

Abstract
Progressive dementias like Alzheimer’s Disease (AD) and other tauopathies are an increasing threat to human health worldwide. Although significant progress has been made in understanding the pathogenesis of these diseases using cell culture and mouse models, the complexity of these diseases has still prevented a comprehensive understanding of their underlying causes. As with other neurological diseases, invertebrate models have provided novel genetic approaches for investigating the molecular pathways that are affected in tauopathies, including AD. This review focuses on transgenic models that have been established in Drosophila melanogaster and Caenorhabtidis elegans to investigate these diseases, and the insights that have been gained from these studies. Also included is a brief description of the endogenous versions of human “disease genes” (like tau and the Amyloid Precursor Protein) that are expressed in invertebrates, and an overview of results that have been obtained from animals lacking or overexpressing these genes. These diverse models can be used to advance our knowledge about how these proteins acquire a pathogenic function and how disrupting their normal functions may contribute to neurological pathologies. They also provide powerful assays for identifying molecular and genetic interactions that are important in developing or preventing the deleterious effects.
Introduction
Alzheimer’s Disease (AD) is the most common form of dementia, accounting for 60–80% of all cases and it is currently the 6th leading cause of death (Alzheimer’s Association; www.alz.org). Worldwide, approximately 26 million people suffer from this devastating disease, and with the increase in life span this number is predicted to quadruple over the next four decades (American Health Assistance Foundation; www.ahaf.org). Starting with slight memory loss and confusion, AD eventually leads to severe mental impairment, often accompanied by changes in personality. Although a number of drugs have been developed for treatment to slow the progression of cognitive decline, there is currently no cure for AD.
Histologically, AD is characterized by the formation of amyloid plaques and neurofibrillary tangles (NFTs). The latter primarily consist of tau, a microtubule-binding protein that, when hyperphosphorylated aggregates into insoluble fibrillar deposits in the form of NFTs (Mandelkow and Mandelkow, 1998). These pathological tau inclusions are also found in several other neurodegenerative diseases, including frontal temporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), corticobasal degeneration, progressive supra nuclear palsy, and Pick’s disease, accordingly this group of diseases is referred to as tauopathies (Gendron and Petrucelli, 2009). In contrast, the main component of amyloid plaques is, as the name implies, Aβamyloid, which are small peptides consisting of 40 or 42 amino acids (Selkoe, 2000). These peptides are cleaved from the larger Amyloid Precursor Protein (APP) by proteases called β-secretase and γ-secretase (De Strooper and Annaert, 2000, Turner et al., 2003). Whereas β-secretase activity is encoded by a single protein, called the β-site APP-cleaving enzyme or BACE,γ-secretase is a protein complex consisting of presenilin (Psn), nicastrin, aph.1, and pen2 (De Strooper, 2003). That the cleavage of APP and the production of Aβ is indeed a crucial factor in AD was shown by the identification of mutations in Psn-1, Psn-2, and APP that promote the production of Aβ and lead to early-onset familial AD (FAD) (Haass and De Strooper, 1999). Alternatively, APP can be cleaved by α- and γ-secretase, which does not result in the production of toxic peptides (De Strooper and Annaert, 2000).
Although studies on patients afflicted with FAD have contributed significantly to our understanding of the pathogenesis of AD, it has also become apparent that other genes contribute to the more prevalent sporadic forms of the disease. However, studies to identify such factors in humans have obvious limitations, due to methodological difficulties and ethical concerns. Therefore, animal models are an essential experimental system to identify and understand the function of candidate genes and delineate the genetic pathways in which they play a role. In addition, animal models can be used to systematically investigate the disease mechanisms from the earliest stages, whereas human studies must rely on postmortem tissue that typically represents late stages of the disease.
Invertebrates, and especially the well-established model organisms Drosophila and Caenorhabditis elegans, provide many experimental advantages for this type of analysis Their anatomy, development, and behavior has been thoroughly studied and they are amenable to a variety of genetic and molecular methods, including the relatively easy generation of transgenic animals and the possibility to perform large-scale mutagenesis screens. Therefore, it is not surprising they have also successfully been used in recent years to investigate the mechanisms of neurodegenerative diseases, including AD and other tauopathies (Bilen and Bonini, 2005, Wu and Luo, 2005, Lu and Vogel, 2009, Teschendorf and Link, 2009).
Modeling Amyloid Toxicity
As noted above, accumulation of Aβ into senile plaques is one of the hallmarks of AD (Glenner and Wong, 1984; Wong et al., 1985) and together with findings that mutations leading to increased Aβ production have been described in familial AD (Chartier-Harlin et al., 1991, Goate et al., 1991, Levy-Lahad et al., 1995, Sherrington et al., 1996) this provided the basis for the amyloid hypothesis, which proposed that Aβ accumulation is the driving factor for disease progression and pathology. For the vast majority of AD cases, however, the cause of amyloid plaque formation is still undefined, and the intrinsic pathogenicity of Aβ peptides remains controversial.
To investigate the toxic function of Aβ peptides in Drosophila, several transgenic fly models have been created that specifically express either Aβ40 or Aβ42 (Finelli et al., 2004, Iijima et al., 2004, Crowther et al., 2005). Both of these peptides are found in human plaques, but the Aβ42 peptide has been found to be the more toxic species (Findeis, 2007). Confirming the increased toxicity of this peptide, only expression of Aβ42 resulted in amyloid deposits and degeneration in the fly eye (Finelli et al., 2004) and brain (Iijima et al., 2004; Crowther et al., 2005). Surprisingly however, both peptides induced defects in an associative olfactory learning assay when expressed pan-neuronally (Iijima et al., 2004). Whereas the performance of young flies expressing either protein was not different from controls, their performance became increasingly worse when aged for 6–7 days, although there was no significant difference between flies expressing Aβ42 versus Aβ40. These experiments clearly showed that Aβ-peptides alone are sufficient to induce AD-like phenotypes. In addition, they suggested that learning defects do not require visible plaque formation, which is similar to results obtained by Crowther et al. in a climbing assay (Crowther et al., 2005). In this study the deficits were also observed before the occurrence of large extracellular deposits and instead correlated with the intracellular accumulation of Aβ (in this case Aβ42 and the arctic mutation which is found in patients with early onset familial AD). More recently, Iijima et al. used the fly model to investigate the effects of different aggregation rates by expressing Aβ42 with the arctic mutation, which increases aggregation, or an artificial mutation shown to decrease aggregation (Iijima et al., 2008). Expressing the arctic mutation resulted in higher levels of Aβ42 oligomers compared to the normal Aβ42, while the artificial mutation reduced the formation of oligomers. Notably, these differences in aggregation tendency correlated with the deleterious effects on life span and locomotion. However, both mutations increased short-term memory deficits compared to flies expressing normal Aβ42, with the artificial mutation causing an even earlier onset than the arctic mutation. This further supported the earlier results by this group, which suggested that the aggregation propensity does not determine the severity of memory deficits. Interestingly, each form of Aβ42 had distinct defects on neuronal degeneration, with the arctic mutation causing mostly vacuoles in the cortex (where all neuronal cell bodies are located, similar to the gray matter in vertebrates). In contrast, the artificial mutation induced vacuolization in the neuropil, which is comprised of neuritis (equivalent to white matter). The specificity of these pathologies correlated with the localization of aggregates, because Aβ42arctic showed large deposits in the cell bodies, whereas the artificial mutation primarily resulted in deposits in neurites. This finding suggested that although aggregation levels can affect some phenotypes, differences in aggregation rates alone do not determine pathogenicity.
Similar results were obtained in C. elegans, where expression of Aβ42 in muscle cells induced the formation of amyloid containing inclusions (Link, 1995). As in flies, however expressing variants of Aβ42 with mutations that prevented deposit formation did not reduce toxicity (Fay et al., 1998), providing additional support for a model in which their pathogenicity is not solely due to the levels of aggregation. Although transgenic worm models expressing Aβ42 in neurons also exhibit amyloid aggregates, the resulting phenotypes are very subtle (Link, 2006, Wu et al., 2006). Although expression of Aβ peptides in these models does certainly not completely recapitulate the disease process, these studies have provided important insights about the toxicity of specific peptide species. However, a caveat of inducing only the expression of Aβ peptides is that features requiring the expression of the entire APP protein cannot be recapitulated nor can these models be used to investigate genetic factors or therapeutic drugs that affect processing.
For these purposes the fly lines that express full-length human APP695 (Fossgreen et al., 1998), the predominant form in the nervous system, might prove more useful. To ensure β-cleavage of APP in this model, a human BACE construct was co-expressed with APP695, whichtogether with endogenous fly γ-secretase produced toxic Aβ fragments (Greeve et al., 2004). Histological analysis revealed the formation of amyloid deposits and age-dependent degeneration in these flies, in addition to a decreased life span. Surprisingly, the same phenotypes were induced after expression of APP695 alone, suggesting that flies posses an endogenous BACE-like enzyme. Indeed Western Blots confirmed the production of an Aβ fragment in these flies, though it was slightly larger than the Aβ produced by co-expression of human BACE. Flies expressing full-length APP were also used to investigate the effects of altering the processing pattern of APP by either genetic or pharmacological means (Greeve et al., 2004). Increasing the levels of endogenous presenilin (dPsn) or a variant of dPsnthat contained mutations linked to familial AD, enhanced the phenotypes in this model, whereas removing one copy of the presenilin gene had the opposite effect. Similarly, treating these flies with BACE or γ-secretase inhibitors ameliorated the phenotypes, consistent with the model that the toxic effects are due to the production of Aβ peptides from the full-length protein.
Drosophila models have also addressed aspects of late onset AD, especially the role of ubiquilin. Genetic variants of human Ubiquilin 1 have been associated with a higher risk for late onset AD and it has been shown that Ubiquilin 1 can bind to Presenilin in cell culture (Bertram et al., 2005, Kamboh et al., 2006). Li et al showed that an RNAi knock-down of Drosophila Ubiquilin (dUbqln) in the brain resulted in neuronal degeneration and shortened life span. When dUbqln was reduced in the eye it enhanced the eye degeneration caused by expression of dPsn while overexpression suppressed the small eye phenotype (Li et al., 2007). In addition, this group reported that co-expression of dUbqln reduced the amount of full-length APP and the AICD. In contrast, Ganguly et al. showed that loss of dUbqln suppressed dPsn induced eye degeneration and that overexpression of dUbqln caused degeneration (Ganguly et al., 2008). They also showed that expression of human Ubiquilin variants associated with increased risk for late onset AD induced a more severe degeneration compared to wild type human Ubiquilin. While the opposing effects on degeneration do not allow mechanistic insights at this point, the results from both groups suggest that Ubiquilin is involved in the regulation of Psn. And indeed the binding of dUbqln to dPsn was verified in this model in vivo (Ganguly et al., 2008).
To identify novel modifiers of AD phenotypes, genetic interaction screens have been performed using both APP/presenilin (van de Hoef et al., 2009) and Aβ42 expressing flies (Cao et al., 2008). Approximately 200 candidates genes were identified from these screens, including genes involved in vesicle transport, protein degradation, stress response, and chromatin structure. The interacting candidates also included the γ-subunit of AMP-activated protein kinase (AMPK) (Cao et al., 2008), a protein complex involved in energy metabolism and cholesterol homeostasis. Interestingly, a mutant in AMPKγ called loechrig (loe) genetically interacts with APPL (Amyloid Precursor Protein-like), the sole APP protein in flies (Luo et al., 1990). Loe mutant flies showed an age-dependent degeneration of the CNS which was significantly increased when combined with a knock-out in APPL (Tschape et al., 2002).
Screens with Aβ42 transgenic worms, have also identified genes associated with stress responses and lifespan as candidate interacting partners (Cohen et al., 2006, Fonte et al., 2008), including orthologues for insulin growth factor 1 and the FOXO transcription factor. Future studies of these candidates identified in flies and worms could yield valuable insights into the pathways affected by Aβ. In addition, they could provide new targets for therapeutic intervention.
Physiological functions of APP proteins
Although the importance of APP in the pathogenesis of AD has been acknowledged for a long time, the normal physiological function of this protein and the potential role that disrupting these functions may play in AD are still not well understood (Anliker and Muller, 2006, Senechal et al., 2006, Jacobsen and Iverfeldt, 2009).
One of the first phenotypes observed in APP transgenic fly models was the occurrence of a blistered wing (Fossgreen et al., 1998). It was therefore proposed that the ectopic expression of APP in the wing disc might interfere with interactions between the cell layers of the wing, supporting studies in cell culture that have suggested that APP proteins are involved in cell adhesion (Jacobsen and Iverfeldt, 2009). Their effects on cell-cell interactions could also be the underlying cause of defects in neuronal outgrowth and post-developmental aborization induced by overexpression of APP and APPL in Drosophila (Li et al., 2004, Leyssen et al., 2005). Likewise, deletion of APPL resulted in decreased neurite outgrowth (Li et al., 2004), further supporting a role for APP proteins in neurite outgrowth. APPL has also been shown to affect synaptogenesis because APPL deficient flies reveal a reduced number of synaptic boutons at the neuromuscular junction, while an increase in APPL levels results in additional synaptic boutons (Torroja et al., 1999b). These structural changes appear to have corresponding functional consequences as well, because flies lacking APPL exhibited smaller excitatory junction potentials (EJPs) compared to wild type (Ashley et al., 2005). Moreover, patch clamp experiments using cultured neurons derived from these flies showed an increase in neuronal excitability, presumably due to the activation of K+ channels (Li et al., 2004). Whether these synaptic defects are a direct cause of the behavioral defects observed in APPL lacking flies (Luo et al., 1992) still needs to be determined. However, these behavioral abnormalities were rescued by expression of human APP695, confirming a functional conservation of human and fly APP/APPL (Luo et al., 1992)
Intriguingly, there is now increasing evidence that synaptic dysfunctions caused by the intracellular accumulation of Aβ is a major aspect of AD (Selkoe, 2008, Nimmrich and Ebert, 2009) and it has also been shown that human APP can affect neuronal excitability (Turner et al., 2003). Therefore, further studies into the functions of invertebrate APP proteins like APPL and APL-1, the sole C. elegans APP protein (Daigle and Li, 1993), might help to elucidate the normal physiological roles of these proteins in addition to their toxic effects when mutated or otherwise affected. Intriguingly, even some of the pathogenic features of human APP appear to be conserved in fly APPL. Despite significant sequence differences in the region corresponding to Aβ, amyloid fragments derived from APPL can accumulate into deposits and induce behavioral deficits and neurodegeneration (Carmine-Simmen et al., 2009). This study also showed that APPL can be cleaved by an endogenousβ-secretase-like enzyme as well as by human BACE1. Therefore, Drosophila can now be used for the initial identification of factors that influence secretase activities without the need and possible complications of ectopically expressed proteins.
Modeling Tauopathies
The second hallmark of AD is the accumulation of hyperphosphorylated forms of tau into Neurofibrillary tangles (NFTs). In fact NFTs correlate more with the progression and severity of AD than amyloid plaques (Arriagada et al., 1992). NFTs are also found in several other neurodegenerative disorders, and although the tau gene appears unaffected in AD, mutations in tau have been shown to cause frontotemporal dementia FTDP-17 (Hutton et al., 1998, Spillantini et al., 1998). Like other microtubule-associated proteins (MAPs), tau binds and regulates the stability and organization of microtubules, a process that is controlled by its phosphorylation level (Lee et al., 2001). Hyperphosphorylation of tau not only interferes with its interaction with tubulin but also increases its ability to form NFTs and to sequester normal tau into these aggregates (Alonso et al., 2008). Moreover, tau mutations in FTDP-17 increase its phosphorylation and impair its ability to bind microtubules (Gendron and Petrucelli, 2009, Iqbal et al., 2009), underscoring the importance of hyperphosphorylation in the toxicity of tau.
The first evidence that Drosophila could provide a model for tau toxicity actually came from experiments in which bovine tau was used as an axonal marker (Murray et al., 1998). Subsequently, Williams et al. found that expressing this construct in sensory neurons resulted in a significant degeneration of their axonal projections within the thoracic ganglia, although they initially grew out normally during development (Williams et al., 2000). Together with findings that the pan-neuronal induction of this construct resulted in disrupted axonal transport (Torroja et al., 1999a), these results set the stage for using flies as a model for tauopathies. To establish such a model, Feany and colleagues expressed both wild type human tau and a variant containing a mutation associated with FTDP-17 (R406W) (Wittmann et al., 2001). While expression of either construct in the CNS led to abnormally phosphorylated tau and induced progressive degeneration and early death, the effects were stronger for the mutant form. Surprisingly, however, neither construct induced NFTs, a result that was also confirmed for another tau FTDP-17 mutation (V337M). In contrast, pan-neuronal expression of FTDP-17 tau mutations in the worm did result in the accumulation of insoluble, phosphorylated tau aggregates (Kraemer et al., 2003). Similar to the fly model, the mutant tau form induced more severe phenotypes than wild type tau, including uncoordinated movement and age-dependent neurodegeneration, although the severity of these phenotypes did not correlate with the levels of tau phosphorylation. Together, these results suggested that while NFTs may be themselves toxic, other non-filamentous forms of tau may also be detrimental. That NFTs do not always correlate with neuropathic phenotypes has also been shown in a mouse model expressing mutant human tau. These animals develop NFTs and show memory defects and neuronal loss, but whereas the neurodegenerative and cognitive defects improved after subsequent suppression of tau, the NFTs continued to accumulate (Santacruz et al., 2005).
Tau phosphorylation and neurotoxicity
Though many studies have connected tau hyperphosphorylation with its toxicity, the importance of specific phosphorylation sites for the development of tauopathies is not clear. A variety of kinases and phosphatases, including glycogen synthase-3 (GSK-3β), cdk5, protein kinase A (PKA), and microtubule-affinity regulating kinase (MARK) have been shown to regulate tau phosphorylation in biochemical studies (Lee et al., 2001). However, their function in vivo is less clear. In fly models, the pathogenicity of tau also appears to be affected by its phosphorylation state. Expressing human tau with mutations in 14 known phosphorylation sites in the eye abolished its neurodegenerative effect (Steinhilb et al., 2007b), while a construct that was pseudophosphorylated at these sites caused a more severe degeneration than wild type human tau (Khurana et al., 2006). In addition, several groups have shown that tau hyperphosphorylation in flies is mediated at least in part by GSK-3β which is encoded by the shaggy (sgg) gene in Drosophila. As described above, pan-neuronal expression of wild-type human tau in flies induced degeneration but did not cause NFTs (Wittmann et al., 2001). However, when tau was co-expressed with sgg, this not only increased the phorphorylation of tau and exacerbated the degenerative phenotype of the eye, but also resulted in the formation of NFTs (Jackson et al., 2002). A similar result was obtained by Chau et al. who showed that co-expression of either sgg or cdk-5 enhanced both tau phosphorylation and its degenerative effect (Chau et al., 2006). In addition, the interaction with sgg was confirmed in studies on axonal transport defects caused by human tau expression in motorneurons. In this case, co-expression of constitutively active SGG enhanced the transport defects, while treatment with GSK-3β inhibitors suppressed the phenotype (Mudher et al., 2004).
Other groups have used the fly model to investigate the role of MARK in tau phosphorylation and toxicity. Overexpressing PAR-1, the fly orthologue of MARK, elevated tau phosphorylation and enhanced its toxic effects, while removing the PAR-1 phosphorylation sites in tau abolished its toxicity (Nishimura et al., 2004). This group also showed that phosphorylation of tau by PAR-1 is a prerequisite for downstream phosphorylation events, most likely including tau phosphorylation by SGG and cdk5. In contrast, Chatterjee et al. recently showed that while mutating the PAR-1 sites in tau did indeed decrease its degenerative effect on photoreceptors, it did not prevent phosphorylation of tau by SGG. In addition, a mutant form of tau that was resistant to SGG phosphorylation retained its deleterious effects, although it did not form aggregates (Chatterjee et al., 2009). Interestingly, mutations in the SGG sites of tau increased its microtubule binding affinity of tau. This suggests that either decreased binding to microtubules (as observed in the case of the FTDP-17 mutations) or increased binding (as described above) can have toxic effect, underscoring the importance of a precisely regulated interaction between tau and microtubule.
In summary, these studies indicate that the mechanisms conferring tau toxicity are quite complex. Hyperphosphorylation certainly appears to be an important factor, whereby tau-associated toxicity may be mediated by the orchestrated phosphorylation of several sites, rather than phosphorylation of a specific single site (Steinhilb et al., 2007a). However, as the results by Chatterjee et al. suggest, hyperphosphorylation seems to be only one mechanism that can confer tau toxicity: This indicates that other modifications that affect the interaction of tau with microtubules can induce neurodegeneration. Furthermore, these effects might not be restricted to interactions between tau and microtubule, as recent studies have shown that tau can also affect the actin cytoskeleton. Phosphorylated tau can induce the accumulation of filamentous F-actin, resembling the Hirano bodies found in patients with AD or Pick’s Disease, while co-expression of tau with actin in the eye increases the severity of its degenerative phenotype (Fulga et al., 2007). It is not yet known whether this interaction is due to a direct effect of tau on actin or an indirect one, possibly mediated by an interaction between the microtubule and actin cytoskeleton (Sider et al., 1999). Intriguingly, another study suggested that phosphorylation and other posttranslational modifications of tau may depend on the cell type. Comparing the phosphorylation pattern of tau expressed in photoreceptors versus CNS neurons revealed that little or no phosphorylation was detected at two SGG/GSK-3β sites (T212/S214) in the retinal tau, whereas these sites were clearly phosphorylated in tau expressed in the CNS (Grammenoudi et al., 2006). Cell type also appeared to affect the levels of specific tau species, because two variants were detected in the eye but only one in CNS neurons. Such cell-specific effects on tau could provide an explanation for the varying sensitivity of different neurons in tauopathy diseases. Using the Drosophila model, which allows selective expression in a variety of different CNS neurons, could therefore provide important insights how different neuronal subtypes process and modify tau.
Genetic interaction studies
One of the biggest advantages of fly and worm models is the comparable ease with which they can be used to identify interacting proteins. Using an RNAi based screen in C. elegans, Kraemer et al. identified 75 putative modifiers that affected the uncoordinated phenotype induced by the expression of human tau (Kraemer et al., 2006). Besides several kinases and phosphatases, including GSK-3β, they identified proteins involved in protein folding, stress response, and protease activity, but also several proteins of unknown function. Two of these, sut-1 and sut-2, were studied in more detail; sut-1 encodes a nematode specific protein that was shown to interact with UNC-34, an enabled protein family member, suggesting that sut-1 may also be involved in the regulation of the cytoskeleton (Kraemer and Schellenberg, 2007). The other candidate, sut-2, encodes a conserved zinc-finger protein that can interact with ZYG-12, a HOOK2 orthologe, which plays a role in the transport of aggregated proteins to the aggresome (Guthrie et al., 2009). Aggresomes have been shown to contain misfolded and aggregated proteins and appear to form when the proteasome pathway is overwhelmed by these proteins. sut-2 might therefore play a role in a cellular defense mechanism against tau toxcicity by transporting aggregated tau to the aggresome.
Excessive protein accumulation can also be regulated by the proteasome pathway or clearance by autophagy. That these pathways also modulate tau levels was shown in the fly model, because the induction of autophagy by rapamycin reduced the degenerative phenotype of wild type or mutant (R406W) human tau when expressed in the eye (Berger et al., 2006). To investigate the role of the proteasome pathway in tau degradation, Blard et al. expressed a dominant-negative form of the 20S proteasome β6 subunit along with wild type human tau and showed that this resulted in increased tau accumulation, including tau that was hyperphosphorylated by SGG/GSK-3β (Blard et al., 2006). Surprisingly, a hyperphosphorylated variant of tau, that was resistant to proteasome degradation, accumulated when a dominant-negative of SGG was co-expressed, indicating that phosphorylation by another kinase results in a degradation resistant variant of tau.
In addition, genetic interaction test have linked tau-induced neurodegeneration to both cell-cycle regulation and oxidative stress responses. Abnormal activation of the cell-cycle accompanied tauR406W or tauV337M-induced retinal degeneration, and co-expression of genes that promote the cell-cycle (Cyclin A, Cyclin B, or Cyclin D) enhanced this phenotype (Khurana et al., 2006). In contrast, blocking cell-cycle progression by co-expressing the cdk2 inhibitor Dacapo (the fly homolog of p21/p27) or the E2F1 inhibitor Rbf1 (Retinoblastoma factor 1) reduced the neurodegenerative effects of tau. Furthermore, the authors showed that these effects are mediated through the TOR (Target Of Rapamycin) kinase pathway, which activates cell-cycle progression in both flies and mammalian cells. Enhanced cell-cycle activation also appears to be the underlying mechanism for how oxidative stress increases neuronal degeneration. Interfering with antioxidant defense mechanisms by removing one copy of the superoxide dismutase (SOD) gene or the thioredoxin reductase (Trxr) gene aggravated the degenerative phenotype induced by tau, although it did not affect its pattern of phosphorylation (Dias-Santagata et al., 2007). However, these genetic manipulations did significantly increase the number of foci that are immunopositive for a marker of cell proliferation (proliferating cell nuclear antigen). Likewise, tau-induced toxicity correlated with the activation of the JNK pathway, a well-characterized response to oxidative damage.
Using the rough eye phenotype, Shulman and Feany performed an unbiased genetic modifier screen for genes that altered the neurotoxic effect of tau (Shulman and Feany, 2003). Several phosphatases and kinases were identified, including PAR-1, which surprisingly suppressed the degenerative phenotype when overexpressed. In addition, they found quite a few novel candidates genes encoding transcription factors, cation transporters, and several unknown proteins. Using an innovative cross-species approach, Karsten et al. first used a microarray experiment to identify differentially expressed genes in wild type mice versus mice expressing mutant tau, and then confirmed their interactions with tau in the fly model (Karsten et al., 2006). One of the genes isolated in this assay was puromycin-sensitive aminopeptidase (PSA), whereby a loss of function mutant form of PSA enhanced tau-induced retinal degeneration. Because co-expression of fly PSA reduced the levels of tau in flies, while human PSA was shown to proteolyze tau in vitro, it was suggested that PSA plays a role in tau degradation.
Cellular processes affected by tau
Although hyperphosphorylation and other abnormalities of tau are known to induce toxic gain of function effects, it is also likely that the loss of its normal function has deleterious consequences. Drosophila tau (dtau) is highly homologous to human tau, and it is expressed both in the developing and adult nervous system, where it is most prominent in photoreceptors (Heidary and Fortini, 2001). Surprisingly, the loss of dtau does not result in lethality, and so far the only phenotype observed in these flies is a defect in oocyte polarity (Tian and Deng, 2009). The lack of more severe phenotypes is possibly due to redundancy with other microtubule-associated proteins, because three other MAPs have been described in flies that may serve overlapping functions. At least one of them, the MAP1B orthologue futsch, is widely expressed in the nervous system and plays a role in dentritic and axonal outgrowth (Hummel et al., 2000). Interestingly, several hypomorphic alleles of futsch (futscholk) show a progressive degeneration in the adult, primarily restricted to the olfactory system (Bettencourt da Cruz et al., 2005). In support of the model that these other MAPs serve redundant functions with tau, expressing tau in the nervous system of futscholk flies suppressed the degenerative phenotype. These studies also indicate that other MAPs, besides tau, could play a role in neurodegenerative diseases.
Overexpressing wild type dtau in the retina resulted in a pattern of degeneration resembling the phenotype caused by expressing wild type human tau (Chen et al., 2007), suggesting that the toxic properties of the fly and human protein are conserved. Likewise, expression of dtau or htau in larval motoneurons resulted in impaired axonal transport and the accumulation of vesicular aggregates accompanied by slower crawling behavior and reduced peristaltic contractions (Ubhi et al., 2007). In addition, both constructs caused a significant reduction in boutons at the larval neuromuscular junction, while htau was also shown to reduce the number of mitochondria in the remaining boutons and perturb synaptic transmission (Chee et al., 2006). In an independent study, Mershin et al. expressed dtau and htau specifically in mushroom bodies, the centers for associative learning and memory in Drosophila (Heisenberg, 2003), and demonstrated that both induced a decrease in olfactory learning by 25–30% (Mershin et al., 2004). Because no neurodegeneration could be detected in the mushroom bodies, these learning defects appear to be a consequence of tau accumulation on neuronal function rather than the induction of neuronal death.
Synergistic effects between tau and APP proteins
In the context of AD, it has long been suggested that tau and APP/Aβ might act synergistically in inducing toxic effects. In Drosophila, support for this hypothesis was provided by studies investigating an interaction between the endogenous APPL protein and bovine tau (Torroja et al., 1999a). Pan-neuronal expression of either tau or APPL alone resulted in the accumulation of vesicles in larval motoneurons (an effect also seen after expression of human APP (Gunawardena and Goldstein, 2001)), while this phenotype became significantly more severe when both were co-expressed. Moreover, these flies exhibited defects in cuticle hardening and wing expansion in 99% of the eclosing adult flies, while malformations of this type were only occasionally observed when only one construct was expressed (app. 7% for APPL, and 0.5% for tau). In other studies, co-expression of Aβ42 with tau in the eye enhanced the degenerative phenotype compared to tau alone, which was accompanied by a substantially greater accumulation of filamentous F-actin (Fulga et al., 2007). Studies in flies have also provided further evidence for the hypothesis that this interaction is mediated by the effects of APP on tau phosphorylation (Bloom et al., 2005). Induction of Aβ42 and tau in motoneurons not only enhanced the bouton phenotypes and larval crawling defects caused by tau alone but also increased the phosphorylation level of tau (Folwell et al., 2009). These histological and behavioral defects were reverted when the animals were treated with LiCl, a common inhibitor of GSK-3β, indicating that Aβ may regulate tau phosphorylation via GSK-3β/sgg. However, similar experiments by Wang et al. suggest that the effects of APP on tau phosphorylation can be mediated by PAR-1 (Wang et al., 2007). Specifically, they showed that PAR-1 is phosphorylated by the tumor suppressor protein LKB-1, which in turn promotes PAR-1 dependent phosphorylation of tau. Expression of full-length APP increased the phosphorylation of both PAR-1 and tau, an effect that was dependent on the presence of LKB-1. Again using eye degeneration as a convenient assay, they also showed that LKB-1 affects the toxic function of tau; knocking-down LKB-1 expression suppressed the degeneration caused by PAR-1 or APP/tau expression, whereas overexpression of LKB-1 enhanced the toxic effects induced by PAR-1 and tau. Together with the observation that expression of APP enhanced the PAR-1 phenotype in an LKB-1 dependent manner, these results suggest a pathway in which APP activates LKB-1, which in turn phosphorylates PAR-1, leading to hyperphosphorylation of tau. As in other models, hyperphosphorylation of tau then results in aggregate formation and degeneration.
Concluding remarks
The successful establishment of fly and worm models that mimic many of the prominent phenotypes described for Alzheimer’s Disease and other tauopathies has opened the door to use invertebrate models and the experimental advantages they offer to delineate the molecular and genetic interactions underlying AD and related tauopathies. In particular, the foregoing studies have demonstrated that many of the neurotoxic effects associated with tau and amyloid peptides can be recapitulated in these organisms. This discovery provides a strong rational for using these models for systematic genetic interaction screens to identify the molecular pathways that are perturbed in these diseases. In turn, promising novel genes and pathways described in invertebrates can then be targeted in mammalian model systems to confirm their relevance in human disease. Combining the advantages of invertebrate and mammalian models can therefore provide a powerful experimental approach for determining the causes of these devastating diseases, and provide new targets for developing novel diagnostic and therapeutic strategies.
Fig. 1.
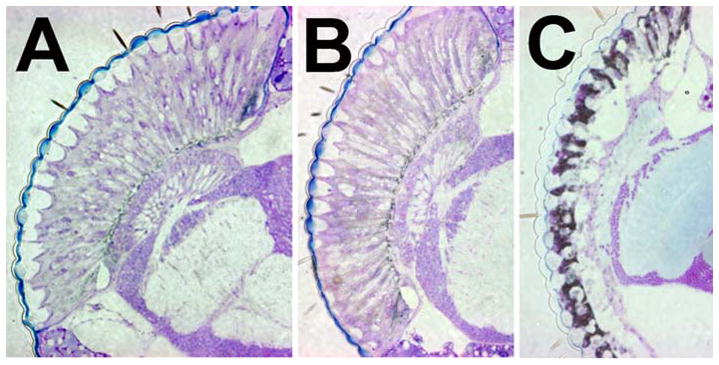
Retinal degeneration caused by human APP695. A) 25d old control fly. B) At 1d of adult life a transgenic fly expressing APP695 in the eye does not show significant degeneration whereas a fly aged for 25d reveals severe retinal degeneration. (C).
Fig. 2.

Transgenic C. elegans expressing tau are uncoordinated. A) A wild type C. elegans shows the characteristic sinusoidal movement which is disrupted in an animal expressing human tauM337V (B). Pictures kindly provided by B. Kraemer, University of Washington.
Acknowledgments
We thank all the fly and worm groups working in the field of Alzheimer’s disease for their excellent work and we apologize for not including certain publications. We are grateful to Philip Copenhaver for critical reading of the manuscript. Work in the authors laboratory is supported by the NIH, the Alzheimer Research Initiative, and the Medical Research Foundation of Oregon.
References
- Alonso AC, Li B, Grundke-Iqbal I, Iqbal K. Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2008;5:375–384. doi: 10.2174/156720508785132307. [DOI] [PubMed] [Google Scholar]
- Anliker B, Muller U. The functions of mammalian amyloid precursor protein and related amyloid precursor-like proteins. Neurodegener Dis. 2006;3:239–246. doi: 10.1159/000095262. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Ashley J, Packard M, Ataman B, Budnik V. Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. J Neurosci. 2005;25:5943–5955. doi: 10.1523/JNEUROSCI.1144-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O’Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, Mullin K, Menon R, Sampson AJ, Hsiao MY, Elliott KJ, Velicelebi G, Moscarillo T, Hyman BT, Wagner SL, Becker KD, Blacker D, Tanzi RE. Family-based association between Alzheimer’s disease and variants in UBQLN1. N Engl J Med. 2005;352:884–894. doi: 10.1056/NEJMoa042765. [DOI] [PubMed] [Google Scholar]
- Bettencourt da Cruz A, Schwarzel M, Schulze S, Niyyati M, Heisenberg M, Kretzschmar D. Disruption of the MAP1B-related protein FUTSCH leads to changes in the neuronal cytoskeleton, axonal transport defects, and progressive neurodegeneration in Drosophila. Mol Biol Cell. 2005;16:2433–2442. doi: 10.1091/mbc.E04-11-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet. 2005;39:153–171. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- Blard O, Frebourg T, Campion D, Lecourtois M. Inhibition of proteasome and Shaggy/Glycogen synthase kinase-3beta kinase prevents clearance of phosphorylated tau in Drosophila. J Neurosci Res. 2006;84:1107–1115. doi: 10.1002/jnr.21006. [DOI] [PubMed] [Google Scholar]
- Bloom GS, Ren K, Glabe CG. Cultured cell and transgenic mouse models for tau pathology linked to beta-amyloid. Biochim Biophys Acta. 2005;1739:116–124. doi: 10.1016/j.bbadis.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Cao W, Song HJ, Gangi T, Kelkar A, Antani I, Garza D, Konsolaki M. Identification of novel genes that modify phenotypes induced by Alzheimer’s beta-amyloid overexpression in Drosophila. Genetics. 2008;178:1457–1471. doi: 10.1534/genetics.107.078394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmine-Simmen K, Proctor T, Tschape J, Poeck B, Triphan T, Strauss R, Kretzschmar D. Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiol Dis. 2009;33:274–281. doi: 10.1016/j.nbd.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Sang TK, Lawless GM, Jackson GR. Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum Mol Genet. 2009;18:164–177. doi: 10.1093/hmg/ddn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau KW, Chan WY, Shaw PC, Chan HY. Biochemical investigation of Tau protein phosphorylation status and its solubility properties in Drosophila. Biochem Biophys Res Commun. 2006;346:150–159. doi: 10.1016/j.bbrc.2006.05.112. [DOI] [PubMed] [Google Scholar]
- Chee F, Mudher A, Newman TA, Cuttle M, Lovestone S, Shepherd D. Overexpression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Biochem Soc Trans. 2006;34:88–90. doi: 10.1042/BST0340088. [DOI] [PubMed] [Google Scholar]
- Chen X, Li Y, Huang J, Cao D, Yang G, Liu W, Lu H, Guo A. Study of tauopathies by comparing Drosophila and human tau in Drosophila. Cell Tissue Res. 2007;329:169–178. doi: 10.1007/s00441-007-0401-y. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, Gubb DC, Lomas DA. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience. 2005;132:123–135. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Daigle I, Li C. apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci U S A. 1993;90:12045–12049. doi: 10.1073/pnas.90.24.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113(Pt 11):1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest. 2007;117:236–245. doi: 10.1172/JCI28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay DS, Fluet A, Johnson CJ, Link CD. In vivo aggregation of beta-amyloid peptide variants. J Neurochem. 1998;71:1616–1625. doi: 10.1046/j.1471-4159.1998.71041616.x. [DOI] [PubMed] [Google Scholar]
- Findeis MA. The role of amyloid beta peptide 42 in Alzheimer’s disease. Pharmacol Ther. 2007;116:266–286. doi: 10.1016/j.pharmthera.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26:365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Folwell J, Cowan CM, Ubhi KK, Shiabh H, Newman TA, Shepherd D, Mudher A. Abeta exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer’s disease. Exp Neurol. 2009 doi: 10.1016/j.expneurol.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Fonte V, Kipp DR, Yerg J, 3rd, Merin D, Forrestal M, Wagner E, Roberts CM, Link CD. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem. 2008;283:784–791. doi: 10.1074/jbc.M703339200. [DOI] [PubMed] [Google Scholar]
- Fossgreen A, Bruckner B, Czech C, Masters CL, Beyreuther K, Paro R. Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci U S A. 1998;95:13703–13708. doi: 10.1073/pnas.95.23.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Feldman RM, Guo M. ubiquilin antagonizes presenilin and promotes neurodegeneration in Drosophila. Hum Mol Genet. 2008;17:293–302. doi: 10.1093/hmg/ddm305. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Grammenoudi S, Kosmidis S, Skoulakis EM. Cell type-specific processing of human Tau proteins in Drosophila. FEBS Lett. 2006;580:4602–4606. doi: 10.1016/j.febslet.2006.07.045. [DOI] [PubMed] [Google Scholar]
- Greeve I, Kretzschmar D, Tschape JA, Beyn A, Brellinger C, Schweizer M, Nitsch RM, Reifegerste R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci. 2004;24:3899–3906. doi: 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- Guthrie CR, Schellenberg GD, Kraemer BC. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum Mol Genet. 2009;18:1825–1838. doi: 10.1093/hmg/ddp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, De Strooper B. The presenilins in Alzheimer’s disease--proteolysis holds the key. Science. 1999;286:916–919. doi: 10.1126/science.286.5441.916. [DOI] [PubMed] [Google Scholar]
- Heidary G, Fortini ME. Identification and characterization of the Drosophila tau homolog. Mech Dev. 2001;108:171–178. doi: 10.1016/s0925-4773(01)00487-7. [DOI] [PubMed] [Google Scholar]
- Heisenberg M. Mushroom body memoir: from maps to models. Nat Rev Neurosci. 2003;4:266–275. doi: 10.1038/nrn1074. [DOI] [PubMed] [Google Scholar]
- Hummel T, Krukkert K, Roos J, Davis G, Klambt C. Drosophila Futsch/22C10 is a MAP1B-like protein required for dendritic and axonal development. Neuron. 2000;26:357–370. doi: 10.1016/s0896-6273(00)81169-1. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Heutink P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Iijima K, Chiang HC, Hearn SA, Hakker I, Gatt A, Shenton C, Granger L, Leung A, Iijima-Ando K, Zhong Y. Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS One. 2008;3:e1703. doi: 10.1371/journal.pone.0001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- Jacobsen KT, Iverfeldt K. Amyloid precursor protein and its homologues: a family of proteolysis-dependent receptors. Cell Mol Life Sci. 2009;66:2299–2318. doi: 10.1007/s00018-009-0020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboh MI, Minster RL, Feingold E, DeKosky ST. Genetic association of ubiquilin with Alzheimer’s disease and related quantitative measures. Mol Psychiatry. 2006;11:273–279. doi: 10.1038/sj.mp.4001775. [DOI] [PubMed] [Google Scholar]
- Karsten SL, Sang TK, Gehman LT, Chatterjee S, Liu J, Lawless GM, Sengupta S, Berry RW, Pomakian J, Oh HS, Schulz C, Hui KS, Wiedau-Pazos M, Vinters HV, Binder LI, Geschwind DH, Jackson GR. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16:230–241. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Burgess JK, Chen JH, Thomas JH, Schellenberg GD. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum Mol Genet. 2006;15:1483–1496. doi: 10.1093/hmg/ddl067. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Schellenberg GD. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum Mol Genet. 2007;16:1959–1971. doi: 10.1093/hmg/ddm143. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003;100:9980–9985. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Leyssen M, Ayaz D, Hebert SS, Reeve S, De Strooper B, Hassan BA. Amyloid precursor protein promotes post-developmental neurite arborization in the Drosophila brain. Embo J. 2005;24:2944–2955. doi: 10.1038/sj.emboj.7600757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Xie Z, Dong Y, McKay KM, McKee ML, Tanzi RE. Isolation and characterization of the Drosophila ubiquilin ortholog dUbqln: in vivo interaction with early-onset Alzheimer disease genes. Hum Mol Genet. 2007;16:2626–2639. doi: 10.1093/hmg/ddm219. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu T, Peng Y, Yuan C, Guo A. Specific functions of Drosophila amyloid precursor-like protein in the development of nervous system and nonneural tissues. J Neurobiol. 2004;61:343–358. doi: 10.1002/neu.20048. [DOI] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD. C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer’s disease. Exp Gerontol. 2006;41:1007–1013. doi: 10.1016/j.exger.2006.06.059. [DOI] [PubMed] [Google Scholar]
- Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol. 2009;4:315–342. doi: 10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Tully T, White K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron. 1992;9:595–605. doi: 10.1016/0896-6273(92)90024-8. [DOI] [PubMed] [Google Scholar]
- Luo LQ, Martin-Morris LE, White K. Identification, secretion, and neural expression of APPL, a Drosophila protein similar to human amyloid protein precursor. J Neurosci. 1990;10:3849–3861. doi: 10.1523/JNEUROSCI.10-12-03849.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998;8:425–427. doi: 10.1016/s0962-8924(98)01368-3. [DOI] [PubMed] [Google Scholar]
- Mershin A, Pavlopoulos E, Fitch O, Braden BC, Nanopoulos DV, Skoulakis EM. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem. 2004;11:277–287. doi: 10.1101/lm.70804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, Asuni AA, Bhat R, Lovestone S. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- Murray MJ, Merritt DJ, Brand AH, Whitington PM. In vivo dynamics of axon pathfinding in the Drosophilia CNS: a time-lapse study of an identified motorneuron. J Neurobiol. 1998;37:607–621. [PubMed] [Google Scholar]
- Nimmrich V, Ebert U. Is Alzheimer’s disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid. Rev Neurosci. 2009;20:1–12. doi: 10.1515/revneuro.2009.20.1.1. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–682. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senechal Y, Larmet Y, Dev KK. Unraveling in vivo functions of amyloid precursor protein: insights from knockout and knockdown studies. Neurodegener Dis. 2006;3:134–147. doi: 10.1159/000094772. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Froelich S, Sorbi S, Campion D, Chi H, Rogaeva EA, Levesque G, Rogaev EI, Lin C, Liang Y, Ikeda M, Mar L, Brice A, Agid Y, Percy ME, Clerget-Darpoux F, Piacentini S, Marcon G, Nacmias B, Amaducci L, Frebourg T, Lannfelt L, Rommens JM, St George-Hyslop PH. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet. 1996;5:985–988. doi: 10.1093/hmg/5.7.985. [DOI] [PubMed] [Google Scholar]
- Shulman JM, Feany MB. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–1242. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sider JR, Mandato CA, Weber KL, Zandy AJ, Beach D, Finst RJ, Skoble J, Bement WM. Direct observation of microtubule-f-actin interaction in cell free lysates. J Cell Sci. 1999;112(Pt 12):1947–1956. doi: 10.1242/jcs.112.12.1947. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, Feany MB. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell. 2007a;18:5060–5068. doi: 10.1091/mbc.E07-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Mulkearns EE, Shulman JM, Biernat J, Mandelkow EM, Feany MB. S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J Neurosci Res. 2007b;85:1271–1278. doi: 10.1002/jnr.21232. [DOI] [PubMed] [Google Scholar]
- Teschendorf D, Link CD. What have worm models told us about the mechanisms of neuronal dysfunction in human neurodegenerative diseases? Mol Neurodegener. 2009;4:38. doi: 10.1186/1750-1326-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian AG, Deng WM. Par-1 and Tau regulate the anterior-posterior gradient of microtubules in Drosophila oocytes. Dev Biol. 2009;327:458–464. doi: 10.1016/j.ydbio.2008.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroja L, Chu H, Kotovsky I, White K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol. 1999a;9:489–492. doi: 10.1016/s0960-9822(99)80215-2. [DOI] [PubMed] [Google Scholar]
- Torroja L, Packard M, Gorczyca M, White K, Budnik V. The Drosophila beta-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci. 1999b;19:7793–7803. doi: 10.1523/JNEUROSCI.19-18-07793.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschape JA, Hammerschmied C, Muhlig-Versen M, Athenstaedt K, Daum G, Kretzschmar D. The neurodegeneration mutant lochrig interferes with cholesterol homeostasis and Appl processing. Embo J. 2002;21:6367–6376. doi: 10.1093/emboj/cdf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Ubhi KK, Shaibah H, Newman TA, Shepherd D, Mudher A. A comparison of the neuronal dysfunction caused by Drosophila tau and human tau in a Drosophila model of tauopathies. Invert Neurosci. 2007;7:165–171. doi: 10.1007/s10158-007-0052-4. [DOI] [PubMed] [Google Scholar]
- van de Hoef DL, Hughes J, Livne-Bar I, Garza D, Konsolaki M, Boulianne GL. Identifying genes that interact with Drosophila presenilin and amyloid precursor protein. Genesis. 2009;47:246–260. doi: 10.1002/dvg.20485. [DOI] [PubMed] [Google Scholar]
- Wang JW, Imai Y, Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neurosci. 2007;27:574–581. doi: 10.1523/JNEUROSCI.5094-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DW, Tyrer M, Shepherd D. Tau and tau reporters disrupt central projections of sensory neurons in Drosophila. J Comp Neurol. 2000;428:630–640. doi: 10.1002/1096-9861(20001225)428:4<630::aid-cne4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- Wu Y, Luo Y. Transgenic C. elegans as a model in Alzheimer’s research. Curr Alzheimer Res. 2005;2:37–45. doi: 10.2174/1567205052772768. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wu Z, Butko P, Christen Y, Lambert MP, Klein WL, Link CD, Luo Y. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci. 2006;26:13102–13113. doi: 10.1523/JNEUROSCI.3448-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]