

Abstract
Glycogen synthase kinase-3 (GSK3) is a critical enzyme in neuronal physiology, however any specific role in presynaptic function is not yet known. We show that GSK3 phosphorylates a key residue on the large GTPase dynamin I (Ser-774) both in vitro and in primary rat neuronal cultures. This is dependent on prior phosphorylation of Ser-778 by cyclin-dependent kinase 5 (cdk5). We found a specific requirement for GSK3 in activity-dependent bulk endocytosis (ADBE), but not clathrin-mediated endocytosis (CME) using both acute inhibition with pharmacological antagonists and silencing of expression using shRNA. Moreover we showed that the specific phosphorylation of Ser-774 on dynamin I by GSK3 is both necessary and sufficient for ADBE. This is the first demonstration of a presynaptic role for GSK3 and reveals that a protein kinase signalling cascade prepares synaptic vesicles (SVs) for retrieval during elevated neuronal activity.
GSK3 is a ubiquitously expressed multifunctional enzyme that plays an essential role in many processes fundamental to cell biology. In addition its dysfunction is implicated in diseases such as cancer and diabetes1. GSK3 has two isoforms, GSK3α and GSK3β that are encoded by two separate genes and display high basal activity in cells2. GSK3β is highly enriched in brain and defects in its function have been implicated in neuronal conditions such as Alzheimer’s disease, schizophrenia and bipolar disorder3,4. GSK3β is also implicated in normal CNS function such as neural tube development5 and the induction of long-term depression6, however it has no known presynaptic role (even though the enzyme is enriched in this region4,6,7).
Neurotransmitter release is dependent on the efficient retrieval of synaptic vesicles (SVs) from the nerve terminal plasma membrane. At least two parallel mechanisms exist to retrieve SVs after exocytosis. Clathrin-mediated endocytosis (CME) generates single SVs and is the dominant SV retrieval mode during mild stimulation8,9. During stronger stimulation additional retrieval capacity is provided by activity-dependent bulk endocytosis (ADBE). ADBE is a rapidly triggered, high capacity endocytic mode that is dominant during elevated neuronal activity10. ADBE invaginates large areas of plasma membrane to form endosomes from which SVs can bud and rejoin the recycling SV pool11-13.
Both CME and ADBE require the activity of the large GTPase dynamin I13,14. However ADBE is also uniquely regulated by a cycle of dynamin I dephosphorylation and rephosphorylation. At a specific activity threshold ADBE is triggered by a calcineurinmediated dephosphorylation of dynamin I on two key sites on its C-terminal proline-rich domain (PRD); Ser-774 and Ser-77813. After stimulation ceases, the rephosphorylation of these residues is dependent on cdk5 activity15, an event that is equally essential for ADBE12. To date, cdk5 is the only protein kinase directly implicated in SV retrieval, despite the fact that the phosphorylation cycles of many endocytic proteins are stimulus-dependent16.
GSK3 is unusual compared to other protein kinases, since mostly it can only phosphorylate its substrates after they are phosphorylated at a nearby site by another protein kinase. This phenomenon is called “priming” and occurs at Ser or Thr residues that are located 4 or 5 amino acids C-terminal to the GSK3 target phosphorylation site1. Cdk5 is one of a small group of “priming protein kinases” that prime GSK3 substrates for phosphorylation17. The major dynamin I phosphorylation sites reside in an ideal consensus sequence motif predictive of GSK3 phosphorylation (RSPTSSPTP773-781). Therefore we postulated that cdk5 may be the priming kinase for Ser-778, allowing GSK3 to phosphorylate Ser-774. If such a priming mechanism took place this would implicate GSK3-dependent dynamin I phosphorylation as a critical event in ADBE, since both cdk5 activity and dynamin I phosphorylation are essential for the process12,13.
We report that cdk5 primes dynamin I for phosphorylation by GSK3 both in vitro and in vivo. Thus GSK3 is a new dynamin I kinase. We also found that GSK3-dependent protein rephosphorylation is required for ADBE, but not CME, in central nerve terminals. Finally we have shown that rephosphorylation of Ser-774 on dynamin I by GSK3 is necessary and sufficient for the triggering and maintenance of ADBE. This is the first demonstration of a role for GSK3 in presynaptic function, and reveals a unique partnership between cdk5 and GSK3 in controlling the majority of neuronal SV retrieval during elevated neuronal activity.
Results
Cdk5 primes dynamin I for phosphorylation by GSK3
The C-terminal PRD of dynamin I contains a predicted consensus motif for GSK3-dependent phosphorylation of Ser-774. This prediction requires that another priming kinase phosphorylates Ser-778. Since we previously established that cdk5 phosphorylates both of these sites in vitro15, it is possible that cdk5 is the priming kinase (Fig 1a).
Figure 1. Dynamin I is an in vitro substrate for GSK3β only after priming by cdk5.

a) Recombinant GST-DynI-PRD was incubated with or without cdk5 and unlabeled ATP for 5 minutes (priming step). Cdk5 was washed out and GSK3β and [γ]32P-ATP were added for 15 minutes in the radiolabelled phosphorylation step. During GSK3β phosphorylation either roscovitine (10 μM) or lithium (20 mM) were added as indicated. 32P-DynI-PRD was visualized by autoradiography following SDS-PAGE. All panels in the lower right image were taken from the same autoradiograph. Residual cdk5 activity was abolished by roscovitine, but not the GSK3 inhibitor lithium. Roscovitine was added during the phosphorylation reaction in all subsequent experiments (panels b-c). A model of cdk5 priming followed by GSK3 phosphorylation is shown (lower left). b) GSK3β phosphorylates cdk5-primed dynamin I at Ser-774. Experiments (in duplicate) were performed as in panel a. The autoradiograph (first panel) shows that GSK3β phosphorylates cdk5-primed dynamin I (lanes 7-8). Total DynI-PRD levels are shown by a Coomassie Brilliant Blue-stained gel (CBB, 2nd panel). Samples were probed by immunoblot with antibodies to either phospho-Ser 774 or phospho-Ser 778 (3rd-4th panel). Panels representative of either 12 (autorad) or 3 (blots) independent experiments. c) Validation that GSK3β phosphorylates dynamin I at Ser-774 only. GST-DynI-PRD (S774A) or (S778A) were treated as in b) and visualized by autoradiography. All increased phosphorylation is abolished by mutagenesis of either the priming site (Ser-778) or the phosphorylation site (Ser-774). Full-length blots/gels are presented in Supplementary Figure 5.
To test this, we performed a series of two-step in vitro phosphorylation experiments (Fig 1a). As the priming step, we first incubated recombinant dynamin I PRD (DynI-PRD) with cdk5 in the presence of unlabelled ATP for a relatively short time of 5 min. For the second phosphorylation step, we removed cdk5 by washing and the DynI-PRD was incubated with or without GSK3β in the presence of radiolabelled [γ]32P-ATP for a further 15 minutes. To ensure that any residual cdk5 activity remaining after washout was eliminated, we included the selective cdk5 antagonist roscovitine for the second [γ]32P-ATP labelling step in all experiments (Fig 1a, bottom right). The GSK3 antagonist lithium had no effect on residual cdk5 activity (Fig 1a). DynI-PRD was a very poor substrate for GSK3β without cdk5 in the priming step, but became an excellent substrate for GSK3β after cdk5 priming (Fig 1b). Lithium abolished this phosphorylation, confirming it was due to GSK3β activity rather than cdk5 (Fig 1b). Thus dynamin I is an in vitro GSK3β substrate only after cdk5 priming.
Dynamin I contains two predicted consensus sites for GSK3 phosphorylation, but only the sequence containing Ser-774 and Ser-778 is phosphorylated in vivo15,18. To determine whether Ser-778 is the cdk5 priming site and Ser-774 is the GSK3 phosphorylation site, we performed immunoblot analysis using phosphosite-specific antibodies15 on DynI-PRD phosphorylated using an identical protocol to described before (Fig 1a). In these experiments, cdk5 specifically phosphorylated Ser-778 during the priming reaction, and GSK3β selectively phosphorylated Ser-774 (Fig 1b, lower two panels).
These results confirm that cdk5 phosphorylates Ser-778 and GSK3 phosphorylates Ser-774 in vitro, but do not rule out the presence of additional phosphorylation sites for GSK3 on dynamin I. We therefore mutated either Ser-774 or Ser-778 to alanine to prevent phosphorylation at either site. Mutation of the GSK3 target site (S774A) abolished GSK3β-dependent phosphorylation of the DynI-PRD (Fig 1c). Importantly, mutation of the cdk5 priming site (S778A) also abolished GSK3β-dependent phosphorylation, even though the GSK3 site (S774) was unaltered. This event is specific to dynamin I, since we found no significant GSK3β-dependent phosphorylation of the ubiquitously expressed dynamin II PRD (which lacks the equivalent S774 site) with or without cdk5 priming (Supplementary Fig 1). Overall, these four independent in vitro approaches reveal that cdk5 primes dynamin I at Ser-778 for subsequent phosphorylation by GSK3 at Ser-774.
We next determined whether GSK3 also phosphorylates dynamin I on Ser-774 in intact neurons. The phosphorylation of both Ser-774 and Ser-778 occurs after prior stimulus-dependent dephosphorylation and is termed, “rephosphorylation”. This event can be visualised by stimulating primary neuronal cultures to dephosphorylate dynamin I, and then monitoring the selective rephosphorylation of either Ser-774 or Ser-778 using site-specific phospho-antibodies12,15 (Fig 2a, 2b). Inhibition of cdk5 activity by the antagonist roscovitine (50 μM) inhibited the rephosphorylation of both Ser-774 and Ser-778, in agreement with previous studies15 (Fig 2). This result would occur whether cdk5 was acting either exclusively at both sites or solely as a priming kinase for GSK3. When GSK3 activity was inhibited using either of the selective antagonists CT99021 (2 μM) or AR-AO14418 (5 μM)19,20 only rephosphorylation of Ser-774 was abolished, whereas Ser-778 was rephosphorylated to the same extent as controls (Fig 2). Thus cdk5 cannot be directly responsible for the rephosphorylation of Ser-774 in vivo, since this site is not rephosphorylated in the absence of GSK3 activity. These experiments confirm that GSK3 is the native protein kinase for Ser-774 on the DynI-PRD, and that this event is dependent on the prior priming phosphorylation of Ser-778 by cdk5. This is the first example of a kinase signalling cascade associated with endocytic proteins.
Figure 2. Dynamin I is selectively phosphorylated on serine 774 by GSK3 in neurons.

a) Cells were incubated in the presence or absence of the following protein kinase antagonists for 15 minutes - roscovitine (50 μM), CT99021 (2 μM) or AR-AO14418 (5 μM). Cells were stimulated with 50 mM KCl (10 sec) and then allowed to repolarise for 7 minutes in the continuous presence of antagonists. Samples were prepared from cultures before stimulation (Basal), directly after KCl stimulation (KCl) or after 7 minutes repolarization (Repol). Lysates were separated by SDS-PAGE and probed for either phospho-Ser774 (P774, b) or phospho-Ser778 (P778, c) on dynamin I by immunoblots. The amount of synaptophysin (SYP) in the lysate was examined as a loading control. Full-length blots/gels are presented in Supplementary Figure 6. Quantitative analysis is shown in the graphs in d-e. These graphs display the extent of phosphorylation of either Ser774 (d) or Ser778 (e), normalised to the amount of SYP, as a percentage of Basal (P774 Ctrl n = 5, Roscovitine n = 3, CT99021 n = 4, AR-AO14418 n = 3; P778 Ctrl n = 7, Roscovitine n = 3, CT99021 n = 3, AR-AO14418 n = 3; one-way ANOVA ** = p < 0.01, *** = p < 0.001 compared to control).
Activity-dependent requirement for GSK3 in SV retrieval
Dynamin I is only dephosphorylated during intense action potential stimulation in central nerve terminals13, and therefore is only rephosphorylated after this event. In agreement, both cdk5 activity and site specific dynamin I rephosphorylation are only required for SV retrieval during high intensity stimulation12,13. Therefore our next aim was to determine whether there was a similar activity-dependent requirement for GSK3-dependent rephosphorylation in SV retrieval. We achieved this by monitoring uptake of the fluorescent dye FM1-43 in cultured neurons using an S2/S1 protocol. This protocol allows the effect of GSK3 inhibition to be directly compared with the control response from the same nerve terminals12,13. Addition of CT99021 during the S2 loading period had no effect on FM1-43 uptake evoked by intense neuronal activity (800 action potentials, 80 Hz, Fig 3a-c). This was expected, since dynamin I is highly phosphorylated at rest and its rephosphorylation should not be affected by GSK3 inhibition either before or during stimulation (Fig 2). We also observed no effect of CT99021 on FM1-43 unloading, ruling out any effect for GSK3 in SV exocytosis (Fig 3a-c). To specifically assess the role of protein rephosphorylation by GSK3, we modified the protocol to include CT99021 throughout the S1 loading period in addition to S2 loading (Fig 3d). This protocol permits the dephosphorylation of dynamin I, but will arrest any GSK3-dependent rephosphorylation (Fig 2). When we performed these experiments, CT99021 strongly inhibited FM1-43 uptake evoked by high intensity stimulation (800 action potentials, 80 Hz, Fig 3e, 3f). This indicates a key role for protein rephosphorylation by GSK3 in SV retrieval during elevated neuronal activity.
Figure 3. GSK3-dependent protein rephosphorylation is required for SV retrieval during high intensity stimulation.

a) Cells were loaded and unloaded with FM1-43 using the protocol displayed. In both S1 and S2, dyes were loaded with 800 action potentials (80 Hz) and then cells were immediately washed. Unloading of the dye was stimulated by 2 sequential stimuli of 400 action potentials (40 Hz). Cells were preincubated with 2 μM CT99021 for either 10 min before and during S2 loading (S2 load) or for 10 min before and during S2 unloading (S2 unload) as indicated. Control (Ctrl) was in the absence of drug at both S1 and S2. b) Cumulative histogram of the effect of CT99021 on FM1-43 loading and unloading in individual nerve terminals (δS2/δS1). c) Mean δS2/δS1 response ± SEM (n = 3 for control and CT99021 S2 load, n = 4 for CT99021 S2 unload, one-way ANOVA). d) Cells were loaded and unloaded with FM1-43 as in (a) with the exception that cells were preincubated with 2 μM CT99021 for all steps prior to and including S2 loading. e) Cumulative histograms display the effect of CT99021 on FM1-43 loading in individual nerve terminals (δS2/δS1). f) Bar graphs display the effect of CT99021 on the average δS2/δS1 value for FM1-43 loading ± SEM. Black symbols represent control and grey symbols represent CT99021 (2 μM). n = 3, Student’s t test ** = p < 0.01.
We next determined whether SV retrieval during mild stimulation conditions also required protein rephosphorylation by GSK3. To test this, we evoked FM1-43 loading with a mild stimulus of 200 action potentials (10 Hz) in the presence of CT99021 throughout the S1 and S2 loading periods (Fig 4a). Under these conditions inhibition of GSK3-dependent rephosphorylation had no significant effect on FM1-43 loading (Fig 4b, 4c) in contrast to the extensive reduction in dye loading during high intensity stimulation (Fig 3d-f). Thus there is an activity-dependent requirement for GSK3-dependent rephosphorylation in SV retrieval.
Figure 4. Activity-dependent requirement for GSK3-dependent protein rephosphorylation in SV retrieval.

a) Experimental protocol. Cells were loaded and unloaded with FM1-43 using the protocol displayed. In both S1 and S2, dyes were loaded with 200 action potentials (10 Hz) and then cells were immediately washed. Unloading of the dye was stimulated by 2 sequential stimuli of 400 action potentials (40 Hz). Cells were preincubated with 2 μM CT99021, when indicated with grey shading, for all steps prior to and including S2 loading. b) Cumulative histograms display the effect of CT99021 on FM1-43 loading in individual nerve terminals (δS2/δS1). c) Bar graphs display the effect of CT99021 on the average δS2/δS1 value for FM1-43 loading ± SEM. d) Experimental protocol. Cells were loaded and unloaded with FM2-10 in an identical manner to a) with the exception that FM2-10 was loaded at S1 and S2 with 800 action potentials (80 Hz). Cumulative histogram (e) and bar graph (f) display the effect of CT99021 on either individual nerve terminal loading (δS2/δS1) or the average δS2/δS1 load (± SEM). Black symbols represent FM1-43 control, open symbols represent FM2-10 control and grey symbols represent CT99021 (2 μM). For all experiments n = 3, Student’s t test.
GSK3 is required for ADBE, but not CME
Since GSK3-dependent protein rephosphorylation is only required during intense neuronal activity, it suggests that it may selectively control ADBE. In support, the GSK3 priming kinase cdk5 is essential for ADBE, but not CME12. To determine whether GSK3 is required for ADBE, we first monitored the uptake of the dye FM2-10 in the continual presence of CT99021 (Fig 4d). FM2-10 shares a similar structure to FM1-43 but does not label ADBE10,11, thus any effect of GSK3 inhibition on ADBE should be “invisible” to this assay. When we stimulated SV turnover by intense stimulation (800 action potentials, 80 Hz), inhibition of GSK3-dependent rephosphorylation had no effect on FM2-10 uptake (Fig 4d-f). This contrasts to the inhibition of FM1-43 uptake during identical stimulation conditions by CT99021 (Fig 3d-f). When the lack of effect of CT99021 is considered regarding either FM2-10 uptake during intense stimulation or FM1-43 uptake during mild stimulation, it suggests that GSK3-dependent protein rephosphorylation is selectively required for ADBE and not CME.
To independently confirm the requirement for GSK3-dependent rephosphorylation in ADBE, we monitored uptake of the large (40 kDa) flourescent dextran, tetramethyrhodamine-dextran, which is too large to be accumulated by single SVs10,21,22. Dextran uptake evoked by intense stimulation (800 action potentials, 80 Hz) was abolished by either of the GSK3 antagonists CT99021 or AR-AO14418, confirming the requirement for GSK3-dependent protein rephosphorylation in ADBE (Fig 5a, 5b). To confirm that these effects were specific to GSK3 inhibition, we silenced GSK3β expression using short hairpin RNA (shRNA, Supplementary Fig 2). Two independent shRNA vectors which reduced GSK3β expression also reduced the uptake of fluorescent dextran when compared to the empty vector (Fig. 5c, 5d). Thus, there is a requirement for GSK3-dependent protein rephosphorylation in ADBE, since either its acute inhibition by pharmacological antagonists or a reduction in its expression using shRNA silencing both retard this SV endocytosis mode.
Figure 5. GSK3-dependent protein rephosphorylation is required for bulk endocytosis of large dextrans.

In all cases neurons were loaded with tetramethyrhodamine-dextran (50 μM) using 800 action potentials (80 Hz). a) Where indicated neurons were incubated with either 2 μM CT99021 or 5 μM AR-AO14418 in depolarizing culture medium for 15 min before a 10 minute repolarization period. Dextran loading in typical fields of view is displayed, either in the absence of antagonists (Ctrl), or in the presence of CT99021 or AR-AO14418. Scale bar represents 15 μM. b) Mean number of dextran puncta per field of view (percentage of control, n = 3 for all experiments; all ± SEM, ** = p < 0.01, one-way ANOVA). c) Cells were transfected with shRNA against GSK3β and loaded with dextran as described in a) 72 hours later. Left panels show transfected neurons with either empty shRNA vector, GSK3β A, or GSK3β B. Middle panels show dextran uptake. Right panels show a merged image with transfected neuron in green and dextran in red. Arrows indicate dextran loading. Scale bar represents 15 μM. d) Mean number of dextran puncta per transfected neuron (percentage of control, n = 8 for empty vector, n = 4 for GSK3β A, n = 5 for GSK3β B, all ± SEM, ** = p < 0.01, one-way ANOVA).
Since GSK3 inhibition has no effect during low intensity stimulation, it suggests GSK3-dependent protein rephosphorylation has no role in CME. To confirm a lack of role for GSK3 activity in CME, we examined both ADBE and CME endocytosis modes in parallel, by monitoring uptake of horse radish peroxidase (HRP) into individual nerve terminals. ADBE is detected as the appearance of large electron-dense endosome-like structures, while CME is detected as electron-dense small SVs10,12,13. On stimulation we observed a marked increase in both the number of HRP-labelled endosomes and HRP-labelled SVs (structures <100 nm in diameter), indicating activation of both ADBE and CME (Fig 6). Inhibition of GSK3-dependent protein rephosphorylation by either CT99021 or AR-AO14418 strikingly reduced the number of HRP-labelled endosomes that were generated during stimulation (Fig 6c), independently confirming the selective requirement for GSK3 in ADBE. In contrast, neither kinase inhibitor had any effect on the number of HRP-labelled SVs, confirming that GSK3 has no role in CME (Fig 6d).
Figure 6. GSK3-dependent protein rephosphorylation is required for HRP uptake into endosomes but not SVs.

a) Experimental protocol. Cells were repolarized for 25 min in incubation medium before a 10 second stimulation with 50 mM KCl to dephosphorylate dynamin I. After a 10 minute repolarization in incubation buffer HRP loading was stimulated by a 2 min stimulus of 50 mM KCl. Cultures were fixed either immediately prior to or after stimulation (shown by the arrowhead symbols in the figure). When GSK3 inhibitors were used, cells were preincubated with either 2 μM CT99021 or 5 μM AR-AO14418 from 15 min before the initial KCl stimulation and all subsequent steps. b) Three panels show HRP-labelled structures in typical fields of view either in the absence of antagonist (Ctrl) or in the presence of either CT99021 or AR-AO14418. Scale bar represents 250 nm in all images. Mean number of HRP-labelled (solid bars) and clear (open bars) endosomes (c) or SVs (d) in either resting (Basal) or stimulated (KCl) nerve terminals (Basal Ctrl, n = 44 nerve terminals; Basal CT99021 n = 30; Basal AR-AO14418 n = 9; KCl Ctrl, n = 28 nerve terminals; KCl CT99021 n = 101; KCl AR-AO14418 n = 51; all ± SEM *** = p < 0.001, one-way ANOVA for HRP-labelled structures).
We have demonstrated a requirement for GSK3 activity in both ADBE and in the rephosphorylation of Ser-774 on dynamin I. However we have not shown that GSK3-dependent rephosphorylation of Ser-774 on dynamin I is required for ADBE. To determine this, we overexpressed dominant negative phosphorylation site mutants of dynamin I23 and examined their effect on fluorescent dextran uptake evoked by intense neuronal activity (800 action potentials, 80 Hz). Overexpression of either phospho-deficient (DynIS774A-mCer) or phospho-mimetic mutants (DynIS774E-mCer) inhibited dextran uptake, in contrast to wild-type dynamin I (DynIWT-mCer) which had no effect (Fig 7). Thus GSK3-dependent rephosphorylation of Ser-774 on dynamin I is a requirement for ADBE to proceed in central nerve terminals.
Figure 7. GSK3β phosphorylation of Ser-774 on dynamin I is required for bulk endocytosis of large dextrans.
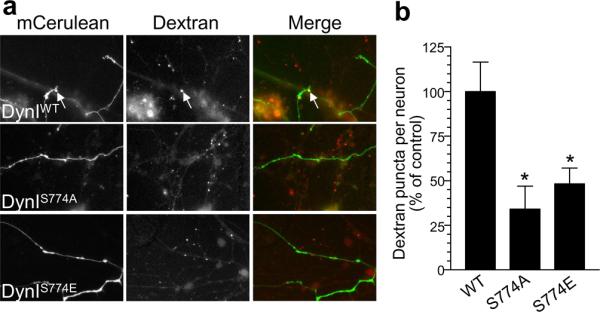
a) Cells were transfected with dynamin overexpresion vectors and after 48 hours were loaded with tetramethyrhodamine-dextran (50 μM) using 800 action potentials (80 Hz). Left panels show neurons transfected with either wild type (DynIWT-mCer), phospho-null (DynIS774A-mCer) or phospho-mimetic (DynIS774E-mCer) dynamin I. Middle panels show dextran uptake. Right panels show a merged image with transfected neuron in green and dextran in red. Arrows indicate dextran loading. Scale bar represents 15 μM. b) Mean number of dextran puncta per transfected neuron (percentage of control n = 7 for WT, n = 3 for S774A, n = 9 for S774E, all ± SEM, * = p < 0.05, one-way ANOVA).
GSK3 inhibition relieves short-term synaptic depression
The perturbation of a number of endocytosis proteins implicated in CME results in an enhanced short-term depression of neurotransmission during high frequency stimulation, presumably from rundown of available SVs within nerve terminals24-26. To determine the function of GSK3-dependent ADBE during high frequency neurotransmission, we examined the effect of CT99021 on synaptic depression evoked by high frequency stimulation (HFS-depression) of the Schaffer collateral inputs onto hippocampal CA1 pyramidal neurons. To eliminate postsynaptic effects of GSK3, we included CT99021 (2 μM) in the ‘internal’ recording solution to inactivate the enzyme. Both control and CT99021-treated slices demonstrated a marked HFS-depression of EPSC amplitudes throughout the duration of the stimulation (Fig 8). However, in the presence of CT99021 the extent of the HFS-depression was significantly reduced at all time points (p < 0.01, 2-way ANOVA, Fig 8). The relief from depression was not due to alterations in SV release probability, since paired pulse facilitation was unaffected by the presence of CT99021 (Supplementary Fig 3). Thus inhibition of GSK3 activity, and by extension ADBE, ameliorates the extent of HFS-depression at a prototypical glutamatergic synapse.
Figure 8. Inhibition of GSK3 relieves HFS-depression of EPSCs in intact slices.

a) Hippocampal slices were incubated with or without CT99021 (2 μM) for 1 hour before being transferred to the recording chamber. Slices were challenged with a prepulse of 600 action potentials (40 Hz) 10 minutes before recording (indicated by arrowheads). b) (i) Example mean EPSCs recorded at four time points during the train of 600 stimuli. The subscript in each set denotes the sweep numbers that were averaged to obtain the trace. The cartoon to the left indicates that in these experiments CT99021 (2 μM) was only present in the pipette-filling solution. (ii) Experiments were performed as in bi but CT99021 (2 μM) was also included in the external recording solution. In bi and bii the first mean EPSC waveforms have been illustrated to have equal heights in order to allow direct comparison of the two recording conditions on the rundown of the amplitudes of the EPSCs. c) Summary plot showing the dependence of the EPSC amplitude (normalized to the amplitude of the first EPSC) on the stimulus number. The numbers above the plot indicate the time-points from which the EPSCs in bi and bii are taken. Two-way ANOVA analysis of the mean EPSC amplitude for all EPSC epochs occurring after the first 80 stimuli indicate that CT99021 (2 μM) significantly reduces the extent of HFS-depression (n = 7 control, n = 11 CT99021; ** p < 0.01).
Discussion
We have demonstrated a novel neuronal function for the multifunctional serine/threonine kinase GSK3 - the phosphorylation of a key residue on dynamin I which is required for ADBE to proceed. In contrast GSK3 activity is not required for CME at the synapse. Thus GSK3 is a key enzyme in the control of SV retrieval modes during times of elevated neuronal activity. This is the first demonstration of a presynaptic role for GSK3 and reveals that a protein kinase signalling cascade prepares SVs for ADBE.
We investigated GSK3 function via use of two independent inhibitors, CT99021 and AR-A014418. Both are highly selective inhibitors, with no action against cdk519,20. This was confirmed by their lack of effect on cdk5-dependent rephosphorylation of Ser778 on dynamin I. The results using these antagonists were corroborated by silencing the expression of GSK3β using shRNA. Knockdown of GSK3β was not complete, due to its long half-life in neurons27. However dextran uptake was still significantly perturbed, confirming the role of GSK3 in ADBE. Thus we have demonstrated a requirement for the enzyme in this key SV retrieval mode using two independent methods to perturb GSK3 function in conjunction with three separate assays of ADBE.
We demonstrate that GSK3 is the in vivo kinase for Ser-774 on the PRD of dynamin I. We originally published that cdk5 rephosphorylated both Ser-774 and Ser-778 both in vitro and in vivo15. We now understand why phosphorylation of Ser-774 by GSK3 was masked in those studies. In vivo inhibition of cdk5 either by either antagonists or overexpression of dominant negative mutants removed priming phosphorylation of Ser-778 and thus GSK3 could not phosphorylate Ser-774. In vitro, cdk5 can phosphorylate Ser-774 during longer incubation times15. However the enzyme has a greater preference for Ser-778, illustrated by a selective phosphorylation of Ser-778 by cdk5 during shorter incubation times (Supplementary Figure 4). Thus cdk5-dependent priming is essential for the downstream phosphorylation of Ser-774 by GSK3.
The activity-dependent dephosphorylation of dynamin I is essential for ADBE but not CME13. In agreement we found no role for GSK3 in CME by three independent approaches (FM1-43 uptake during mild stimulation, FM2-10 uptake during intense stimulation and HRP uptake into SVs during intense stimulation). Since GSK3 exclusively controlled rephosphorylation of Ser-774 on dynamin I, and GSK3 activity was required for ADBE, this raised the possibility that Ser-774 phosphorylation status might be the key regulator of ADBE. This was confirmed by overexpression of phosphorylation site mutants of Ser-774. A key role for the GSK3β-dependent rephosphorylation of Ser-774 in ADBE is supported by studies showing that its phosphorylation exclusively controls an interaction with the endocytosis protein syndapin23. This is a critical point, since both syndapin13,28 and the phospho-dependent dynamin-syndapin interaction13 are essential for ADBE, highlighting the significance of this signalling cascade in nerve terminal physiology. This is the first demonstration that the phosphorylation status of a single key residue of an endocytosis protein can control a specific SV retrieval mode.
There is an obligatory requirement for cdk5-dependent phosphorylation of Ser-778 before GSK3 can phosphorylate Ser-774 in vivo. However the converse is not true and opens possibilities for the regulation of ADBE by multiple signalling cascades. Both cdk5 and GSK3 are constitutively active, however they can be inhibited by either proteolysis (for cdk529 or via phosphorylation by upstream protein kinases including Akt (for GSK31), or by protein-protein interactions (such as Wnt signalling1). It is unlikely that cdk5 proteolysis is a key regulatory step, since the cleaved form of the kinase can prime the DynI-PRD for GSK3 phosphorylation equally well to the non-cleaved form in vitro (data not shown). However it is possible that an upstream signalling cascade could selectively control ADBE by regulating GSK3 activity. In support of this, overexpression of constitutively active Akt inhibits FM1- 43 uptake during elevated neuronal activity in our cultures (MAC unpublished data). Thus there is great potential for the fine control of ADBE by multiple signalling cascades upstream of the key effector GSK3.
Inhibition of GSK3 relieved HFS-depression in hippocampal slices, suggesting that triggering of ADBE may exacerbate rundown of neurotransmission by reducing the number of SVs that are available for release in the short term. This has implications for learning and memory, since selective inhibitors that arrest ADBE may increase synaptic strength during tetanic stimulation, resulting in increased cognitive performance. In addition these results suggest that ADBE may be a key neuroprotective mechanism during neurological conditions such as epilepsy, since its triggering will reduce the extent of neurotransmitter release, and thus neuronal excitability, during seizure activity.
In summary we have demonstrated the first specific presynaptic function for GSK3, regulation of the major SV retrieval mode that occurs during high neuronal activity - ADBE. This highlights for the first time a key role for GSK3 in presynaptic physiology and identifies GSK3 as a new dynamin kinase. Our observation that cdk5 primes dynamin I for phosphorylation by GSK3 reveals a unique partnership between these kinases in controlling SV retrieval during elevated neuronal activity. These results identify GSK3 signalling as a key regulator of SV traffic in central synapses and suggest that GSK3 inhibitors may have a therapeutic role in reducing synaptic transmission under conditions of high synaptic transmission, such as occurs during seizure in humans.
Methods
Materials
FM2-10, FM1-43, tetramethyrhodamine-dextran, penicillin / streptomycin, phosphate buffered salts, foetal calf serum and Minimal Essential Medium, were obtained from Invitrogen (Paisley, U.K.). Synaptophysin antibody was from Synaptic Systems (Germany). Glutaraldehyde and osmium tetroxide were from Agar Scientific (Essex, UK). The highly selective inhibitors of GSK3, CT99021 and AR-A014418, were synthesised as described previously30. cdk5/p35NCK was purchased from Cell Signalling Technology (Arundel, Queensland, Australia). GSK3β was purchased from Calbiochem (Kilsyth, Victoria, Australia). All other reagents were from Sigma (Poole, UK). Phospho-ser774 / phospho-Ser778 antibodies have been previously described15. The PRD of dynI (rat, amino acids 746-864, long splice variant DynIxa) was amplified from a GFP-tagged dynamin I plasmid and subcloned into pGEX4T-123. DynII-GFP (rat 2aa isoform, GFP positioned at the C-terminus) in pEGFP-N1 was provided by M. A. McNiven (Mayo Clinic, Rochester, MN). GST-DynII-PRD (740-870) was generated by PCR amplification of the DynII-PRD using the primers (TGGGGAATTCAGCACCAGCACCGTGTCCACGC and CGGGAATTCCCTAGTCGAGCAGGGACGG) and inserting into pGEX4T-1. Full length rat dynamin Ixa was fused to the fluorescent protein mCerulean at its C terminus13,23. Full length dynamin I Ser774 phospho-site mutants were generated by site directed mutagenesis23. shRNA against GSK3β was designed using the pSUPER vector system (pSUPER neo-GFP, Oligoengine, USA), using the following oligonucleotides: GSK3β A - CCAACAAGGGAGCAAATTA; GSK3β B - GGAAGCTTGTGCACATTCA. The pSUPER vector was engineered to express mCerulean by removing GFP with the enzymes AgeI and BsrGI and replacing with predigested mCerulean.
In vitro phosphorylation
GST-DynI-PRD (606 nM, wildtype or mutant) attached to glutathione beads was phosphorylated with cdk5/p35NCK (9.5 nM) in buffer containing 30 mM Tris-HCl, pH 7.4, 5 mM MgSO4, 1 mM EGTA and 80 μM unlabelled ATP for 5 minutes at 37°C. Reactions were terminated by cooling and beads were washed. A second kinase reaction was then initiated in which cdk5 activity was inhibited by addition of roscovitine (10 μM), then GSK3β (21.3 nM) and 40 μM [γ]32P-ATP (10 μCi) was added for a further 15 minutes at 37°C. In selected samples, GSK3β activity was inhibited by 20 mM lithium. Reactions were terminated by addition of SDS, subjected to SDS-PAGE, and either Coomassie Blue-stained and subjected to autoradiography or transferred to nitrocellulose membrane for Western blot analysis.
Primary neuronal cultures, transfections and immunofluorescence studies
Transfections, immunofluorescence studies and preparation of cerebellar granule neuron cultures were performed15. In all experiments neurons were used between 8 - 10 days in vitro. GSK3β expression was monitored by measuring the immunofluorescence intensity in the cell body of transfected neurons. Fluorescence intensity was expressed as a percentage of the untransfected neurons in the same field of view (after subtraction of background fluorescence). At least three independent experiments were performed, with at least three fields of view being assessed for each experiment.
Fluorescence imaging of SV turnover using styryl dyes
Cultures were removed from culture medium and left for 10 min in incubation medium (170 mM NaCl, 3.5 mM KCl, 0.4 mM KH2PO4, 20 mM TES (N-tris[hydroxy-methyl]-methyl-2-aminoethane-sulphonic acid), 5 mM NaHCO3, 5 mM glucose, 1.2 mM Na2SO4, 1.2 mM MgCl2, 1.3 mM CaCl2, pH 7.4). Cultures were then mounted in a Warner (Hamden, CT, USA) imaging chamber (RC-21BRFS). Invaginating membrane was loaded with either FM2-10 (100 μM) or FM1-43 (10 μM) by evoking SV turnover with a brief train of action potentials (80 or 10 Hz, 100 mA, 1 ms pulse width, delivered using platinum wires embedded in the imaging chamber). Dye was washed from the cultures immediately after termination of stimulation with incubation medium (S1 loading). After a 10 min rest period, accumulated dye was unloaded from nerve terminals using a train of 400 action potentials (40 Hz). This provides an estimate of the total number of SVs turned over during stimulation (δS1). After a 20 min rest period the S1 protocol was repeated (S2 loading and unloading). Thus for any selected nerve terminal, the δS2 response has a matched individual internal control (δS1). The GSK3 antagonist CT99021 (2 μM) was present throughout the S1 loading protocol and up to and including S2 loading unless otherwise stated. Results are represented as either cumulative histograms (δS2/δS1 for individual nerve terminals) or averaged data (average δS2/δS1). Dye unloading was visualised using a Nikon (Tokyo, Japan) Diaphot-TMD epifluorescence microscope and 20 × air objective at 480 nm excitation and > 510 nm emission. Images were visualised using a Hamamatsu (Hamamatsu City, Japan) Orca-ER CCD digital camera and offline imaging software (Simple PCI, Compix Imaging Systems, USA). At least 70 nerve terminals were selected for each experiment and at least 3 independent experiments for each experimental condition.
Fluorescence imaging of dextran uptake
Uptake of tetramethyrhodamine-dextran (40 kDa) was monitored10,13. Briefly, cells were left for 10 min in incubation medium and then stimulated with a train 800 action potentials (80 Hz 10 s) in the presence of tetramethyrhodamine-dextran. The GSK3 antagonists CT99021 (2 μM) or AR-AO11418 (5 μM) were present for 15 minutes in the culture medium and at all steps up to and including action potential stimulation. Dextran loading was determined by the number of fluorescent puncta in a defined field of view (130 μM X 130 μM) using a 20 × air objective at 550 nm excitation and > 575 nm emission. Thresholding analysis was performed to discount regions too large to represent individual nerve terminals (diameter greater than 2 μM). The average number of dextran puncta per field for each experiment (usually 10 fields of view per experiment) were averaged for the same conditions and subtracted from background fluorescence. To ensure the density of nerve terminals was consistent between fields and experimental conditions, experiments were performed on the same set of cultures. Experiments using neurons transfected with either GSK3β shRNA or dynamin I mutants were performed in the same manner (48 hours post transfection for dynamin I overexpression and 72 hours for shRNA). At least three independent experiments were performed, with at least three neurons assessed for each experiment.
Labelling of endocytosis pathways by horse radish peroxidase (HRP)
Neurons were processed as described10,13. Neurons were transferred to incubation medium and after 25 min they were stimulated with 50 mM KCl for 10 seconds. Cells were then repolarized in incubation medium for 15 minutes before a second stimulation with 50 mM KCl medium supplemented with HRP (10 mg / ml). Cells were fixed in a 2% solution of glutaraldehyde in phosphate buffered saline for 30 min at 37°C either directly before or after stimulation. After washing with 100 mM Tris (pH 7.4) cells were exposed to 0.1% diaminobenzidine and 0.2% H2O2 in 100 mM Tris. On development of colour, they were washed with 100 mM Tris then stained with 1% osmium tetroxide for 30 min. After washing, they were post-stained with 2% uranyl acetate for 15 min and then dehydrated using ethanol series and polypropylene oxide and embedded using Durcupan. Samples were sectioned, mounted on grids and viewed using a FEI Tecnai 12 transmission electron microscope (Oregon, USA). Where indicated, cells were incubated with GSK3 antagonists CT99021 (2 μM) or AR-AO11418 (5 μM) for 15 min prior to the first KCl stimulus. HRP-labelled intracellular structures that were less than 100 nm in diameter were arbitrarily designated to be SVs, whereas larger structures were designated to be endosomes.
Assays of dynamin I rephosphorylation in vivo
Cells were washed and left for 10 min in incubation medium. They were then preincubated with or without the GSK3β antagonists CT99021 (2 μM) or AR-AO11418 (5 μM) for a further 15 minutes. Cells were then stimulated with 50 mM KCl for 10 seconds and then repolarised in incubation medium for 7 minutes in the presence and absence of GSK3β antagonists. Neurons were lysed using SDS sample buffer15 either before, during or 7 minutes after KCl stimulation. Lysate was quickly removed and boiled for subsequent analysis by SDS-PAGE and Western blotting. The intensity of signal from phospho-dynamin blots was normalised against the amount of synaptophysin and expressed as a percentage of control.
Mass spectrometric separation of monophosphorylated dynamin I peptides
Phosphorylated GST-DynI-PRD was washed twice with 0.1M triethylammonium bicarbonate (TEAB) and then incubated for 12 hours in 0.1M TEAB supplemented with 200 ng trypsin. After digestion phosphopeptides were enriched using TiO2 chromatography31 and resuspended in 0.1% formic acid (FA). Mass spectrometry (MS) analysis used an nHPLC-ESI-MS/MS system consisting of an LC Packings Ultimate HPLC system (Dionex, Netherlands) preceding a QSTAR XL quadrupole-TOF-MS (QqTOF-MS, Applied Biosystems). Phosphopeptides were loaded onto a fused-silica precolumn in 0.1% FA in water for 5 min. They were eluted through a 12 cm fused-silica analytical column (i.d. 50 μM) using a gradient from 100% solvent A (H2O/FA, 100:0.1, v/v) to 40% solvent B (H2O/acetonitrile /FA, 20:80:0.1, v/v/v) in 35 min, then 40–65% solvent B in 2 min and 65-100% in 0.1 min. The eluate was sprayed through a 10 μM i.d. distal coated SilicaTip (FS360-20-10-D-20, New Objective). The MS was set to constantly select and fragment m/z 569.3 over the entire experiment, corresponding to MH2+ of the singly phosphorylated DynI-PRD peptide SPTSSPTPQR. Precursor selection was set to a wide m/z setting (2–3 units) using consecutive 1 s MS/MS scans with a collision energy of 32 eV. The MS/MS spectra of individual chromatographic peaks was summed and manually annotated and identified using GPMAW version 8.0 (Lighthouse Data, Denmark).
Synaptic depression in acute hippocampal slices
For whole-cell voltage-clamp experiments horizontal hippocampal slices (400 μm thick) were prepared from P14/15 male Sprague-Dawley rats. Slices were allowed to recover for at least 1 hour prior to making electrophysiological recordings. In addition, when testing the effects of CT99021 on HFS-depression of EPSC amplitudes, slices were stored in a solution containing the inhibitor (2 μM) for a minimum of 1 h before being transferred to the recording chamber. In such experiments the slices were continuously perfused (1.5 ml min−1) with the external recording solution containing this same concentration of CT99021. Slices were challenged with a prepulse of 600 action potentials (40 Hz) 10 minutes before EPSC recording.
Patch-pipettes were filled with an internal recording solution contain (in mM): Cs-methylsulphonate, 130; CsCl, 10; HEPES, 10; EGTA, 0.1; glucose, 10, Na-phosphocreatine, 10; Mg-ATP, 4; Mg-GTP, 0.5; QX-314, 5 (pH 7.3 with CsOH; 290-300 mOsm). When filled with this solution, the patch-pipettes had resistances of 3 – 5 MΣ. CT99021 (2 μM) was added to this solution to directly deliver this inhibitor to the postsynaptic neuron. Thus while both ‘control’ and ‘+CT99021’ recording have the inhibitor in the internal recording solution, it is only the experiments in the latter set where whole slices (and therefore presynaptic terminals) were exposed to CT99021 (2 μM). The external recording solution contained (in mM): NaCl, 124; NaH2PO4, 1.2; KCl, 2.5; NaHCO3, 25; glucose, 20; CaCl2, 2; MgCl2, 1; picrotoxin, 0.05 (pH 7.3 with 95% O2/5% CO2; 310-320 mOsm). Whole-cell recordings were made at 32 °C from visually identified CA1 pyramidal neurons held at −70 mV using a Molecular Devices 700B amplifier. Series resistances were monitored throughout each experiment and were typically <15 MΣ. Only experiments where this varied <15% for the duration of the recording were included for analysis. Whole-cell currents were filtered at 2 kHz and digitized at 10 kHz. A surgical cut was made to isolate the CA1 and CA3 regions and monosynaptic glutamatergic EPSCs were evoked by placing a stimulating electrode in stratum radiatum (100 – 150 μm from stratum pyramidale). Constant current (15 – 50 μA for 50 – 200 μs) stimuli were delivered at 40 Hz for 15 s (600 pulses in total). The mean amplitudes of 10 consecutive EPSC epochs were measured using custom-written software and normalized to the peak amplitude of the initial EPSC recorded in the train of 600 stimuli. Normalized EPSC amplitudes were pooled (n = 7 for ‘control’; n = 11 for ‘+CT99021’) and plotted against stimulus number in order to assess activity-evoked run-down of synaptic currents.
Statistical Analysis
Two sample sets were compared using a Student’s t test, whereas those samples sets >2 were compared using a one-way ANOVA as indicated. HFS-depression of EPSC amplitudes in the presence and absence of CT99021 was compared using a two-way ANOVA.
Supplementary Material
Acknowledgements
This work was supported by grants from the Wellcome Trust (Ref: 070569 & 084277), Epilepsy Research UK (0503) and the Australian National Health and Medical Research Council. CS is supported by the Alzheimer’s Research Trust (ART/PG2007/4) and the MRC (88158). AC is supported by an Australian NH&MRC Peter Doherty Fellowship (454886). We wish to thank Dr. Alan Prescott and Mr. John James (both University of Dundee) for excellent technical assistance.
Reference List
- 1.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem.J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol.Med. 2002;8:126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- 4.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J.Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma YC, et al. Regulation of motor neuron specification by phosphorylation of neurogenin 2. Neuron. 2008;58:65–77. doi: 10.1016/j.neuron.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peineau S, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad-Annuar A, et al. Signaling across the synapse: a role for Wnt and Dishevelled in presynaptic assembly and neurotransmitter release. J.Cell Biol. 2006;174:127–139. doi: 10.1083/jcb.200511054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y, Xu J, Heinemann SF. Two pathways of synaptic vesicle retrieval revealed by single-vesicle imaging. Neuron. 2009;61:397–411. doi: 10.1016/j.neuron.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clayton EL, Evans GJ, Cousin MA. Bulk synaptic vesicle endocytosis is rapidly triggered during strong stimulation. J.Neurosci. 2008;28:6627–6632. doi: 10.1523/JNEUROSCI.1445-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards DA, Guatimosim C, Betz WJ. Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron. 2000;27:551–559. doi: 10.1016/s0896-6273(00)00065-9. [DOI] [PubMed] [Google Scholar]
- 12.Evans GJ, Cousin MA. Activity-dependent control of slow synaptic vesicle endocytosis by cyclin-dependent kinase 5. J Neurosci. 2007;27:401–411. doi: 10.1523/JNEUROSCI.3809-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clayton EL, et al. The phospho-dependent dynamin-syndapin interaction triggers activity-dependent bulk endocytosis of synaptic vesicles. J.Neurosci. 2009;29:7706–7717. doi: 10.1523/JNEUROSCI.1976-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newton AJ, Kirchhausen T, Murthy VN. Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc.Natl.Acad.Sci.U.S.A. 2006;103:17955–17960. doi: 10.1073/pnas.0606212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan TC, et al. Cdk5 is essential for synaptic vesicle endocytosis. Nat.Cell Biol. 2003;5:701–710. doi: 10.1038/ncb1020. [DOI] [PubMed] [Google Scholar]
- 16.Cousin MA, Robinson PJ. The dephosphins: Dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci. 2001;24:659–665. doi: 10.1016/s0166-2236(00)01930-5. [DOI] [PubMed] [Google Scholar]
- 17.Cole AR, et al. Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J.Biol.Chem. 2006;281:16591–16598. doi: 10.1074/jbc.M513344200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graham ME, et al. The in vivo phosphorylation sites of rat brain dynamin I. J.Biol.Chem. 2007;282:14695–14707. doi: 10.1074/jbc.M609713200. [DOI] [PubMed] [Google Scholar]
- 19.Bhat R, et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J.Biol.Chem. 2003;278:45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- 20.Wagman AS, Johnson KW, Bussiere DE. Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes. Curr.Pharm.Des. 2004;10:1105–1137. doi: 10.2174/1381612043452668. [DOI] [PubMed] [Google Scholar]
- 21.Holt M, Cooke A, Wu MM, Lagnado L. Bulk membrane retrieval in the synaptic terminal of retinal bipolar cells. J.Neurosci. 2003;23:1329–1339. doi: 10.1523/JNEUROSCI.23-04-01329.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teng H, Lin MY, Wilkinson RS. Macroendocytosis and Endosome Processing in Snake Motor Boutons. J Physiol. 2007;582:243–262. doi: 10.1113/jphysiol.2007.130989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anggono V, et al. Syndapin I is the phosphorylation-regulated dynamin I partner in synaptic vesicle endocytosis. Nat Neurosci. 2006;9:752–760. doi: 10.1038/nn1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shupliakov O, et al. Synaptic vesicle endocytosis impaired by disruption of dynamin-SH3 domain interactions. Science. 1997;276:259–263. doi: 10.1126/science.276.5310.259. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, et al. Formation of an endophilin-Ca2+ channel complex is critical for clathrin-mediated synaptic vesicle endocytosis. Cell. 2003;115:37–48. doi: 10.1016/s0092-8674(03)00726-8. [DOI] [PubMed] [Google Scholar]
- 26.Koh TW, et al. Eps15 and Dap160 control synaptic vesicle membrane retrieval and synapse development. J.Cell Biol. 2007;178:309–322. doi: 10.1083/jcb.200701030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hongisto V, Vainio JC, Thompson R, Courtney MJ, Coffey ET. The Wnt pool of glycogen synthase kinase 3beta is critical for trophic-deprivation-induced neuronal death. Mol.Cell Biol. 2008;28:1515–1527. doi: 10.1128/MCB.02227-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersson F, Jakobsson J, Low P, Shupliakov O, Brodin L. Perturbation of syndapin/PACSIN impairs synaptic vesicle recycling evoked by intense stimulation. J.Neurosci. 2008;28:3925–3933. doi: 10.1523/JNEUROSCI.1754-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhavan R, Tsai LH, Tsai LH. A decade of cdk5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 30.Bain J, et al. The selectivity of protein kinase inhibitors: a further update. Biochem.J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol.Cell Proteomics. 2005;4:873–886. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.