S. C., Aridor M., Weisz O. A. Distinct Golgi populations of phosphatidylinositol 4-phosphate regulated by phosphatidylinositol 4-kinases. J. Biol. Chem. 2005;280:10501–10508. doi: 10.1074/jbc.M414304200. [DOI] [PubMed] [Google Scholar]
- Wyles J. P., McMaster C. R., Ridgway N. D. Vesicle-associated membrane protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify export from the endoplasmic reticulum. J. Biol. Chem. 2002;277:29908–29918. doi: 10.1074/jbc.M201191200. [DOI] [PubMed] [Google Scholar]
- Yeaman C., Ayala M. I., Wright J. R., Bard F., Bossard C., Ang A., Maeda Y., Seufferlein T., Mellman I., Nelson W. J., Malhotra V. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat. Cell Biol. 2004;6:106–112. doi: 10.1038/ncb1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B., Jeong J. H., Asara J. M., Yuan Y. Y., Granter S. R., Chin L., Cantley L. C. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell. 2009;33:237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.