

Abstract
We prepared ultrafine Fe–Pt alloy nanoparticle colloids by UV laser solution photolysis (KrF excimer laser of 248 nm wavelength) using precursors of methanol solutions into which iron and platinum complexes were dissolved together with PVP dispersant to prevent aggregations. From TEM observations, the Fe–Pt nanoparticles were found to be composed of disordered FCC A1 phase with average diameters of 0.5–3 nm regardless of the preparation conditions. Higher iron compositions of nanoparticles require irradiations of higher laser pulse energies typically more than 350 mJ, which is considered to be due to the difficulty in dissociation of Fe(III) acetylacetonate compared with Pt(II) acetylacetonate. Au colloid preparation by the same method was also attempted, resulting in Au nanoparticle colloids with over 10 times larger diameters than the Fe–Pt nanoparticles and UV–visible absorption peaks around 530 nm that originate from the surface plasmon resonance. Differences between the Fe–Pt and Au nanoparticles prepared by the KrF excimer laser solution photolysis are also discussed.
Keywords: Nanoparticle, Excimer laser, Laser solution photolysis, Precursor, Fe–Pt alloy, Au
Introduction
Recently, nanomaterials have been researched due to their diverse application potentials. Particular attention has focused on nanoparticles of Fe–Pt alloys because, they demonstrate potentials for ultra-high density recording media [1] of which materials require a high magnetocrystalline anisotropy for thermal stability of magnetization, biomedical applications [2,3] of which materials require chemical stability and biocompatibility, catalysts for fuel cells with high poisoning resistance [4], and other magnetic application potentials [5]. Besides the preparation method for well-defined self assembly of Fe–Pt nanoparticles [6], precise deposition control of nanoparticles employing Langmuir–Blodgett method has also been reported [7,8].
Processes for nanoparticle production by light irradiations, which are clean, one-step processes different from conventional physical or chemical ones, have been proposed [9-17]. Syntheses of gold nanoparticles by UV light irradiation to gold chloride solutions, referred to as “photolysis”, have long been known [9] and “laser photolysis” using UV laser light has also been applied for syntheses of gold nanoparticles [10,11] and iron based nanoparticles from ferrocene and iron(II) acetylacetonate that are UV-absorbing complexes [12,13]. In addition to photolysis, laser using processes under other generation principles such as “laser pyrolysis” based on thermal decomposition of gas phase sources by far-infrared laser irradiation [14,15] and “laser ablation in liquid phase” based on laser ablation phenomena in solutions resulted in monodisperse nanoparticles of target materials submerged in solutions [16,17], has also been reported.
In the present study, we prepared ultrafine Fe–Pt alloy nanoparticles of 0.5–3 nm diameters dispersed in methanol solvent by KrF excimer laser solution photolysis for the first time, employing precursors of methanol solutions into which iron and platinum complexes were dissolved together with polymer dispersant of polyvinylpyrrolidone, PVP. Au nanoparticles with diverse application potentials [18] were also prepared using the same preparation technique. The differences between the results of Au and Fe–Pt nanoparticle colloids by this method are discussed in this article.
Experimental
The employed experimental set up for KrF excimer laser irradiation to solutions is schematically shown in Fig. 1. UV sample irradiation was carried out using a KrF excimer pulsed laser generation system (COMPex205, Lambda Physik, λ = 248 nm). The emitted laser lights were introduced to the surface of precursor solution filled in a 100 mL-sized beaker from the top of the beaker by passing through a mirror. The laser conditions of power, pulse energy, and pulse frequency were varied in the range of 0.32–31.5 W, 160–630 mJ, 2–50 Hz, respectively. The irradiation time was fixed to 30 min. The laser beam is rectangular-shaped with a size of 2.4 × 0.9 cm (2.16 cm2) and a pulse duration of 25 ns.
Figure 1.
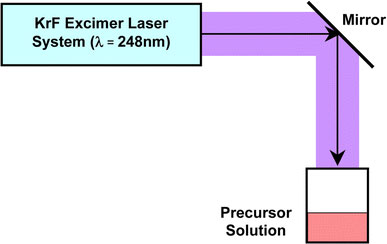
Experimental set up of KrF excimer laser irradiation to precursor solution
Precursor solutions for Fe–Pt were methanol (CH3OH, Wako 99.8+% dehydrated) solutions in which iron(III) acetylacetonate (Fe(III)(C5H7O2)3, Aldrich 99.9+%), denoted by Fe(III)(acac)3, and platinum(II) acetylacetonate (Pt(II)(C5H7O2)2, Aldrich 97%), denoted by Pt(II)(acac)2were completely dissolved. Polyvinylpyrrolidone ((C6H9NO)n, Aldrich average molecular weight ~10,000), denoted by PVP, was dissolved in all cases to prevent aggregation. Concentrations for Fe(III)(acac)3, Pt(II)(acac)2, and PVP were varied while keeping constant the sum of both metal complex concentrations and PVP ones to 3 mM and 6 mM, respectively. The combination of ferrocene (Fe(II)(C5H5)2, Aldrich 98%), denoted by Fe(II)Cp2, and Pt(II)(acac)2has also been investigated while keeping constant the sum of both metal complex concentrations and PVP ones to 10 mM and 50 mM, respectively. For Au nanoparticle preparation, water solutions into which chloroauric acid (HAu(III)Cl4 · H2O, Aldrich 99.999%) and PVP were completely dissolved with concentrations of 0.5 mM and 1.0 mM, respectively. After laser irradiation, the resulting solutions were centrifuged at 3,000 rpm for 10 min for both the Fe–Pt and Au cases. In the case of Fe–Pt, irradiated solutions were dissolved into hexane for removal of decomposed and undecomposed matter.
An HF2000 (Hitachi, 200 kV) was used for transmission electron microscopic (TEM) observations and Vantage (Noran, a minimum analytical beam size of ϕ1 nm) attached to the TEM apparatus enabled energy dispersive X-ray spectroscopy (EDXS) measurement. TEM and EDXS measurements were performed on the samples of C-supported Cu grids on which colloids were dropped and allowed to dry. UV3600 (Shimadzu) and Zetasizer Nano (Malvern) were used for measurements of absorbance spectra in the UV–visible light region and dynamic light scattering (DLS) intensities as a function of Zeta potentials, respectively. Quartz or polystyrene cells (a path length of 10 mm) were used for these optical measurements.
Results and Discussion
Fe–Pt Nanoparticles
After laser irradiation, the red-colored precursor solution for Fe–Pt changed color to black, similar to that of platinum colloids. We investigated the absorbance spectrum change in the UV–visible light region before and after laser irradiation as shown in Fig. 2, which includes UV–visible absorbance spectra of the precursor methanol solution with Fe(III)(acac)3/Pt(II)(acac)2 = 2.4/0.6 mM, and methanol solutions of Fe(III)(acac)3 and Pt(II)(acac)2 with a concentration of 3 mM for reference. The measured solutions were diluted with methanol by a factor of 100 due to absorbance saturations in the UV region. It was found that the contribution of Fe(III)(acac)3 absorbance, which has a main peak at around 273 nm due to the π–π* transition of acetylacetonate ligand [19], is dominant for the precursor spectrum before irradiation. We assumed that the precursor main absorbance peak close to the KrF excimer laser wavelength of 248 nm allowed efficient photolysis of the complexes. The laser irradiation did not cause a new absorbance band but only a general reduction in peak intensities, which is the same tendency as the reported laser photolysis of Fe(II) acetylacetonate in 2-propanol solvent [13].
Figure 2.
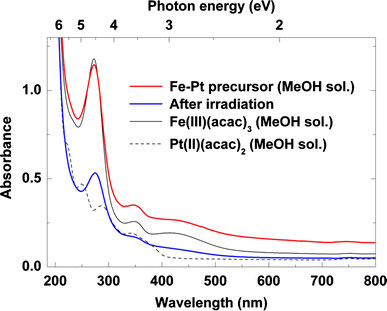
UV–visible absorption spectra of Fe–Pt precursor (Fe(III)(acac)3/Pt(acac)2 = 2.4/0.6 mM) before and after laser irradiation (20 W, 400 mJ, 50 Hz). Spectra of Fe(III)(acac)3and Pt(II)(acac)2MeOH solutions (3 mM) are also indicated with black solid lines and dashed lines, respectively, for comparison
Figure 3 shows EDXS spectrum measured on the Fe–Pt nanoparticles with a precursor of Fe(III)(acac)3/Pt(II)(acac)2 = 2.0/1.0 mM and laser conditions of 31.5 W (630 mJ, 50 Hz) for 30 min. From the appearance of peaks attributed to iron and platinum elements on several points of the nanoparticles, alloying of Fe–Pt alloy in the nanoparticles can be confirmed. Except carbon and copper from the grids, the existence of other element such as silicon, which was reported for the laser photolysis with a different experimental configuration [13], was not confirmed. Figure 4 shows typical TEM images with different magnifications (a), (b) and an electron beam diffraction pattern (c) for Fe50Pt50 nanoparticles with a precursor of Fe(III)(acac)3/Pt(II)(acac)2 = 2.4/0.6 mM and a laser conditions of 15 W (300 mJ, 50 Hz) for 30 min. From the images, diameters of Fe–Pt nanoparticles are found to be 0.5–3 nm. Fringes characteristic for crystallinity in nanoparticles were partly observed on the Fe–Pt nanoparticles in Fig. 4a. Assemblies or aggregations of nanoparticles are partly observed together with well-dispersed nanoparticles spread in the larger areas (Fig. 4b). The electron beam diffraction pattern shows continuous diffuse rings assigned to (111), (200), (220), (311) and (331) planes of disordered A1 phase of FePt, which correspond to the ultrafine microstructure in the TEM image. Figure 5 shows TEM images of Fe50Pt50 nanoparticles with a precursor concentration of Fe(III)(acac)3/Pt(II)(acac)2 = 2.4/0.6 mM (a) and Fe21Pt79 nanoparticles with a precursor of Fe(II)Cp2/Pt(II)(acac)2 = 4.0/6.0 mM (b). The employed laser condition is 15 W (300 mJ, 50 Hz) in both cases. Nanoparticles prepared from a combination of Fe(II)Cp2/Pt(II)(acac)2 are found to have almost the same size range but more diffuse particle images compared with the Fe(III)(acac)3/Pt(II)(acac)2 complex combination, which implies an insufficient crystallinity in the nanoparticles. FePt alloying was also confirmed from micro EDXS measurements for the Fe(II)Cp2/Pt(II)(acac)2 combination. Size distributions of the Fe–Pt nanoparticles, which were obtained from the TEM images, are shown in Fig. 6. TEM observation results revealed that all the Fe–Pt nanoparticles obtained were ultrafine with the same diameter range of 0.5–3 nm regardless of the precursor conditions (kinds or concentrations of metal complexes) or the laser conditions (pulse energies or irradiation time). The comparison of size distributions in Fig. 6 shows that the Fe(II)Cp2/Pt(II)(acac)2 precursor case is found to have a maximum at a smaller diameter compared with the Fe(III)(acac)3/Pt(II)(acac)2 case.
Figure 3.
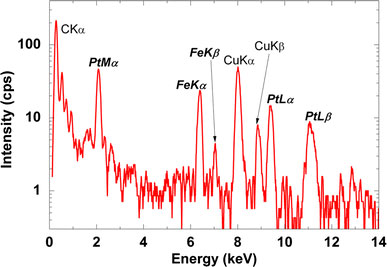
EDXS spectrum measured on Fe–Pt nanoparticles with a precursor of Fe(III)(acac)3/ Pt(II)(acac)2 = 2.0/1.0 mM (31.5 W, 630 mJ, 50 Hz)
Figure 4.

TEM images with different magnifications (a), (b), and electron beam diffraction pattern (c) for Fe50Pt50nanoparticles with a precursor of Fe(III)(acac)3/Pt(II)(acac)2 = 2.4/0.6 mM (15 W, 300 mJ, 50 Hz)
Figure 5.

TEM images of Fe50Pt50nanoparticles with a precursor of Fe(III)(acac)3/Pt(II)(acac)2 = 2.4/0.6 mM (15 W, 300 mJ, 50 Hz) (a) and Fe21Pt79nanoparticles with a precursor of Fe(II)Cp2/Pt(II)(acac)2 = 4.0/6.0 mM (15 W, 300 mJ, 50 Hz) (b)
Figure 6.
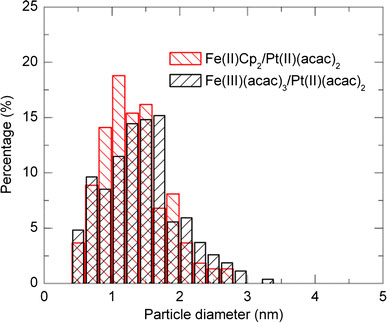
Size distributions of FePt nanoparticles with a precursor of Fe(III)(acac)3/Pt(II)(acac)2 = 2.4/0.6 mM (15 W, 300 mJ, 50 Hz) and that with a precursor of Fe(II)Cp2/Pt(II)(acac)2 = 4.0/6.0 mM (15 W, 300 mJ, 50 Hz)
Increase of Fe(II)Cp2 concentration in precursors did not cause increase of iron concentration in generated nanoparticles, which may be considered to be attributable to a difficulty in photolysis of Fe(II)Cp2 compared with Fe(III)(acac)3. Ouchi et al. reported the investigation results of Fe-based nanoparticle formation by ArF laser solution photolysis of Fe(II)Cp2 in hexane, including its very low quantum yield <10−3[20]. Thus, the low iron concentration in Fe–Pt nanoparticles with Fe(II)Cp2/Pt(II)(acac)2 complex combination might be related to the reported low quantum yield of Fe(II)Cp2 solution photolysis. The harder photolysis of Fe(II)Cp2 can be also explained from the mass difference between the ligands of Cp and acac as follows: frequency of vibration ω is known to be proportional to (k/m)1/2, where k is the elastic constant and m is the reduced mass on the iron–ligand bond. Thus, ω of Fe(II)Cp2 can be estimated to be higher than that of Fe(III)(acac)3 because Fe(II)Cp2 has a Cp ligand lighter than an acac of Fe(III)(acac)3 if the same value of k is assumed. We think that the higher ω of Fe(II)Cp2 would be one of the possible reason for its harder photolysis.
Adiabatic dissociation energies of metal–ligand bonds in iron complexes including Fe(CO)5, Fe(II)Cp2 and Fe(III)(acac)3 were reported to be nearly equal to 6.0 eV from their photodissociation and thermodynamic investigation [21]. In particular, Fe(II)Cp2 has been investigated due to its unusual photochemical behavior [22]. The dissociation energy of nearly 6.0 eV is not sufficient for a single 248 nm photon energy of 5.0 eV, and hence two photon dissociation can be considered for these dissociations. The dissociation energy of nearly 6.0 eV is only for the cleavage of metal–ligand bonds and solvent effect such as the scavenging effect in alcohols [23], energies for cleaved ion reduction to zero-valent iron and the formation scheme of alloy nanoparticles are not taken into consideration. Therefore, further investigations are required to elucidate nanoparticle formation by UV laser solution photolysis of Fe and Pt complex solutions.
For applications of Fe–Pt nanoparticles, iron-rich concentrations greater than the equiatomic ones are necessary due to the steep dissipation of magnetization in the Pt-rich side of concentrations [5]. Thus, we investigated the influence of irradiated laser powers on Fe–Pt compositions in order to explore the controllability of Fe–Pt compositions through adjustment of laser pulse energies. Figure 7 shows evaluated Fe compositions as a function of irradiated pulse laser energies for Fe–Pt nanoparticles with precursor concentrations of Fe(III)(acac)3/Pt(II)(acac)2 = 2.0/1.0 mM and 2.4/0.6 mM. The Fe compositions are found to have a tendency to increase with irradiated laser pulse energy. Thus, higher laser pulse energies typically more than 350 mJ (185 mJ/cm2 for fluence) are required in order to obtain Fe–Pt nanoparticles with higher Fe compositions, which might be attributable to harder dissociation of Fe(III) acetylacetonate than that of Pt(II) acetylacetonate. From the investigation results of UV laser solution photolysis of Fe(II)(acac)2 in i-propanol [20], Pola et al. proposed a deep photolysis from the complex directly into zero-valent elemental Fe(0) and organic photofragments without passing through an intermediate product such as the case of Cu(II) acetylacetonate [24]. This can be considered to be a multiphoton dissociation process of Fe(II)(acac)2 through cleavage of acetylacetonate ligands as shown in the following photolysis (Eq. 1).
| (1) |
Figure 7.
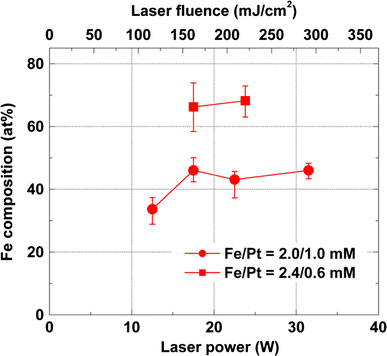
Evaluated Fe compositions as a function of irradiated laser powers for Fe–Pt nanoparticles with precursors of Fe(III)(acac)3/Pt(II)(acac)2 = 2.0/1.0 mM and 2.4/0.6 mM
The absorbance spectra in Fig. 2 show only the reduced source spectrum and no new absorbance band after laser irradiation. Therefore, as shown in the following simplified Eq. 2, we may estimate that the photolysis for Fe–Pt alloy nanoparticle formation mechanism of Fe(III)(acac)3 and Pt(II)(acac)2 in methanol is based on a multiphoton dissociation for both the complexes, which is similar to the above-mentioned deep photolysis of Fe(II)(acac)2, even though the intermediate process between the initiation of dissociation for each complex and the formation completion of alloy nanoparticles (FePt)n has not yet been clarified.
| (2) |
Figure 8 shows DLS intensity as a function of Zeta potential for Fe–Pt colloid solutions with varying laser pulse energies from 250 to 630 mJ (12.5–31.5 W, 50 Hz). Despite the fairly low zeta potentials ranging from −8 to −2 mV, the colloid solutions are mostly stable for several weeks. We consider that the aggregation is mainly prevented not by the repulsive force of nanoparticle charges but by the steric hinderance of dispersing agent of PVP. PVP concentrations less than two times concentrations of the sum of Fe and Pt complex ones were found to cause precipitations after several days of their syntheses.
Figure 8.
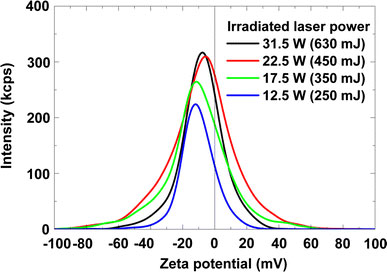
DLS intensities as a function of Zeta potentials for Fe–Pt colloids with varying laser powers from 12.5 W (250 mJ, 50 Hz) to 31.5 W (630 mJ, 50 Hz)
Au Nanoparticles
We investigated the laser solution photolysis of HAu(III)Cl4 precursors varying laser pulse frequency from 2 to 20 Hz while keeping constant pulse energy to 160 mJ and irradiation time to 30 min. Figure 9 shows TEM images and electron beam diffraction patterns of Au nanoparticles with varying laser pulse frequencies from 2 Hz (a), 5 Hz (b), 10 Hz (c) and 20 Hz (d) (160 mJ, 0.32–3.2 W) while keeping constant irradiation time to 30 min. Nonspherical particles including rods or triangular or pentagonal ones were partly observed, which have also been reported for Au nanoparticles prepared by photochemical or other synthetic methods [25-27]. Synthesis of nonspherical particles (nano-rods or nano-wires) of WO3 by laser pyrolysis and analysis of their formation by the solid-vapor-solid (SVS) mechanism have also been reported [28]. Diffraction rings attributed to (111), (200), (220), (311) (222), (400) and (331) planes of FCC Au can be found in the obtained diffraction images. When compared with other laser conditions of Fig. 9a, b, d, the ring of Fig. 9c (10 Hz) shows more continuous and diffuse characteristics and less diffraction spot especially in the higher order rings compared with other pulse frequency cases, which implies less crystallinity than the nanoparticles with other laser pulse frequencies.
Figure 9.

TEM images and electron beam diffraction patterns of Au nanoparticles with laser pulse frequencies of 2 Hz (a), 5 Hz (b), 10 Hz (c) and 20 Hz (d) (160 mJ, 0.32–3.2 W)
Size distributions of the Au nanoparticles, which were obtained from the TEM images, are summarized with histograms in Fig. 10. The nanoparticles with lower pulse frequencies of 2 and 5 Hz are found to have main size distributions of ~10–50 nm (Fig. 10a) while the nanoparticles with higher pulse frequencies of 10 and 20 Hz (Fig. 10b) show sharper distributions of 10–30 nm even though a small amount of agglomerated larger particles exist for the 20 Hz frequency case. In addition to the nanoparticles having 10 nm diameters, smaller particles with several nanometers were also observed. Figure 11shows the comparison of size distributions between the typical Fe–Pt (Fe(II)Cp2/Pt(II)(acac)2complexes) and Au nanoparticles (pulse laser frequency = 2 Hz). The Au nanoparticles are found to have larger particle diameters of tens nm for all the cases compared with the Fe–Pt nanoparticles as shown in Fig. 11.
Figure 10.

Size distributions of Au nanoparticles with laser pulse frequencies of 2 Hz and 5 Hz (a), and 10 Hz and 20 Hz (b) (160 mJ, 0.32–3.2 W)
Figure 11.
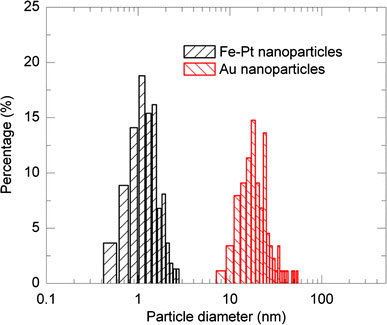
Comparison of size distributions between Fe–Pt nanoparticles with a precursor of Fe(II)Cp2/Pt(II)(acac)2 = 4.0/6.0 mM (300 mJ, 50 Hz, 15 W) and Au nanoparticles with a laser pulse frequency of 2 Hz
Kurihara et al. proposed the photoreduction scheme for Au nanoparticle formation by HAu(III)Cl4 UV laser solution photolysis as shown in the following Eqs. 38 [10]. It consists of reduction of trivalent Au(III) ion to zero-valent elemental Au(0) through the formation of a caged divalent gold Au(II) complex followed by its dissociation and disproportionation, and finally resulted in Au nanoparticles (Au(0))n after accumulation of Au(0). Recently, Nakazato et al. confirmed the dynamic process of this photoreduction scheme by the single-shot near-field heterodyne transient grating (NF-HD-TG) method and also reported that PVP dispersant concentrations affect the photoreduction process [29].
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
As mentioned in the section “Fe–Pt nanoparticles”, Fe or Fe–Pt nanoparticle formation by UV laser photolysis of iron and platinum complexes is considered to be based on multiphoton dissociation of metal complexes, which might be the reason for relatively high-laser pulse energies for generation of Fe–Pt nanoparticles. Conversely, the laser powers for Au nanoparticle formation by UV solution photolysis are relatively low compared with the metal complex case. It has been known that UV incoherent light of relatively low intensity compared with laser light is sufficient for Au nanoparticle formation in chloride solutions, which may be considered to originate from the above-mentioned difference in decomposition mechanism between the multiphoton dissociation of metal complexes and the photoreduction of gold chloride ions.
Figure 12 shows UV-visible absorption spectra of the Au nanoparticles prepared with varying laser pulse frequency from 2 to 20 Hz (160 mJ, 0.32–3.2 W). The absorption spectrum for gold precursor, which has a peak around 294 nm due to the ligand to metal charge transfer (LMCT) band of AuCl4− ion [30], is also shown for comparison. Absorption peaks ranging from 532 to 538 nm that originate from surface plasmon resonance of Au nanoparticles were observed for each sample. All the samples also exhibit peaks attributed to the precursor solution due to the existence of unreduced AuCl4− ions in the obtained solutions. Increase in size or nonspherical shapes including nanorods, ellipsoids, triangular prism, and tetrahedrons have been known to influence the absorption spectra of Au nanoparticles from both empirical investigations and numerical simulations using the extended Mie theory or discrete dipole approximation, DDA [31-33]. From the small amount of nonspherical nanoparticles, however, we can consider that the spectrum broadening and the lesser magnitude of absorbance for the 10 Hz pulse frequency case are attributable to the lesser crystallinity that can be confirmed from the diffuse diffraction ring in Fig. 9c compared with the other laser frequency cases.
Figure 12.
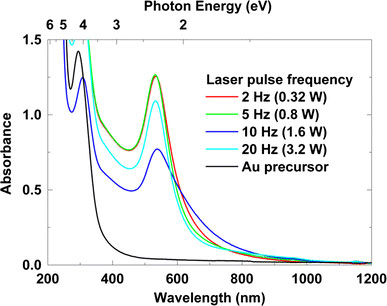
UV–visible absorption spectra of Au nanoparticle solutions with varying laser pulse frequencies from 2 Hz (160 mJ, 0.32 W) to 20 Hz (160 mJ, 3.2 W)
In order to check the stability of colloids, we measured Zeta potential properties of the Au colloids. Figure 13shows DLS intensity as a function of Zeta potential for the Au nanoparticle colloids with varying laser pulse frequency from 2 to 20 Hz with the pulse energy of 160 mJ. Although the obtained absolute values of Zeta potentials are less than 20 mV, which are larger than those of the Fe–Pt colloids, the Au colloids are not particularly stable compared with the Fe–Pt colloids. This is considered to be due to the fact that the stability of Au nanoparticles is dominated mainly by the steric hinderance of PVP dispersant as described in the section “Fe–Pt nanoparticles”.
Figure 13.

DLS intensities as a function of Zeta potentials for Au colloids with varying laser pulse frequencies from 2 Hz (160 mJ, 0.32 W) to 20 Hz (160 mJ, 3.2 W)
Conclusion
Fe–Pt and Au nanoparticles were prepared by KrF excimer laser solution photolysis. TEM observations revealed that the Fe–Pt nanoparticles are composed of FCC A1 phase and are ultrafine with diameters of 0.5–3 nm. From EDXS analyses, compositions of Fe–Pt nanoparticles are found to be mainly influenced by irradiated laser powers, which implies that Fe acetylacetonate is harder to decompose compared with Pt acetylacetonate. Although the Zeta potentials are lower than those of the Au colloids, the Fe–Pt colloids are stable for longer time periods than the case of Au colloids due to the steric hinderance of PVP. The Au nanoparticles are over 10 times larger than those of Fe–Pt nanoparticles.
Acknowledgments
We are grateful for Mr. T. Miyazaki of Tohoku University for his skilled TEM observations and EDXS analyses and Mr. K. Tamura for his assistance with sample preparations.
References
- Weller D, Moser A, Folks L, Best ME, Lee W, Toney MF, Schwickert M, Thiele J-U, Doerner MF. IEEE Trans. 2000. p. 10. COI number [1:CAS:528:DC%2BD3cXhvFaksbY%3D]; Bibcode number [2000ITM....36...10W] [DOI]
- Taton TA. Nat. 2003. p. 73. COI number [1:CAS:528:DC%2BD3sXns1CrsQ%3D%3D]; Bibcode number [2003NatMa...2...73T] [DOI] [PubMed]
- Hong R, Fischer NO, Emrick T, Rotello VM. Chem. 2005. p. 4617. COI number [1:CAS:528:DC%2BD2MXmvFCnu7s%3D] [DOI]
- Yano H, Kataoka M, Yamashita H, Uchida H, Watanabe M. Langmuir. 2007. p. 6438. COI number [1:CAS:528:DC%2BD2sXkt1Oqur0%3D] [DOI] [PubMed]
- Watanabe M. Recent Research Developments in Applied Physics, vol. 6. Transworld Research Network, Kerala; 2003. p. 81. [Google Scholar]
- Sun S, Murray CB, Weller D, Folks L, Moser A. Science. 2000. p. 1989. COI number [1:CAS:528:DC%2BD3cXhvF2rt7g%3D]; Bibcode number [2000Sci...287.1989S] [DOI] [PubMed]
- Wang Y, Ding B, Li H, Zhang X, Cai B, Zhang Y. J. 2007. p. 108. COI number [1:CAS:528:DC%2BD28XhtVOntLvE]; Bibcode number [2007JMMM..308..108W] [DOI]
- Takamura H, Sugai H, Watanabe M, Kasahara T, Kamegawa A, Okada M. J. 2006. p. 741. COI number [1:CAS:528:DC%2BD2sXns1eisg%3D%3D] [DOI]
- Hada H, Yonezawa Y, Yoshida A, Kurakake A. J. 1976. p. 2728. COI number [1:CAS:528:DyaE2sXhtVGqsg%3D%3D] [DOI]
- Kurihara K, Kizling J, Stenius P, Fendler JH. J. 1983. p. 2574. COI number [1:CAS:528:DyaL3sXhs1OksbY%3D] [DOI]
- Bronstein L, Chernyshov D, Valetsky P, Tkachenko N, Lemmetyinen H, Hartmann J, Förster S. Langmuir. 1999. p. 83. COI number [1:CAS:528:DyaK1cXnvValuro%3D] [DOI]
- Powell JA, Logan SR. J Photochem. 1974. p. 189. COI number [1:CAS:528:DyaE2MXpvFCk] [DOI]
- Pola J, Maryško M, Vorlíček V, Bakardjieva S, Šubrt J, Bastl Z, Ouchi A. J. 2008. p. 156. COI number [1:CAS:528:DC%2BD1cXhtVGjtr7J] [DOI]
- Haggerty JS, Cannon WR. Laser-induced Chemical Processes. Plenum Press, New York; 1981. [Google Scholar]
- Kambe N. Scr. 2001. p. 1671. COI number [1:CAS:528:DC%2BD3MXksVSktLY%3D] [DOI]
- Liang C, Shimizu Y, Masuda M, Sasaki T, Koshizaki N. Chem. 2004. p. 963. COI number [1:CAS:528:DC%2BD2cXhsFyhs7Y%3D] [DOI]
- Ishikawa Y, Kawaguchi K, Shimizu Y, Sasaki T, Koshizaki N. Chem. 2006. p. 426. COI number [1:CAS:528:DC%2BD28XptVCgurk%3D]; Bibcode number [2006CPL...428..426I] [DOI]
- Teranishi T, Miyake M. Encyclopedia of Nanoscience and Nanotechnology. American Scientific Publishers, Los Angeles; 2004. [Google Scholar]
- Kishi K, Ikeda S, Hirota K. J. 1967. p. 4384. COI number [1:CAS:528:DyaF1cXktlCqtw%3D%3D] [DOI]
- Ouchi A, Tsunoda T, Bastl Z, Maryško M, Vorlíček V, Boháček J, Vacek K, Pola J. J. 2005. p. 251. COI number [1:CAS:528:DC%2BD2MXjtFOltrw%3D] [DOI]
- Nagano Y, Achiba Y, Kimura K. J. 1986. p. 1288. COI number [1:CAS:528:DyaL28Xhtl2itr8%3D] [DOI]
- Ray U, Hou HQ, Zhang Z, Schwarz W, Vernon M. J. 1989. p. 4248. COI number [1:CAS:528:DyaK3cXjs1SqtA%3D%3D]; Bibcode number [1989JChPh..90.4248R] [DOI]
- Hada H, Yonezawa Y, Yoshida A, Kurakake A. J. 1976. p. 2728. COI number [1:CAS:528:DyaE2sXhtVGqsg%3D%3D] [DOI]
- Pola J, Ouchi A, Bakardjieva S, Urbanová M, Boháček J, Šubrt J. J. 2007. p. 84. COI number [1:CAS:528:DC%2BD2sXht1ajsbjK] [DOI]
- Huang WC, Chen YC. J. 2008. p. 697. COI number [1:CAS:528:DC%2BD1cXisFCqsLY%3D] [DOI]
- Chu HC, Kuo CH, Huang MH. Inorg. 2006. p. 808. COI number [1:CAS:528:DC%2BD2MXhtlWmurjL] [DOI] [PubMed]
- Porel S, Singh S, Radhakrishnan TP. Chem. 2005. p. 2387. [DOI] [PubMed]
- Mwakikunga BW, Forbes A, Sideras-Haddad E, Arendse C. Nanoscale Res. 2008. p. 372. COI number [1:CAS:528:DC%2BD1cXhsVyhtrrE]; Bibcode number [2008NRL.....3..372M] [DOI]
- Nakazato Y, Okuda M, Katayama K. Chem. 2008. p. 271. COI number [1:CAS:528:DC%2BD1cXlvFyiu7s%3D]; Bibcode number [2008CPL...457..271N] [DOI]
- Gangopadhayay AK, Chakravorty A. J. 1961. p. 2206. Bibcode number [1961JChPh..35.2206G] [DOI]
- Gans R. Ann. 1912. p. 881. [DOI]
- Link S, El-Sayed MA. J. Phys. Chem. B. 1999. p. 8410. COI number [1:CAS:528:DyaK1MXlslGntrs%3D] [DOI]
- Kelly KL, Coronado E, Zhao LL, Schatz GC. J. Phys. Chem. B. 2003. p. 668. COI number [1:CAS:528:DC%2BD38Xps1Ghur0%3D] [DOI]