

Abstract
Polycystic liver diseases (PCLDs) are genetic disorders with heterogeneous etiologies and a range of phenotypic presentations. PCLD exhibits both autosomal or recessive dominant pattern of inheritance and is characterized by the progressive development of multiple cysts, isolated or associated with polycystic kidney disease, that appear more extensive in women. Cholangiocytes have primary cilia, functionally important organelles (act as mechanosensors) that are involved in both normal developmental and pathological processes. The absence of polycystin-1, 2, and fibrocystin/polyductin, normally localized to primary cilia, represent a potential mechanism leading to cyst formation, associated with increased cell proliferation and apoptosis, enhanced fluid secretion, abnormal cell–matrix interactions, and alterations in cell polarity. Proliferative and secretive activities of cystic epithelium can be regulated by estrogens either directly or by synergizing growth factors including nerve growth factor, IGF1, FSH and VEGF.
The abnormalities of primary cilia and the sensitivity to proliferative effects of estrogens and different growth factors in PCLD cystic epithelium provide the morpho-functional basis for future treatment targets, based on the possible modulation of the formation and progression of hepatic cysts.
Keywords: Cystic epithelium, Estrogen, FSH, IGF-1, Polycystic liver, Polycystin, Primary cilium, VEGF
1. Introduction
Polycystic liver diseases (PCLDs) are genetic disorders characterized by the progressive development of multiple cysts (Fig. 1) that initially bud from biliary epithelium and subsequently lack communication with the biliary tree [1].
Fig. 1.
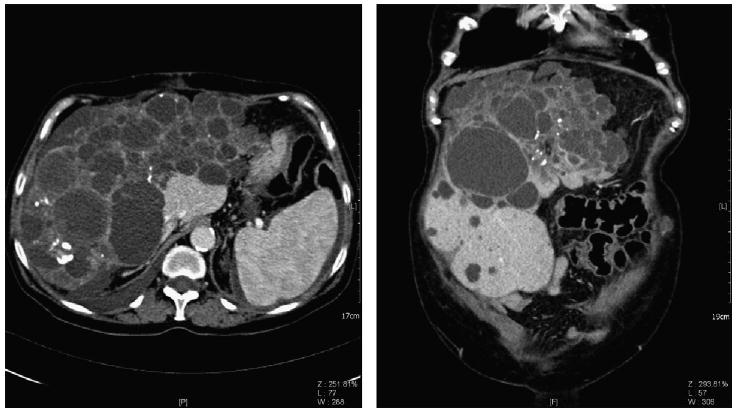
Abdominal computed tomography (axial and frontal orientation respectively) of an adult with ADPCLD showing extensive cyst formation in liver parenchyma (images kindly provided by Prof. C. Catalano and Dr. V. Cardinale).
PCLD is frequent observed in association with the presence of autosomal dominant polycystic kidney disease (ADPKD) or autosomal recessive polycystic kidney disease (ARPKD) [2,3]. ADPKD is the most common life-threatening monogenic human renal disease, with a prevalence of between 1:400 and 1:1000 [4]. It is characterized by progressive development and enlargement of fluid-filled cysts originating from only 3% of nephrons, leading ultimately to renal failure in 50% of affected individuals. More than 85% of ADPKD cases are caused by mutations in the PKD1 gene, with almost all remaining cases associated with PKD2 gene mutations [5] (Fig. 2). Liver cysts are the most common extra-renal manifestation of ADPKD [6] and are found in 60–75% of ADPKD patients on dialysis [7]. They appear at a later age than renal cysts but are most extensive in subjects with the worst renal function and most severe renal cystic disease [8]. Although the development of liver failure in ADPKD is unusual, cystic liver disease is responsible for significant morbidity and accounts for 10% of deaths of ADPKD patients on dialysis [7]. Severe cystic liver disease in ADPKD primarily affects females and is more extensive in women who have had many pregnancies or prior exposure to female sex hormones [8]. Partial hepatectomy is performed as a palliative procedure for such patients [9].
Fig. 2.
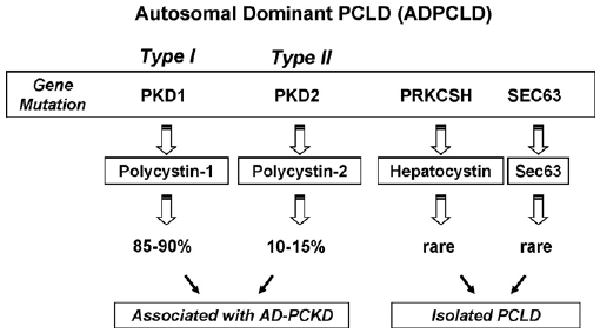
Diagram of the genetics of polycystic liver disease in its autosomic dominant form, with mutation on the genes PKD1, PKD2, PRKCSH and SEC63.
ARPKD is a rare condition that occurs in 1:40,000 live births [10] with a high mortality rate. In surviving patients liver disease is the major cause of morbidity and mortality and is characterized by biliary dysgenesis associated with congenital hepatic fibrosis (CHF), bile duct dilatation, and cyst formation [11]. It is caused by mutations of the polycystic kidney and hepatic disease 1 gene (PKHD1), which encodes the protein fibrocystin/polyductin [12,13] (Fig. 3). The typical disease presentation is of greatly enlarged kidneys detected in utero or in the perinatal period, with 30% of cases dying shortly after birth because of respiratory difficulties due to enlarged kidneys [14]. The most common complications of ARPKD include hypertension (60–100%), portal hypertension owing to severe hepatic fibrosis or Caroli's disease (30–75%), and chronic lung disease (approximately 11%) [15]. In addition, growth retardation intracranial aneurysms [16] and adrenal insufficiency [17] can be seen in patients with ARPKD.
Fig. 3.
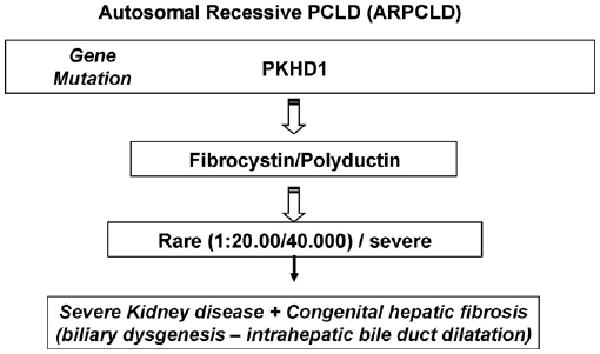
Diagram of the genetic of polycystic liver disease in its subtype autosomic recessive linked to modifications in the gene PKHD1 that codes for the protein fibrocystin.
In general, hepatic cysts are rare in children. Their frequency increases with age and may have been underestimated by ultrasound and CT studies [18]. Normally, PCLD is asymptomatic, but it becomes more frequent as the lifespan of ADPKD patients has lengthened with dialysis and transplantation. Symptoms may result from mass effect or from complications related to the cysts. They typically caused by massive enlargement of the liver and include dyspnoea, early satiety, gastro-oesophageal reflux and mechanical low back pain. Other complications caused by mass effect include hepatic venous outflow obstruction, inferior vena cava and portal vein compression, or bile duct compression. Symptomatic cyst complications include cyst haemorrhage, infection, and rarely torsion or rupture [19].
2. Polycystic liver disease: genetics
PCLD demonstrates in two main clinical presentations: (i) polycystic liver associated with autosomal dominant polycystic kidney disease and (ii) isolated polycystic liver disease. Both of these forms of polycystic liver disease exhibit an autosomal dominant pattern of inheritance [1]. Autosomal dominant PCLD (ADPLD) displays no renal involvement when is caused by a mutation of the gene protein kinase substrate 80K-H (PRKCSH), which encodes the protein hepatocystin [20]. Whereas ADPLD shows renal involvement when is linked to mutations in PKD1, encoding polycystin-1 (PC1) or PKD2, encoding polycystin-2 (PC2) [21]. In PCLD associated with polycystic kidney disease, cell proliferation is one of the major mechanisms of cystogenesis, whereas in isolated PCLD, disrupted cell adhesion may be more important in cyst progression [22]. In summary, the polycystic liver and kidney diseases are disorders with heterogeneous etiologies and a range of phenotypic presentations. ADPKD is associated with renal and liver cystogenesis that clinically manifests in adulthood, often leading to dialysis and renal transplantation. Regarding the genotype–phenotype correlation in ADPKD, several studies in adults have shown that patients with PKD1 mutation have more renal cysts, larger cysts, more prevalent hypertension and faster progression to end-stage of renal disease than the patients with PKD2 mutation. Similarly, in children with PKD1 mutation, more and larger renal cysts have been found than in children with PKD2 at a similar age [23].
ARPKD can present in neonates with massive renal cysts, causing respiratory failure secondary to abdominal competition that subsequently leads to infant demise, although milder forms can present later in life. Whilst ARPKD is always found in conjunction with CHF, it is less commonly associated with hepatic cystogenesis [11,24]. Whereas the products of PKD1, PKD2, and PKHD1 are present in multiple subcellular locations, one common denominator that they all share is expression in the cilium. This has led to the theory that cystogenesis is due, at least in part, to ciliary dysfunction [25].
3. Function of the polycystin proteins
3.1. Polycystin-1 (PC1)
PC1 is a 4302-amino acid type I membrane glycoprotein containing a long N-terminal extracellular domain of over 3000 amino acids, 11 transmembrane domains and a short cytoplasmic C-terminus (198 amino acids) [26]. PC1 interacts with PC2 via its C-terminus to form a heterodimeric complex. This primary interaction is critical for the functional regulation of both proteins [27]. A direct role of PC1 in mediating cell–cell adhesion via homophilic interactions of the PKD domains has been demonstrated [28]. Of interest, the entire N-terminal ectodomain has the biomechanical properties of a mechanosensor [29]. Potentially it could sense laminar flow or the force of coupling between cells or cell–matrix attachments during tubulogenesis. These force-triggered events could in turn activate a PC2-dependent Ca2+ signal and/or PC1-dependent signals. In addition to Ca2+, the latter could include activation of monomeric GTPase proteins (Ras, Rho), heterotrimeric G proteins (Gi and Go), JAK/STAT, phosphatidylinositol 3-kinase (PI3-K)/Akt/mammalian target of rapamycin (mTOR) and activator protein 1 (AP-1)/mitogen-activated protein kinases (MAPK) pathways [30].
3.2. Polycystin-2 (PC2)
PC2 is a Ca2+ channel of 968-amino acid type II membrane glycoprotein with six transmembrane domains and intracellular N- and C-termini. PC2 (or TRPP2) has significant homology to the transient receptor potential (TRP) family of store-operated calcium channels and is likely to function similarly as a non-selective calcium channel [31]. The subcellular location of PC2 has been an area of controversy with immunolocalisation to the endoplasmic reticulum (ER), lateral plasma membrane and primary cilia. A second phosphorylation site for PC2 within its N-terminal domain (Ser 76) is critical for its localisation in the lateral plasma membrane but not in the primary cilia [32].
3.3. Hepatocystin and SEC63
Two separate genes, PRKCSH and SEC63, have been identified to cause familiar PCLD. Multiple cysts arise from progressive dilatation of abnormal ducts in biliary hamartomas [33], the result of a ductal plate malformation at the level of the small intrahepatic bile ducts. These ducts have lost continuity with the remaining biliary tree, which explain the non-communicating nature of the cysts in polycystic liver disease [34]. PRKCSH is located on chromosome 19p13.2 and encodes hepatocystin [35]. The exact function of this protein remains to be elucidated, but it is thought that it acts as the regulatory subunit of glucosidase II and is involved in the folding and quality control of newly synthesized glycoproteins [20]. SEC63 is located on chromosome 6q21 and encodes SEC63p, which is probably involved in the protein translocation machinery in the end oplasmic reticulum [36,37]. PCLD is different from ADPKD at both the phenotype and genotype level. Patients with PCLD do not have polycystic kidneys, whereas up to 60% of patients suffering from ADPKD may have polycystic livers in addition to polycystic kidneys [8].
3.4. Fibrocystin/polyductin
Fibrocystin/polyductin (FPC) is an integral membrane protein with unknown functions, encoded by PKHD1. This gene consists of at least 86 exons spanning 470 kb on chromosome 6p12 and produces a 16-kb transcript. The longest open reading frame is predicted to include 66 exons and to encode the 4074–amino acid membrane-associated receptor-like protein FPC [13,38]. FPC has a signal peptide, and structural predictions indicate a large extracellular region with multiple copies of the TIG domain (an immunoglobulin-like fold), a single transmembrane region, and a short cytoplasmic tail [39]. A homologous gene, not involved in renal cystic disease, PKHDL1, has recently been described [40]. It was shown that FPC is associated with the basal bodies/primary cilia of epithelial cells [41] and co-localizes with PC2 within the cell [42]. These observations suggest the possibility that FPC and PC2 may function in a common molecular pathway in vivo.
4. The primary cilium
The primary cilium is a common, solitary, non-motile, long, tubular organelle extending from the apical plasma membrane of the cell [43–45]. Cholangiocytes are ciliated cells that show a primary cilium consisting of the microtubule-based axoneme that has a 9 + 0 pattern and the basal body, a centriole-derived, microtubule-organizing centre, from which the axoneme emerges [46]. Cholangiocytes cilia extending from the apical plasma membrane into the bile duct lumen are ideally positioned to detect changes in bile flow, composition and osmolarity [46] (Fig. 4). In mouse biliary tree the large bile ducts the ciliary axonemes are 7.35 ± 1.32 μm in length, approximately two times longer than in the small bile ducts with a inner diameter of 50 μm [47]. Several lines of evidence suggest that the major physiologic role of primary cilia is to function as sensory organelles.
Fig. 4.
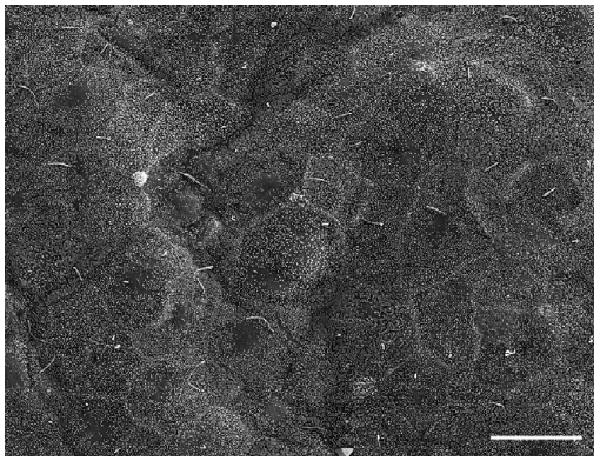
SEM of luminal surface of a small hepatic cyst of ADPKD liver. The epithelium lining small cyst (1 cm maximum diameter) shows a carpet of regular microvilli and typical primary cilia comparable with a normal biliary epithelium. Bar 10 μm.
In fact in recent years it has been proposed that primary cilia sense and transduce multiple stimuli, such as fluid flow, signals initiated by hormones, morphogens, growth factors and other physiologically active substances present [48,49]. Changes in flow are communicated to other cellular response elements via changes in intracellular Ca2+ and cAMP concentrations. Increased flow causes a cilium-dependent rise in intracellular Ca2+ followed by a decrease in cAMP via a Ca2+ inhibitable adenylyl cyclase (AC6) that, interestingly, is also localized within the cilium [50]. These data also suggest that components present in cyst fluid that bathes the apical surface may modulate electrolyte and compensatory fluid secretion into the cyst [51]. Interest of biomedical scientists in these organelles has increased only recently triggered by three critical observations; (i) primary cilia in the node of gastrulation-stage embryos are essential for the determination of left-right asymmetry of the body [52]; (ii) the two most frequent lethal genetic disorders (i.e. ADPKD and ARPKD), are cilia-related diseases [24,53,54]; and (iii) Bardet–Biedl syndrome (patients with kidney failure, loss of eyesight, obesity and diabetes) is a result of mutations in genes which determine ciliary structure and function [55,56]. Thus, it has become evident that primary cilia are functionally important organelles that are involved in both normal developmental and pathological processes.
PC1 and PC2 are normally localized to primary cilia. FPC is also localized to primary cilia. When these genes are mutated, the absence of their protein products results in ciliary dysfunction and cyst formation. Defects in ciliary structure and their integrated sensory/transducing functions appear to result in decreased [Ca2+]i and increased cAMP, causing cholangiocyte hyperproliferation, abnormal cell–matrix interactions, and altered fluid secretion/absorption, capable of resulting in cystogenesis [46].
Recently, Torrice et al. have been showed the central role of PC1 and PC2 in the modulation of cholangiocyte growth suggesting that primary cilia may act as sensors of cell injury activating a proliferative response to trigger the reparative processes. Moreover, they demonstrated that different pharmacological agents may block cholangiocyte proliferation by affecting different pathways of PC1 activation, degradation and processing. These agents are currently under evaluation in clinical trials for different pathological conditions caused by intracellular accumulation of pathological proteins [57].
In recent years, considerable new information has been generated regarding the physiological functions of primary cilia in renal epithelia [58,59]. In MDCK cells, a cultured cell line derived from the collecting duct of canine kidney, bending of a single cilium by a micropipette or by alterations in perfusate flow rates increases [Ca2+]i, whilst pharmacological removal of cilia abolishes the flow-induced [Ca2+]i increase [60,61]. Finally, an acute increase in tubular fluid flow rates in microperfused cortical collecting ducts of the rabbit and mouse nephrons also led to an increase in [Ca2+]i with the involvement of primary cilia [59]. In rat liver, mutations in genes encoding ciliary-associated proteins cause a broad spectrum of clinically and genetically heterogeneous human disorders, referred to as ciliopathies [62]. Ciliopathies affecting liver may more appropriately be called “cholangiociliopathies” [22] since cholangiocytes are the only epithelial cells in the liver that contain primary cilia. Based on these observations and given the importance of both [Ca2+]i and cAMP signalling in biliary epithelia, cholangiocyte cilia may also act as mechanosensors that monitor and transmit luminal bile flow stimuli into integrated intracellular Ca2+ and cAMP signalling [63–65]. The mechanosensory function of cholangiocyte cilia was addressed directly by using a microperfused rat intrahepatic bile duct unit model (IBDU) that allows controlled manipulation of luminal fluid flow rates, detection of both [Ca2+]i and cAMP levels [66,67].
The morphology of cholangiocyte cilia has been elucidated by SEM demonstrating that apical surface of cyst epithelium showed heterogeneous abnormalities depending on cyst size. In rodent models of PKD, diverse changes in cilia structure have been noted. These include shortened, malformed cilia as well as cilia which are unusually long or entirely absent [68]. In BALB/c-cpk/cpk mouse, cystic epithelium shows a range of ciliary length, both shorter and longer, compared to those seen in wild-type ductal epithelia. This may contribute to the observed differences in cellular morphology and/or cellular ion transport observed in the cystic epithelia. Regarding the ion transport, Muchatuta et al. have used, for the first time, an ex vivo tissue to demonstrate ion secretory flux, which is consistent with cyst enlargement via compensatory fluid movement, suggesting that the components of the cyst fluid may contribute to cyst enlargement [51]. In humans, the epithelial cells lining small cysts show an apical surface with adequately represented cilia and which is similar to normal biliary epithelium. In contrast, the apical surface of medium cysts show rare and shortened cilia (Fig. 5). These features further worsen in large hepatic cysts that totally lack primary cilia. Probably, these morphological abnormalities of primary cilia are linked to a mechanical consequence of enhanced endoluminal pressure during cyst growth [69].
Fig. 5.
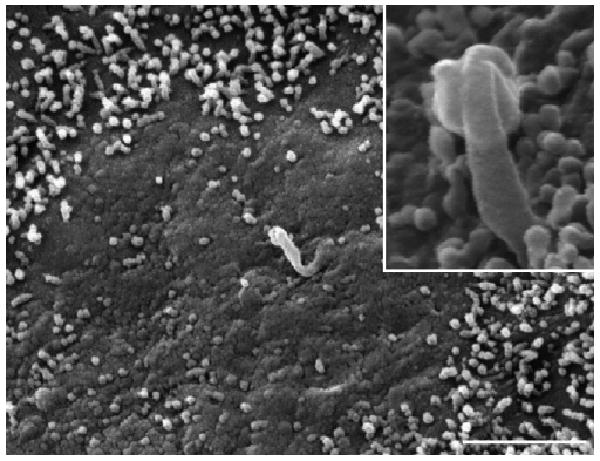
SEM of epithelial surface of a cyst with a 2–2.5 cm maximum diameter shows less dense microvilli and clear areas become visible. The cilium is absent or shorter than the typical structure and often showed alteration of the apical zone. In the box, at higher magnification a short and abnormal cilium is evident. Bar 2 μm.
5. Cystogenesis
Human and experimental data suggest several potential mechanisms that could lead to cyst formation: (i) increased cell proliferation and apoptosis; (ii) enhanced fluid secretion; (iii) abnormal cell–matrix interactions; (iv) alterations in cell polarity, and (v) abnormal ciliary structure or function.
A primary increase in tubular proliferative activity, e.g. through the transgenic expression of oncogenes or growth factors, can lead to cyst formation [70]. PKD1 cystic epithelial cells are more sensitive to the mitogenic effect of growth factors and cAMP in vitro—these effects are dependent on Ras and Raf activity [71]. In addition, in patients with ADPKD, the tubular epithelium appears to switch from an absorptive to a secretory phenotype [72]. ADPKD cyst epithelia sit on an expanded basement membrane and abnormal increases in basement membrane components (laminin, fibronectin, type IV collagen, and heparan sulphate proteoglycan), and interstitial type I collagen have been reported. These changes in basement membrane composition could contribute to cyst initiation or expansion. Many cystoproteins have since been immunolocalised to primary cilia or centrosomes (which give rise to primary cilia) and some have been associated with structural or functional abnormalities of these organelles. But cilia are [73], among other things, signal transducers capable of sending messages via a variety of pathways, the aforementioned discovery of mutations in genes encoding ciliary proteins has led to speculation that abnormal ciliary function leads to disordered signal transduction, altered gene expression, and/or alterations in cell–cell and cell–matrix adhesion [74].
Furthermore, the type of dramatic cystic degeneration seen in human disease is only evident in mice in which both PKD alleles are disrupted. By contrast, heterozygous PKD KO mice develop only a few cysts by adulthood. These observations were reconciled by the demonstration that “two hits” are required for development of the cystic phenotype [75]. Although ADPKD is phenotypically autosomal dominant, at the cellular level it is likely to be a “molecular recessive” disease, since it requires a second somatic mutation. The “two hits” model needs an initial germline mutation in either PKD1 or PKD2 (first hit), followed by a somatic mutation in the remaining allele, that initiates cell proliferation and cyst formation in the individual cell that have received the second hit [76,77]. Since the second hit would be predicted to be a rare and stochastic event, affected cells would probably be few and far between and cysts would be borne from individual cells rather than epithelial fields. However, more recent studies in chimeric mice suggest that part of the cyst wall can be formed by epithelial cells retaining the heterozygous state, raising the possibility that environmental cues elaborated by one group of cells may influence the phenotype of neighbouring cells in the nephron [78] (Fig. 6). In experimental models, homozygous PKD1 or PKD2 null mice die shortly after birth. Whilst, heterozygous animals with a recombination-sensitive second allele, that allows development of spontaneous null mutations, results in age-related formation of renal and liver cysts, similar to human disease [3]. This model has not been investigated in isolated PCLD.
Fig. 6.
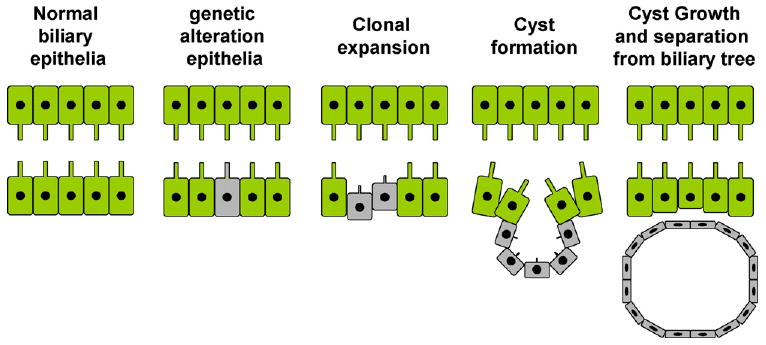
Diagram depicting the genetic basis of cyst formation in ADPCLD. The biliary epithelium in a normal person has both alleles of PKD genes. In patients with ADPCLD, there is a germ-line mutation of one of the alleles (‘first hit’). Some cells within the duct acquire a mutation in the second allele (‘second hit’), resulting in increased proliferative activity in these cells, leading ultimately to cyst formation. The cells lining these cysts are monoclonal and are homozygous for PKD mutation.
6. Experimental models
Different animal models are using to study the polycystic liver disease, in particular about ARPKD have been developed, over time; many of them display distinct liver pathology [79,80]. The cpk model was the first to be described [81], and probably the most extensively characterized. Mutants develop massive renal cystic disease and progressive renal insufficiency in a pattern that strongly resembles human ARPKD. The BALB/c polycystic kidneys (bpk) mutation arose spontaneously on the BALB/c inbred background. Like cpk, the bpk mutation is transmitted as a fully penetrant, recessive trait [82]. Affected homozygotes develop both cystic dilatation of the renal collecting ducts as well as biliary dysgenesis, and the genetic background modulates disease progression. Death ensues within 4 wk of birth, presumably due to renal insufficiency. The extra-renal defects in the BALB/c-cpk/cpk mice included pancreatic cystic dysplasia, and intrahepatic biliary duct cysts with periductal hyperplasia. The identification of the human ADPKD genes, PKD1 and PKD2, prompted the characterisation and targeted mutagenesis of their mouse orthologs, Pkd1 and Pkd2. Both null and hypomorphic alleles have been generated [83,84]. Heterozygous mice develop renal, biliary, and pancreatic cysts between 4 and 19 mo of age. Homozygous mutants develop renal and pancreatic cysts, coincident with the induction of Pkd1 and Pkd2 expression in normal maturing tubular epithelia. Disease progression is rapid, with embryonic lethality occurring in most homozygous mutants. These data, demonstrating that loss of Pkd1 or Pkd2 is sufficient to cause renal cysts, support the two-hit model of cystogenesis proposed for ADPKD [79].
Compared with the mouse, where different genetic mechanisms have given rise to a large number of PKD models, only a few heritable rat PKD models have been described. The Han:SPRD-cy rat is well characterized and has been studied extensively as a model of ADPKD [85]. The mutation arose spontaneously in the Sprague–Dawley strain, and initial analysis indicated inheritance as an autosomal dominant trait. In heterozygotes, the renal cystic lesion is evident within the first few weeks of life, primarily involves the proximal tubules, and progresses slowly. The Wistar polycystic kidneys (wpk) mutation arose spontaneously in an outbred Wistar strain [86]. Homozygous mutants develop nephromegaly, hypertension, proteinuria, impaired urinary concentrating capacity, and uremia, resulting in death at 4 wk of age, developing cysts at E19. Whilst wpk mutants exhibit renal histopathology that is strikingly similar to human ARPKD, the biliary ductal plate malformation invariably associated with the human disease is not evident [79,86]. The gene, Pkhd1, that causes kidney and liver disease in these rats is orthologous to the human PKHD1 [13]. But, more important, the development of the PCK rat was initiated by sibling mating of the female offspring, and continuous sibling mating since 1996 has led to the establishment of this rat model, which is now in its twelfth generation. In a preliminary study with mating experiments, Sanzen et al. [80] described that the liver pathology in the PCK rats up to 4 months of age was characterized by progressive liver enlargement and multiple saccular and segmental dilatations of the intrahepatic bile ducts. Under normal conditions, FPC is expressed in primary cilia of cholangiocytes. In the PCK rat, a splicing mutation of Pkhd1 results in structural and functional ciliary abnormalities. These modifications may lead to abnormalities in cell proliferation and biliary tree differentiation, ultimately resulting in significant bile duct dilatation and cyst formation [87]. PCK rat has several features that resemble human ARPKD, as in the human, polycystic disease in the PCK rat is inherited in an autosomal recessive manner and severity of liver cystic lesions is age dependent.
Studies of the mechanisms of liver cyst formation in ARPKD have been limited by the lack of suitable cell lines. Thus, to explore the mechanisms of cyst growth and expansion in ARPKD, it has been developed a cholangiocyte cell line derived from the intrahepatic bile ducts of the PCK rat which is designated PCK-CCL (i.e. PolyCystic Kidney Cholangiocyte Cell Line) [88]. Perrone et al. have also developed the first human cell lines from ADPKD patients [89]. These liver cyst-derived epithelial (LCDE) cell lines display the distinctive features of biliary epithelium and lack of expression of hepatocyte specific markers. LCDE cells exhibit pH regulatory pathways that are similar to those found in normal intrahepatic biliary epithelium. Impaired alkalinisation in response to Cl− substitution is suggestive of decreased function or abundance of a Cl−/HCO, anion exchanger and could account for the failure of ADPKD hepatic cysts to secrete HCO, in response to secretin [90] (Table 1).
Table 1.
Experimental models used to study polycystic liver and kidney diseases.
| Models | Gene | Protein | Human PKD |
|---|---|---|---|
| cpk | Cys1 | Cystin | AR-PKD |
| bpk | Bicc1 | Bicaudal C | AR-PKD |
| jcpk | Bicc1 | Bicaudal C | AD-PKD |
| orpk | TgN737Rpw | Polaris | AR-PKD |
| inv | Invs | Inversin | AR-PKD |
| jck | Nek8 | Nek8 | AD-PKD |
| kat | Nek1 | Nek1 | AD-PKD |
| pcy | NI | NI | AD-PKD |
| Han:SPRD-cy | NI | NI | AD-PKD |
| wpk | NI | NI | AR-PKD |
| pck | Pkkd1 | Fibrocystin | AR-PKD |
NI: not identified.
AD-PKD: autosomic dominant polycystic kidney disease.
AR-PKD: autosomic recessive polycystic kidney disease.
7. Regulator factors in PCLD growth
The presence of an aberrant vascularisation is associated with cyst formation in liver, kidney and lung [91,92] where angiogenesis appears to regulate the development of epithelial and endothelial tissues [92,93]. In fact, treatment of human microvascular endothelial cells (HMEC-1) with human liver cyst fluid (huLCF) induced a rapid increase in vascular endothelium growth factor receptor 2 (VEGFR2) phosphorylation, indicating that factors secreted by liver cyst epithelia can activate VEGF signalling pathways and induce endothelial cell proliferation and differentiation [93]. Many reports showing expression of VEGF by rat cholangiocytes [94–99] suggesting that angiogenic factors, a series of multifunction cytokines capable to regulate the morphogenesis and growth of the vascular system [100] might play a role in cyst development and growth. Cholangiocyte co-expression of VEGF, angiopoietins and their cognate receptors, the strong correlation between VEGF expression on abnormal biliary structures and the surrounding vasculature [97] in ADPKD indicates that the aberrant production of angiogenic factors by biliary cysts may, on one side, promote the vascularisation of the cysts through a paracrine effect on endothelia, and, on the other side, stimulate the growth of biliary cysts through an autocrine effect on cholangiocytes. For that reason, it has been also suggested that VEGF inhibitors might be used to treat patients with PCLD [101]. Previous morphological studies underlined the immature aspect of the cystic biliary epithelium characterized by the expression of VEGF, VEGF receptors as well as Tie-2 on cystic epithelium of ADPKD, since they are features indicating the lack of maturation of the biliary epithelium in this developmental cholangiopathy, together NCAM, an adhesion molecule typically present in immature cholangiocytes and strongly expressed by the cystic epithelium [102]. In addition to that, it has been showed that PC1 and PC2 are localized in cholangiocyte cilia where they may be involved in the regulation of bile secretion as mechanoreceptors [103] and also influence the transcription of a number of regulatory proteins. A speculation may be that altered function of polycystins in cholangiocyte cilia can cause a lack of differentiating signals favouring the maintenance of an immature phenotype by biliary epithelial cells. This is consistent with the hypothesis that in the presence of polycystin mutations, the transcription of a number of foetal genes fails to be switched off and thereby allows the postnatal expression of developmental proteins [104]. Accumulated evidence suggests that two intracellular signalling mediators cAMP and Ca2+, regulate proliferation in different cell types [105–107], including cholangiocytes [97,108,109]. Thus, cAMP levels are increased in cholangiocytes of the PCK rat and octreotide, a somatostatin analogue known to inhibit cAMP, decreases hepatic cyst volume, hepatic fibrotic scores, and mitotic indices [110]. Another somatostatin analogue has been studied, lanreotide, that after 6 months of treatment is able to reduce liver volume in patients with PCLD. It has also been shown that in renal cells from ADPKD patients, intracellular Ca2+ modulates cAMP-dependent proliferation. Moreover, these cells were characterized by decreased [Ca2+]i and exhibited higher rates of cell proliferation in response to cAMP, whilst experimental restoration of [Ca2+]i inhibited their cAMP-stimulated growth [105]. FPC, the protein mutated in ARPKD, is known to form functional complexes within primary cilia with proteins involved in intracellular Ca2+ signalling, such as PC2 and Ca2+-modulating cyclophilin1 ligand, suggesting a possible role for FPC in Ca2+ signalling [111]. In fact, preincubation of kidney cells with antibodies against FPC was reported to abolish a flow-induced increase in [Ca2+]i [112]. Disappearance of FPC from primary cilia was observed in cholangiocytes from the PCK rat and also in renal cysts of ARPKD patients [103]. Thus, the absence of FPC from cilia may lead to its inability to form the aforementioned functional complexes and, subsequently, to maintain baseline [Ca2+]i in cystic cells. Furthermore, other Ca2+ channels present in cholangiocyte cilia (such as TRPV4) may also affect the influx of extracellular Ca2+ into the cells [113,114]. In addition to the intracellular pathways involved in this cystic growth, a number of studies have implicated or demonstrated the presence of a several specific cytokines and growth factors within human liver cyst fluid, which can support its development [115]. Recently, it has been investigated the protein composition of hepatic cyst fluid in an orthologous animal model of ARPKD, heterozygous (BALB/c-cpk/1) mice by a proteomic analysis of cyst fluid to identify 303 proteins, related with enhanced inflammatory cell processes, cellular proliferation, and basal laminar fibrosis associated with the growth of hepatic bile duct cysts [116]. Cytokines and growth factors secreted by the cyst-lining epithelia have been suggested to initiate autocrine/paracrine signalling and promote cyst growth. CXCR2 agonists, including IL-8, epithelial neutrophil-activating peptide (ENA-78), growth-related oncogene-α (GRO-α), are potent proliferative agents that were found at high levels in liver but not kidney cyst fluids [115] (Table 2). CXCR2 agonists were of specific interest since they can not only attract inflammatory cells into diseased tissues [117] but can also promote angiogenesis in the absence of preceding inflammation [118] and induce proliferation of epithelial cells [119]. Furthermore, recent evidence indicates that the intrahepatic biliary epithelium is the target of GH/insulin-like growth factor 1 (IGF1) axis [120] and is particularly sensitive to the proliferative effects of IGF1 and estrogens [121–123]. Estrogens exert their proliferative effects either directly or by synergising growth factors including nerve growth factor and IGF1. In addition, estrogens induce the expression and secretion of growth factors (IGF1, VEGF) by epithelial cells [124] (Fig. 7). A number of clinical observations suggest that estrogens play a role in development and progression of hepatic cysts in ADPKD. In fact, women with ADPKLD have more possibility to develop hepatic cysts with respect to men [125]. Nulliparous women have less probability to develop hepatic cysts with respect to pluripare [8]. Finally, oral contraceptives or postmenopausal hormonal substitutive therapy increases the number and size of the hepatic cysts [126]. In the same context, previous data provide the first direct evidence of an obligatory role for FSH in the induction of ovarian follicular cysts in the rat and indicate that unabated, combined stimulation by FSH and LH/hCG is sufficient for the induction of ovarian cysts in this animal. Further, the observation that pre-treatment with hCG prior to FSH + hCG injections had no effect on the timing of the onset of follicular cyst development suggests that FSH stimulation results in the expression of an ovarian factors that plays an obligatory role in the induction of follicular cysts [127]. To evaluate directly the role of estrogens, VEGF, FSH, IGF1 and their receptors in the modulation of proliferation, our group have used the immortalized cell line LCDE obtained from the epithelium of liver cysts of ADPKD patients (Fig. 7). LCDE cells constitutively express ER-α and -β, IGF1, IGF1-R, FSH and FSHR (Fig. 8). 17β-Estradiol, IGF1 and FSH induce a rate of proliferation which was completely inhibited by two highly specific ER (Ici 182,780), IGF1-R (α-IR3) antagonists [128] and partially inhibited by pre incubation with PD98059 (a MEK inhibitor), that interfere with the cAMP pathway activated by FSH. Although IGF1-induced proliferation was completely inhibited by the specific IGF-R blocking antibody (α-IR3), that induced by 17β-estradiol was only partially inhibited by ER antagonist, Ici 182,780, which also partially blocked the proliferative effect of IGF1. Conversely, 17β-estradiol-induced proliferation was also partially inhibited by the IGF1-R antagonist and completely blocked only when LCDE cells were exposed to both ER and IGF1-R blockers. This indicates that IGF1-R is directly involved in estrogen-induced proliferation because the specificity of α-IR3 for IGF1 is absolute and nonspecific interference of this blocking antibody could be excluded [69]. In the end, ADPKD epithelium is sensitive to the proliferative effects of estrogens, FSH and IGF1. Estrogens act not only directly but also by promoting the synthesis and release of growth factors from the cyst epithelium. In addition, FSH plays a key role in sustaining cholangiocyte proliferation by an autocrine mechanism mediated by the activation of cAMP-dependent signalling pathways [129] (Fig. 9). In conclusion, hyperproliferation of cystic epithelia is obviously linked to abnormalities in cell cycle progression and also to microRNA expression. Progression of cells through the cell cycle is controlled by a family of dual specificity phosphatases, Cdc25, that activate cyclin-dependent kinases (Cdks) [130]. In PCLD patients and PCK rats, Cdc25A protein (one of the three known isoforms of Cdc25) is overexpressed in cholangiocytes lining liver cysts. Cdc25A upregulation in diseased livers is accompanied by downregulation of one of the miRNAs (i.e. miR-15a). MicroRNAs are small noncoding RNAs that posttranscriptionally inhibit target messenger RNA transcripts via sequence-specific base paring [131]. Thus, overexpression of miR-15a in cystic epithelia inhibits cholangiocyte proliferation [132].
Table 2.
Liver cysts fluid composition.
| IL-8 | IL-6 | ENA-78 | Ang | VEGF | MCP-1 | |
|---|---|---|---|---|---|---|
| ADPCLD | ++ | + | ++ | ++ | + | ++ |
| Non ADPCLD | − | − | − | ++ | + | + |
| ARPCLD | −/+ | −/+ | −/+ | −/+ | −/+ | −/+ |
ADPCLD: autosomic dominant polycystic liver disease; ARPCLD: autosomic recessive polycystic liver disease; IL: interleukin; ENA; epithelial neutrophil-activating peptide; Ang: angiotensin; VEGF: vascular endothelial growth factor; MCP: monocyte chemotactic protein-1.
Fig. 7.
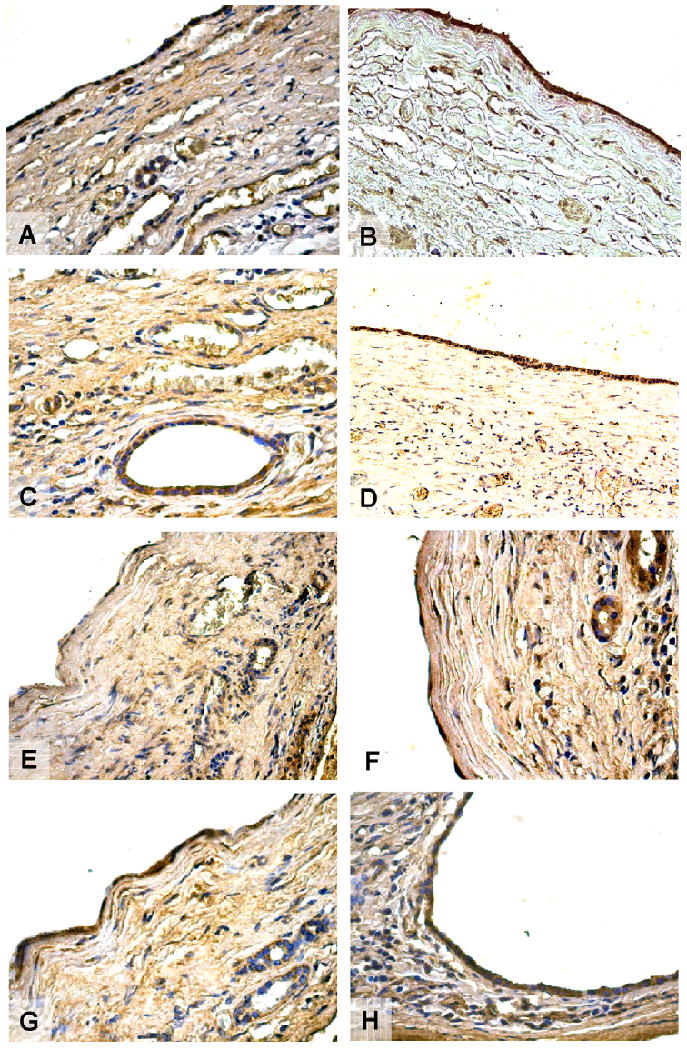
Immunohistochemistry for ER-α (A), ER-β (B), IGF-1 (C), IGF1-R (D), FSH (E), FSHR (F), VEGF-A (G), and VEGF-C (H) in hepatic cysts from patients with ADPCLD. Cholangiocytes of reactive bile ducts close to the cysts are positive for both estrogen receptors. The epithelium lining small and large cysts showed strong positivity for ER-α and ER-β located at cytoplasmic levels. Both IGF1 and its receptor showed positive immunolocalisation in biliary epithelium. The expression of the hormone FSH is also present in both small and large cysts. A stronger immunolocalisation is found for FSHR in the biliary epithelium that lines hepatic cysts. Furthermore, biliary epithelium of small and large cysts shows an high positivity for VEGF-A and VEGF-C, that play an important role in the growth of cysts and in the progression of PCLD. Original magnification 20 ×.
Fig. 8.
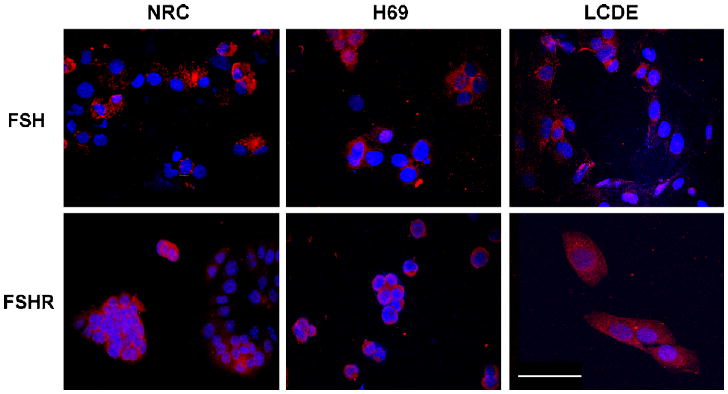
Immunofluorescence in immortalized cell cultures from normal rat cholangiocytes (NRC), normal human cholangiocytes (H69) and cholangiocytes from hepatic cysts (LCDE) shows the presence of positivity for FSH and FSHR. Original magnification 40×. Bar 200 μm.
Fig. 9.
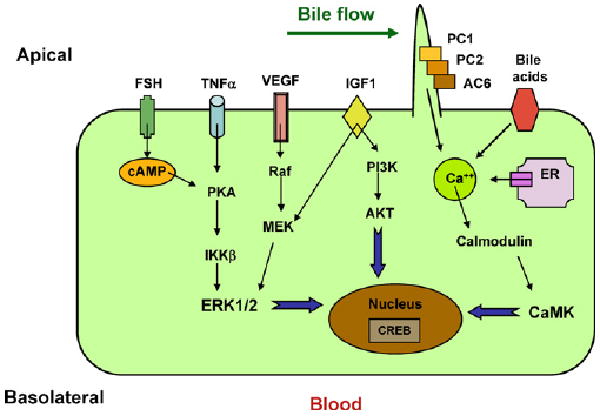
Diagram depicting putative pathways in polycystic liver disease. Dysregulation of [Ca2+]i, increased concentrations of cAMP and upregulation of FSH, IGF1, VEGF, and TNFα occur in cells bearing PKD mutations. Increased accumulation of cAMP in polycystic livers may result from disruption of the polycystin complex, as PC1 may act as a Gi protein-coupled receptor and stimulation of Ca2+ inhibitable AC6. Increased cAMP levels contribute to cystogenesis by stimulating chloride and fluid secretion. In addition, cAMP stimulates mitogen-activated protein kinase/extracellular regulated kinase (MAPK/ERK) signalling in cyst-derived cells.
8. Treatment targets: future perspectives
Therapy for PCLD is invasive, expensive and has disappointing long-term results. In view of its stimulatory effects on cell proliferation and fluid secretion, a major approach has been to target cAMP production. Vasopressin antagonism was associated with a decrease in kidney cAMP concentrations. The role of vasopressin action in modifying cyst expansion in ADPKD has been given added weight by recent studies which indirectly suppressed (high fluid intake) [133] or enhanced its action (endothelin B receptor blockade) [79] on the collecting duct. Similarly, somatostatin treatment (which can inhibit cAMP-stimulated chloride secretion) led to a slowing in the rate of cyst volume expansion in a small study of ADPKD patients and in PCK rats [134]. In fact, it has been demonstrated that treatment with lanreotide a somatostatin analogue, slowed kidney growth in patients with PKD and reduced liver and kidney volume in a PKD rodent model [135]. Seen the sensitivity of cystic epithelium to the proliferative effects of estrogens, IGF1 and angiogenic factors, another therapeutic strategy might be the use of specific inhibitors of these growth factors or the modulation of the same receptors to block ADPKD liver cyst vascularisation and growth. Other features associated with cyst enlargement such as cystic epithelium apoptosis, inflammation or interstitial fibrosis could be alternative treatment targets, as well as miRNAs: for example overexpression of downregulated miRNAs or knock down of overexpressed miRNAs may help reverse many abnormal cellular functions.
Acknowledgments
This work was supported by University Federate Athenaeum funds from University of Rome “La Sapienza” and PRIN 2007 to Prof. Gaudio, University funds to P. Onori and the NIH grant DK58411.
Footnotes
Conflict of interest statement: All the authors declare that there are no conflicts of interest.
References
- 1.Everson GT, Helmke SM, Doctor B. Advances in management of polycystic liver disease. Expert Rev Gastroenterol Hepatol. 2008;2:563–76. doi: 10.1586/17474124.2.4.563. [DOI] [PubMed] [Google Scholar]
- 2.Masyuk T, LaRusso N. Polycystic liver disease: new insights into disease pathogenesis. Hepatology. 2006;43:906–8. doi: 10.1002/hep.21199. [DOI] [PubMed] [Google Scholar]
- 3.Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40:774–82. doi: 10.1002/hep.20431. [DOI] [PubMed] [Google Scholar]
- 4.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 5.Xu C, Rossetti S, Jiang L, et al. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2007;292:F930–45. doi: 10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. Mayo Clin Proc. 1990;65:1020–5. doi: 10.1016/s0025-6196(12)65165-9. [DOI] [PubMed] [Google Scholar]
- 7.Grunfeld JP, Albouze G, Jungers P, et al. Liver changes and complications in adult polycystic kidney disease. Adv Nephrol Necker Hosp. 1985;14:1–20. [PubMed] [Google Scholar]
- 8.Gabow PA, Johnson AM, Kaehny WD, et al. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033–7. doi: 10.1002/hep.1840110619. [DOI] [PubMed] [Google Scholar]
- 9.Newman KD, Torres VE, Rakela J, et al. Treatment of highly symptomatic polycystic liver disease. Preliminary experience with a combined hepatic resection–fenestration procedure. Ann Surg. 1990;212:30–7. doi: 10.1097/00000658-199007000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sessa A, Meroni M, Righetti M, et al. Autosomal recessive polycystic kidney disease. Contrib Nephrol. 2001:50–6. doi: 10.1159/000060211. [DOI] [PubMed] [Google Scholar]
- 11.Shneider BL, Magid MS. Liver disease in autosomal recessive polycystic kidney disease. Pediatr Transpl. 2005;9:634–9. doi: 10.1111/j.1399-3046.2005.00342.x. [DOI] [PubMed] [Google Scholar]
- 12.Onuchic LF, Furu L, Nagasawa Y, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–17. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–69. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 14.Roy S, Dillon MJ, Trompeter RS, et al. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol. 1997;11:302–6. doi: 10.1007/s004670050281. [DOI] [PubMed] [Google Scholar]
- 15.Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003;111:1072–80. doi: 10.1542/peds.111.5.1072. [DOI] [PubMed] [Google Scholar]
- 16.Lilova MI, Petkov DL. Intracranial aneurysms in a child with autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2001;16:1030–2. doi: 10.1007/s004670100019. [DOI] [PubMed] [Google Scholar]
- 17.Yonemura K, Yasuda H, Fujigaki Y, et al. Adrenal insufficiency due to isolated adrenocorticotropin deficiency complicated by autosomal recessive polycystic kidney disease. Ren Fail. 2003;25:485–92. doi: 10.1081/jdi-120021162. [DOI] [PubMed] [Google Scholar]
- 18.Bae KT, Zhu F, Chapman AB, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64–9. doi: 10.2215/CJN.00080605. [DOI] [PubMed] [Google Scholar]
- 19.Bleeker-Rovers CP, Vos FJ, Corstens FH, et al. Imaging of infectious diseases using [18F] fluorodeoxyglucose PET. Q J Nucl Med Mol Imaging. 2008;52:17–29. [PubMed] [Google Scholar]
- 20.Drenth JP, Martina JA, Te Morsche RH, et al. Molecular characterization of hepatocystin, the protein that is defective in autosomal dominant polycystic liver disease. Gastroenterology. 2004;126:1819–27. doi: 10.1053/j.gastro.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 21.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–68. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masyuk T, Masyuk A, LaRusso N. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Curr Opin Gastroenterol. 2009;25:265–71. doi: 10.1097/MOG.0b013e328328f4ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fencl F, Janda J, Blahova K, et al. Genotype–phenotype correlation in children with autosomal dominant polycystic kidney disease. Pediatr Nephrol. 2009;24:983–9. doi: 10.1007/s00467-008-1090-9. [DOI] [PubMed] [Google Scholar]
- 24.Tahvanainen E, Tahvanainen P, Kaariainen H, et al. Polycystic liver and kidney diseases. Ann Med. 2005;37:546–55. doi: 10.1080/07853890500389181. [DOI] [PubMed] [Google Scholar]
- 25.Ibraghimov-Beskrovnaya O, Bukanov N. Polycystic kidney diseases: from molecular discoveries to targeted therapeutic strategies. Cell Mol Life Sci. 2008;65:605–19. doi: 10.1007/s00018-007-7362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes J, Ward CJ, Peral B, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–60. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 27.Newby LJ, Streets AJ, Zhao Y, et al. Identification, characterization, and localization of a novel kidney polycystin-1–polycystin-2 complex. J Biol Chem. 2002;277:20763–73. doi: 10.1074/jbc.M107788200. [DOI] [PubMed] [Google Scholar]
- 28.Streets AJ, Newby LJ, O'Hare MJ, et al. Functional analysis of PKD1 transgenic lines reveals a direct role for polycystin-1 in mediating cell–cell adhesion. J Am Soc Nephrol. 2003;14:1804–15. doi: 10.1097/01.asn.0000076075.49819.9b. [DOI] [PubMed] [Google Scholar]
- 29.Qian F, Wei W, Germino G, et al. The nanomechanics of polycystin-1 extracellular region. J Biol Chem. 2005;280:40723–30. doi: 10.1074/jbc.M509650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67:1234–47. doi: 10.1111/j.1523-1755.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 31.Giamarchi A, Padilla F, Coste B, et al. The versatile nature of the calcium-permeable cation channel TRPP2. EMBO Rep. 2006;7:787–93. doi: 10.1038/sj.embor.7400745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Streets AJ, Moon DJ, Kane ME, et al. Identification of an N-terminal glycogen synthase kinase 3 phosphorylation site which regulates the functional localization of polycystin-2 in vivo and in vitro. Hum Mol Genet. 2006;15:1465–73. doi: 10.1093/hmg/ddl070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qian Q, Li A, King BF, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164–71. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 34.Lazaridis KN, Strazzabosco M, LaRusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology. 2004;127:1565–77. doi: 10.1053/j.gastro.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 35.Drenth JP, te Morsche RH, Smink R, et al. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33:345–7. doi: 10.1038/ng1104. [DOI] [PubMed] [Google Scholar]
- 36.Davila S, Furu L, Gharavi AG, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–7. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 37.Drenth JP, Martina JA, van de Kerkhof R, et al. Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol Med. 2005;11:37–42. doi: 10.1016/j.molmed.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 38.Bergmann C, Frank V, Kupper F, et al. Functional analysis of PKHD1 splicing in autosomal recessive polycystic kidney disease. J Hum Genet. 2006;51:788–93. doi: 10.1007/s10038-006-0022-4. [DOI] [PubMed] [Google Scholar]
- 39.Bork P, Doerks T, Springer TA, et al. Domains in plexins: links to integrins and transcription factors. Trends Biochem Sci. 1999;24:261–3. doi: 10.1016/s0968-0004(99)01416-4. [DOI] [PubMed] [Google Scholar]
- 40.Hogan MC, Griffin MD, Rossetti S, et al. PKHDL1, a homolog of the autosomal recessive polycystic kidney disease gene, encodes a receptor with inducible T lymphocyte expression. Hum Mol Genet. 2003;12:685–98. [PubMed] [Google Scholar]
- 41.Wang S, Luo Y, Wilson PD, et al. The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J Am Soc Nephrol. 2004;15:592–602. doi: 10.1097/01.asn.0000113793.12558.1d. [DOI] [PubMed] [Google Scholar]
- 42.Zhang MZ, Mai W, Li C, et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci USA. 2004;101:2311–6. doi: 10.1073/pnas.0400073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wheatley DN. Primary cilia in normal and pathological tissues. Pathobiology. 1995;63:222–38. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- 44.Praetorius HA, Spring KR. A physiological view of the primary cilium. Annu Rev Physiol. 2005;67:515–29. doi: 10.1146/annurev.physiol.67.040403.101353. [DOI] [PubMed] [Google Scholar]
- 45.Davenport JR, Yoder BK. An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol. 2005;289:F1159–69. doi: 10.1152/ajprenal.00118.2005. [DOI] [PubMed] [Google Scholar]
- 46.Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease. Dev Dyn. 2008;237:2007–12. doi: 10.1002/dvdy.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang BQ, Masyuk TV, Muff MA, et al. Isolation and characterization of cholangiocyte primary cilia. Am J Physiol Gastrointest Liver Physiol. 2006;291:G500–9. doi: 10.1152/ajpgi.00064.2006. [DOI] [PubMed] [Google Scholar]
- 48.Christensen ST, Pedersen LB, Schneider L, et al. Sensory cilia and integration of signal transduction in human health and disease. Traffic. 2007;8:97–109. doi: 10.1111/j.1600-0854.2006.00516.x. [DOI] [PubMed] [Google Scholar]
- 49.Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 50.Masyuk AI, Masyuk TV, Splinter PL, et al. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–20. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muchatuta MN, Gattone VH, 2nd, Witzmann FA, et al. Structural and functional analyses of liver cysts from the BALB/c-cpk mouse model of polycystic kidney disease. Exp Biol Med (Maywood) 2009;234:17–27. doi: 10.3181/0807-RM-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yost HJ. Left-right asymmetry: nodal cilia make and catch a wave. Curr Biol. 2003;13:R808–9. doi: 10.1016/j.cub.2003.09.051. [DOI] [PubMed] [Google Scholar]
- 53.Pazour GJ, San Agustin JT, Follit JA, et al. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol. 2002;12:R378–80. doi: 10.1016/s0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- 54.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–16. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 55.Pan J, Wang Q, Snell WJ. Cilium-generated signaling and cilia-related disorders. Lab Invest. 2005;85:452–63. doi: 10.1038/labinvest.3700253. [DOI] [PubMed] [Google Scholar]
- 56.Vogel G. News focus: betting on cilia. Science. 2005;310:216–8. doi: 10.1126/science.310.5746.216. [DOI] [PubMed] [Google Scholar]
- 57.Torrice A, Cardinale V, Gatto M, et al. Polycystins play a key role in the modulation of cholangiocyte proliferation. Dig Liver Dis. doi: 10.1016/j.dld.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 58.Liu W, Xu S, Woda C, et al. Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol. 2003;285:F998–1012. doi: 10.1152/ajprenal.00067.2003. [DOI] [PubMed] [Google Scholar]
- 59.Liu W, Murcia NS, Duan Y, et al. Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. Am J Physiol Renal Physiol. 2005;289:F978–88. doi: 10.1152/ajprenal.00260.2004. [DOI] [PubMed] [Google Scholar]
- 60.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71–9. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 61.Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol. 2003;191:69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- 62.Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–93. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 63.Alpini G, Phinizy JL, Glaser S, et al. Development and characterization of secretin-stimulated secretion of cultured rat cholangiocytes. Am J Physiol Gastrointest Liver Physiol. 2003;284:G1066–73. doi: 10.1152/ajpgi.00260.2002. [DOI] [PubMed] [Google Scholar]
- 64.Alpini G, Glaser S, Alvaro D, et al. Bile acid depletion and repletion regulate cholangiocyte growth and secretion by a phosphatidylinositol 3-kinase-dependent pathway in rats. Gastroenterology. 2002;123:1226–37. doi: 10.1053/gast.2002.36055. [DOI] [PubMed] [Google Scholar]
- 65.Alpini G, Glaser SS, Rodgers R, et al. Functional expression of the apical Na+-dependent bile acid transporter in large but not small rat cholangiocytes. Gastroenterology. 1997;113:1734–40. doi: 10.1053/gast.1997.v113.pm9352879. [DOI] [PubMed] [Google Scholar]
- 66.Masyuk AI, Gong AY, Kip S, et al. Perfused rat intrahepatic bile ducts secrete and absorb water, solute, and ions. Gastroenterology. 2000;119:1672–80. doi: 10.1053/gast.2000.20248. [DOI] [PubMed] [Google Scholar]
- 67.Splinter PL, Masyuk AI, LaRusso NF. Specific inhibition of AQP1 water channels in isolated rat intrahepatic bile duct units by small interfering RNAs. J Biol Chem. 2003;278:6268–74. doi: 10.1074/jbc.M212079200. [DOI] [PubMed] [Google Scholar]
- 68.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–8. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 69.Alvaro D, Onori P, Alpini G, et al. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172:321–32. doi: 10.2353/ajpath.2008.070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calvet JP. Molecular genetics of polycystic kidney disease. J Nephrol. 1998;11:24–34. [PubMed] [Google Scholar]
- 71.Parker E, Newby LJ, Sharpe CC, et al. Hyperproliferation of PKD1 cystic cells is induced by insulin-like growth factor-1 activation of the Ras/Raf signalling system. Kidney Int. 2007;72:157–65. doi: 10.1038/sj.ki.5002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grantham JJ, Ye M, Gattone VH, 2nd, et al. In vitro fluid secretion by epithelium from polycystic kidneys. J Clin Invest. 1995;95:195–202. doi: 10.1172/JCI117638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Motta PM. The three-dimensional microanatomy of the liver. Arch Histol Jpn. 1984;47:1–30. doi: 10.1679/aohc.47.1. [DOI] [PubMed] [Google Scholar]
- 74.Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. quiz 55. [DOI] [PubMed] [Google Scholar]
- 75.Qian F, Watnick TJ, Onuchic LF, et al. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–87. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 76.Wu G, D'Agati V, Cai Y, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–88. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 77.Colgin LM, Hackmann AF, Emond MJ, et al. The unexpected landscape of in vivo somatic mutation in a human epithelial cell lineage. Proc Natl Acad Sci USA. 2002;99:1437–42. doi: 10.1073/pnas.032655699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nishio S, Hatano M, Nagata M, et al. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. J Clin Invest. 2005;115:910–8. doi: 10.1172/JCI22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guay-Woodford LM. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol. 2003;285:F1034–49. doi: 10.1152/ajprenal.00195.2003. [DOI] [PubMed] [Google Scholar]
- 80.Sanzen T, Harada K, Yasoshima M, et al. Polycystic kidney rat is a novel animal model of Caroli's disease associated with congenital hepatic fibrosis. Am J Pathol. 2001;158:1605–12. doi: 10.1016/S0002-9440(10)64116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fry JL, Jr, Koch WE, Jennette JC, et al. A genetically determined murine model of infantile polycystic kidney disease. J Urol. 1985;134:828–33. doi: 10.1016/s0022-5347(17)47448-9. [DOI] [PubMed] [Google Scholar]
- 82.Nauta J, Ozawa Y, Sweeney WE, Jr, et al. Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr Nephrol. 1993;7:163–72. doi: 10.1007/BF00864387. [DOI] [PubMed] [Google Scholar]
- 83.Muto S, Aiba A, Saito Y, et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum Mol Genet. 2002;11:1731–42. doi: 10.1093/hmg/11.15.1731. [DOI] [PubMed] [Google Scholar]
- 84.Pennekamp P, Karcher C, Fischer A, et al. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol. 2002;12:938–43. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 85.Gretz N, Kranzlin B, Pey R, et al. Rat models of autosomal dominant polycystic kidney disease. Nephrol Dial Transpl. 1996;11(Suppl. 6):46–51. doi: 10.1093/ndt/11.supp6.46. [DOI] [PubMed] [Google Scholar]
- 86.Nauta J, Goedbloed MA, Herck HV, et al. New rat model that phenotypically resembles autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2000;11:2272–84. doi: 10.1681/ASN.V11122272. [DOI] [PubMed] [Google Scholar]
- 87.Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell. 2003;112:19–28. doi: 10.1016/s0092-8674(02)01283-7. [DOI] [PubMed] [Google Scholar]
- 88.Muff MA, Masyuk TV, Stroope AJ, et al. Development and characterization of a cholangiocyte cell line from the PCK rat, an animal model of Autosomal Recessive Polycystic Kidney Disease. Lab Invest. 2006;86:940–50. doi: 10.1038/labinvest.3700448. [DOI] [PubMed] [Google Scholar]
- 89.Perrone RD, Grubman SA, Rogers LC, et al. Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium. Am J Physiol Gastrointest Live Physiol. 1995;269:G335–45. doi: 10.1152/ajpgi.1995.269.3.G335. [DOI] [PubMed] [Google Scholar]
- 90.Perrone RD, Grubman SA, Murray SL, et al. Autosomal dominant polycystic kidney disease decreases anion exchanger activity. Am J Physiol Gastrointest Live Physiol. 1997;272:C1748–56. doi: 10.1152/ajpcell.1997.272.5.C1748. [DOI] [PubMed] [Google Scholar]
- 91.Bello-Reuss E, Holubec K, Rajaraman S. Angiogenesis in autosomal-dominant polycystic kidney disease. Kidney Int. 2001;60:37–45. doi: 10.1046/j.1523-1755.2001.00768.x. [DOI] [PubMed] [Google Scholar]
- 92.Jakkula M, Le Cras TD, Gebb S, et al. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L600–7. doi: 10.1152/ajplung.2000.279.3.L600. [DOI] [PubMed] [Google Scholar]
- 93.Brodsky KS, McWilliams RR, Amura CR, et al. Liver cyst cytokines promote endothelial cell proliferation and development. Exp Biol Med (Maywood) 2009;234:1155–65. doi: 10.3181/0903-RM-112. [DOI] [PubMed] [Google Scholar]
- 94.Gaudio E, Barbaro B, Alvaro D, et al. Administration of r-VEGF-A prevents hepatic artery ligation-induced bile duct damage in bile duct ligated rats. Am J Physiol Gastrointest Liver Physiol. 2006;291:G307–17. doi: 10.1152/ajpgi.00507.2005. [DOI] [PubMed] [Google Scholar]
- 95.Gaudio E, Barbaro B, Alvaro D, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–82. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 96.Amura CR, Brodsky KS, Groff R, et al. VEGF receptor inhibition blocks liver cyst growth in pkd2(WS25/-) mice. Am J Physiol Cell Physiol. 2007;293:C419–28. doi: 10.1152/ajpcell.00038.2007. [DOI] [PubMed] [Google Scholar]
- 97.Fabris L, Cadamuro M, Fiorotto R, et al. Effects of angiogenic factor overex-pression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43:1001–12. doi: 10.1002/hep.21143. [DOI] [PubMed] [Google Scholar]
- 98.Gaudio E, Franchitto A, Pannarale L, et al. Cholangiocytes and blood supply. World J Gastroenterol. 2006;12:3546–52. doi: 10.3748/wjg.v12.i22.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mancinelli R, Onori P, Gaudio E, et al. Taurocholate feeding to bile duct ligated rats prevents caffeic acid-induced bile duct damage by changes in cholangiocyte VEGF expression. Exp Biol Med (Maywood) 2009;234:462–74. doi: 10.3181/0808-RM-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ross MA, Sander CM, Kleeb TB, et al. Spatiotemporal expression of angiogenesis growth factor receptors during the revascularization of regenerating rat liver. Hepatology. 2001;34:1135–48. doi: 10.1053/jhep.2001.29624. [DOI] [PubMed] [Google Scholar]
- 101.Spirli C, Okolicsanyi S, Fiorotto R, et al. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fabris L, Strazzabosco M, Crosby HA, et al. Characterization and isolation of ductular cells coexpressing neural cell adhesion molecule and Bcl-2 from primary cholangiopathies and ductal plate malformations. Am J Pathol. 2000;156:1599–612. doi: 10.1016/S0002-9440(10)65032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Masyuk TV, Huang BQ, Ward CJ, et al. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–10. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 104.Wilson PD. Polycystin: new aspects of structure, function, and regulation. J Am Soc Nephrol. 2001;12:834–45. doi: 10.1681/ASN.V124834. [DOI] [PubMed] [Google Scholar]
- 105.Yamaguchi T, Hempson SJ, Reif GA, et al. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17:178–87. doi: 10.1681/ASN.2005060645. [DOI] [PubMed] [Google Scholar]
- 106.Yamaguchi T, Wallace DP, Magenheimer BS, et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–30. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 107.Glaser SS, Gaudio E, Miller T, et al. Cholangiocyte proliferation and liver fibrosis. Expert Rev Mol Med. 2009;11:e7. doi: 10.1017/S1462399409000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alpini G, Ueno Y, Glaser SS, et al. Bile acid feeding increased proliferative activity and apical bile acid transporter expression in both small and large rat cholangiocytes. Hepatology. 2001;34:868–76. doi: 10.1053/jhep.2001.28884. [DOI] [PubMed] [Google Scholar]
- 109.Alpini G, Glaser SS, Ueno Y, et al. Bile acid feeding induces cholangiocyte proliferation and secretion: evidence for bile acid-regulated ductal secretion. Gastroenterology. 1999;116:179–86. doi: 10.1016/s0016-5085(99)70242-8. [DOI] [PubMed] [Google Scholar]
- 110.Masyuk TV, Masyuk AI, Torres VE, et al. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–16. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 111.Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12:3496–511. doi: 10.3748/wjg.v12.i22.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang S, Zhang J, Nauli SM, et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol. 2007;27:3241–52. doi: 10.1128/MCB.00072-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gradilone SA, Masyuk AI, Splinter PL, et al. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci USA. 2007;104:19138–43. doi: 10.1073/pnas.0705964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Banales JM, Masyuk TV, Gradilone SA, et al. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2009;49:160–74. doi: 10.1002/hep.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Amura CR, Brodsky KS, Gitomer B, et al. CXCR2 agonists in ADPKD liver cyst fluids promote cell proliferation. Am J Physiol Cell Physiol. 2008;294:C786–96. doi: 10.1152/ajpcell.00457.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lai X, Blazer-Yost BL, Gattone VH, 2nd, et al. Protein composition of liver cyst fluid from the BALB/c-cpk/+ mouse model of autosomal recessive polycystic kidney disease. Proteomics. 2009;9:3775–82. doi: 10.1002/pmic.200800379. [DOI] [PubMed] [Google Scholar]
- 117.Mitsuyama K, Toyonaga A, Sasaki E, et al. IL-8 as an important chemoattractant for neutrophils in ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1994;96:432–6. doi: 10.1111/j.1365-2249.1994.tb06047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Addison CL, Daniel TO, Burdick MD, et al. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165:5269–77. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- 119.Wang Y, Yang J, Gao Y, et al. Regulatory effect of e2, IL-6 and IL-8 on the growth of epithelial ovarian cancer cells. Cell Mol Immunol. 2005;2:365–72. [PubMed] [Google Scholar]
- 120.Alvaro D, Metalli VD, Alpini G, et al. The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis. J Hepatol. 2005;43:875–83. doi: 10.1016/j.jhep.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 121.Alvaro D, Alpini G, Onori P, et al. Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats. Gastroenterology. 2000;119:1681–91. doi: 10.1053/gast.2000.20184. [DOI] [PubMed] [Google Scholar]
- 122.Alvaro D, Macarri G, Mancino MG, et al. Serum and biliary insulin-like growth factor I and vascular endothelial growth factor in determining the cause of obstructive cholestasis. Ann Intern Med. 2007;147:451–9. doi: 10.7326/0003-4819-147-7-200710020-00003. [DOI] [PubMed] [Google Scholar]
- 123.Onori P, Alvaro D, Floreani AR, et al. Activation of the IGF1 system characterizes cholangiocyte survival during progression of primary biliary cirrhosis. J Histochem Cytochem. 2007;55:327–34. doi: 10.1369/jhc.6R7125.2006. [DOI] [PubMed] [Google Scholar]
- 124.Koduri S, Goldhar AS, Vonderhaar BK. Activation of vascular endothelial growth factor (VEGF) by the ER-alpha variant, ERDelta3. Breast Cancer Res Treat. 2006;95:37–43. doi: 10.1007/s10549-005-9028-4. [DOI] [PubMed] [Google Scholar]
- 125.Chapman AB. Cystic disease in women: clinical characteristics and medical management. Adv Ren Replace Ther. 2003;10:24–30. doi: 10.1053/jarr.2003.50005. [DOI] [PubMed] [Google Scholar]
- 126.Sherstha R, McKinley C, Russ P, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26:1282–6. doi: 10.1002/hep.510260528. [DOI] [PubMed] [Google Scholar]
- 127.Bogovich K. Follicle-stimulating hormone plays a role in the induction of ovarian follicular cysts in hypophysectomized rats. Biol Reprod. 1992;47:149–61. doi: 10.1095/biolreprod47.2.149. [DOI] [PubMed] [Google Scholar]
- 128.Surmacz E. Growth factor receptors as therapeutic targets: strategies to inhibit the insulin-like growth factor I receptor. Oncogene. 2003;22:6589–97. doi: 10.1038/sj.onc.1206772. [DOI] [PubMed] [Google Scholar]
- 129.Mancinelli R, Onori P, Gaudio E, et al. Follicle-stimulating hormone increases cholangiocyte proliferation by an autocrine mechanism via cAMP-dependent phosphorylation of ERK1/2 and Elk-1. Am J Physiol Gastrointest Liver Physiol. 2009;297:G11–26. doi: 10.1152/ajpgi.00025.2009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 130.Karlsson-Rosenthal C, Millar JB. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006;16:285–92. doi: 10.1016/j.tcb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 131.Lee SO, Masyuk T, Splinter P, Banales JM, Masyuk A, Stroope A, Larusso N. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118(11):3714–24. doi: 10.1172/JCI34922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Masyuk T, Masyuk A, LaRusso N. MicroRNAs in cholangiociliopathies. Cell Cycle. 2009;8:1324–8. doi: 10.4161/cc.8.9.8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nagao S, Nishii K, Katsuyama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol. 2006;17:2220–7. doi: 10.1681/ASN.2006030251. [DOI] [PubMed] [Google Scholar]
- 134.Ruggenenti P, Remuzzi A, Ondei P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68:206–16. doi: 10.1111/j.1523-1755.2005.00395.x. [DOI] [PubMed] [Google Scholar]
- 135.Keimpema LV, Nevens F, Vanslembrouck R, et al. Lanreotide reduces the volume of polycystic liver: A randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.07.052. [DOI] [PubMed] [Google Scholar]