

Abstract
Hirano bodies are cytoplasmic inclusions predominantly found in the central nervous system associated with various conditions including aging and Alzheimer's disease (AD). Since most studies of Hirano bodies have been performed in post-mortem samples, the physiological roles of Hirano bodies have not been investigated. Astrocytoma H4 cells were employed to test the hypothesis that Hirano bodies interact with and modulate signaling by the C-terminal fragment of amyloid-β precursor protein (AICD). We demonstrated by immunofluorescence and immunoprecipitation that model Hirano bodies accumulate AICD. Since stimulation of transcription by AICD is dependent on its interaction with the nuclear adaptor protein Fe65, we examined localization of Fe65, and employed a dual luciferase reporter assay to test the effects of Hirano bodies on AICD- and Fe65-dependent modulation of gene expression. We find that both AICD and Fe65 are co-localized in model Hirano bodies. Model Hirano bodies also down-regulate both AICD-dependent apoptosis and AICD- and Fe65-dependent transcriptional activity. Thus, association of AICD and Fe65 with Hirano bodies impedes their function in promoting apoptosis and modulating transcription.
Keywords: AICD, Amyloid Precursor Protein, Fe65, Hirano Body, Actin Filaments, Alzheimer's Disease, neurodegenerative disease, apoptosis
1. Introduction
Hirano bodies are intracellular inclusions found in a number of neurodegenerative diseases including Alzheimer's disease (AD), Amyotrophic Lateral Sclerosis (ALS), Parkinson's disease, and other conditions in humans and animals producing persistent injury or stress such as diabetes and chronic alcoholism (Gibson and Tomlinson, 1977; Hirano, 1994; Laas and Hagel, 1994; Mitake et al., 1997; Mori et al., 1986; Okamoto et al., 1982; Schmidt et al., 1989; Yamamoto and Hirano, 1985). Although Hirano bodies are often reported in neurons of the central nervous system (CNS), particularly the Ammon's horn (CA1) in the hippocampus, they have been reported in a variety of cells such as glial cells, peripheral nerve axons, and in skeletal muscle (Hirano, 1994). These inclusions are also observed in normal elderly individuals without obvious underlying neurodegeneration (Gibson and Tomlinson, 1977; Ogata et al., 1972). The fundamental organization of Hirano bodies consists of highly ordered, eosinophilic, paracrystalline interlacing arrays of 6-10 nm parallel filaments which show a lattice or herringbone-like arrangement of fibrillary structure (Hirano, 1994; Schochet and McCormick, 1972). They have been reported to contain actin filaments and actin-associated proteins (Galloway et al., 1987; Goldman, 1983; Maciver and Harrington, 1995), microtubule associated proteins including tau (Davis et al., 2008; Galloway et al., 1987; Peterson et al., 1988), and a variety of signaling molecules and transcriptional regulators including protein kinase C (Shao et al., 2006), inducible nitric oxide synthase (Lee et al., 1999), FAC1 (Jordan-Sciutto et al., 1998), and the cytoplasmic fragment of the amyloid precursor protein (Munoz et al., 1993). The latter observation is particularly interesting, since the carboxy-terminal fragment of the amyloid precursor protein (AICD or γ-CTF) is released by presenilin-dependent gamma secretase cleavage of the amyloid precursor protein. AICD forms multimeric complexes with the nuclear adaptor protein Fe65, the histone acetyltransferase Tip60, or CP2/LSF/LBP1 transcription factor, and these complexes potently stimulate gene expression and induce apoptosis (Cao and Südhof, 2001, 2004; Kim et al., 2003; Kim et al., 2004; King and Turner, 2004; Kinoshita et al., 2002; Kinoshita et al., 2002; von Rotz et al., 2004; Xu et al., 2007).
The physiological roles of Hirano bodies are not understood, because most studies of Hirano bodies have been performed in post-mortem samples from patients with neurodegenerative disease. Model Hirano bodies were first induced in Dictyostelium by expression of modified forms of a 34 kDa actin bundling protein with activated actin binding activity (Maselli et al., 2002; Maselli et al., 2003). More recently, we have demonstrated formation of model Hirano bodies in cultured mammalian cells, and compared these model Hirano bodies to authentic Hirano bodies in the brain by rigorous immunohistochemical and ultrastructural criteria providing firm evidence that this system provides a good model for studies of Hirano bodies (Davis et al., 2008). In this paper, we have utilized this model to test the hypothesis that Hirano bodies interact with and modulate signaling by the C-terminal fragment of amyloid-β precursor protein (AICD). We report that expression in human H4 cells of GFP fused to the carboxy-terminus of a 34 kDa actin binding protein (CT-GFP) with activated actin binding activity induces the formation of model Hirano bodies that contains sites for accumulation of a C-terminal fragment of amyloid-β precursor protein (AICD) and a neuronal adaptor protein Fe65. The model Hirano bodies efficiently decrease AICD-induced apoptotic cell death determined by cell viability and caspase-3 activity. Moreover, we demonstrate that model Hirano bodies down-regulate the transcriptional activity of APP-Gal4 and APPct-Gal4 reporter constructs in the presence of Fe65. Therefore, our results suggest that a physiological effect of Hirano bodies on mammalian cells is to impede and down-regulate AICD-induced gene transactivation, and AICD-mediated apoptosis.
2. Methods
2.1 Generation of expression construct and cell culture conditions
For generation of the expression construct for model Hirano bodies (Lim et al., 1999b; Maselli et al., 2002), truncated CT fragment (amino acids 124-295) encoding 34 kDa actin bundling protein from Dictyostelium was subcloned into the BamHI site of the pEGFP-N1 (Clontech, Palo Alto, CA) at the carboxyl-terminus to express a fusion protein of CT with GFP (CT-GFP) (Davis et al., 2008). H4 cells (American Type Culture Collection, Manassas, VA) and HEK293T (CRL-1573, ATCC) cells derived from human embryonic kidney cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 U/ml penicillin/streptomycin and 10% fetal bovine serum. Transient transfection of cells was performed using FuGene 6 (Roche Applied Science, Nutley, NJ) or LipofectAMINE PLUS (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. To generate stable cell lines expressing pEGFP-N1 or CT-GFP in H4 and HEK 293 cells, cells were transfected following the manufacturer's instructions with 10 μg of plasmid DNA and either 50 μL of LipofectAMINE PLUS or FuGene 6 per 100 mm culture dish. Stable cell lines were generated through G418 selection (800 μg/ml final concentration) for 14 days, and clones were isolated and sorted by MoFlo cell sorter (DakoCytomation, Ft. Collins, Colorado).
2.2 Immunofluorescence
Immunofluorescence of transiently transfected cells was performed 24–48 h post-transfection. The cells were fixed in 3.7% formaldehyde for 10 min, washed three times with PBS, permeabilized with 0.2% Triton X-100 for 5 min, and blocked with PBS containing 5 % bovine serum albumin for 1 h at room temperature. The cells were incubated with appropriate primary antibodies for 1 h at room temperature: mouse anti-HA.11 monoclonal antibody (Covance, Princeton, NJ; 1:1000) to label HA-Fe65, rabbit anti-c-Myc A-14 (Santa Cruz Biotechnology, Santa Cruz, CA; 1:200) to label APPc58-myc. The cells were washed three times with PBS and labeled with Cy5-conjugated anti-mouse or anti-rabbit IgG antibody (Jackson Immunoresearch Laboratories, West Grove, PA) or TRITC-conjugated anti-mouse or anti-rabbit IgG antibody (Sigma-Aldrich, St. Louis, MO) and Hoechst 33342 (Sigma-Aldrich, St. Louis, MO) for labeling nuclei for 1 h at room temperature. The cells were washed three times with PBS, mounted with Crystal Mount (Biomeda, Foster City, CA), and examined with a Leica TCS SP2 spectral confocal microscope with coherent Ti:sapphire multiphoton laser (Mira Optima 900-F) (Leica Microsystems Inc, PA).
2.3 Assessment of cell viability and caspase-3/7 activity
H4 cells stably expressing CT-GFP or EGFP were plated at 4 × 105 cells in 60-mm culture dishes. Cells were transiently transfected with 0, 0.5 or 1.0 μg of APPc58-myc (generously provided by Dr. Bradley T. Hyman, Harvard Medical School, Charlestown, Massachusetts). At 24 h post-transfection, cells were stained directly with a mixture of 10 μg Hoechst 33342 and 10 μg propidium iodide. Stained cells were visualized and counted using the confocal microscope with a 40× water immersion objective in five random fields from each culture dish. More than one hundred cells were counted per field in triplicate for each condition. To calculate the cell death ratio, the number of propidium iodide stained cells was divided by the total number of Hoechst stained cells. Caspase activity was measured by fluorometric Apo-One Homogeneous Caspase-3/7 assay (Promega, Madison, WI). Cells were transiently co-transfected with APPc58-myc and pCS-βgal constructs in 60-mm culture dishes. At 24 h post-transfection, cells were washed twice with PBS and lysed with 500 μL of 1× reporter lysis buffer at room temperature for 15 min. After centrifugation, protein concentrations in the supernatant were determined by Bradford protein assay (Bio-Rad, Richmond, CA, USA) using BSA as a standard. Cell lysates (20 μg of protein) and caspase-3/7 substrate (Z-DEVD-R110) were added to 96-well clear-bottom plates in triplicate wells and incubated at 37°C for 1 h to measure caspase-3/7 activity. Reporter lysis buffer without cells was used to measure background fluorescence. Fluorescence of the rhodamine 110 leaving group was measured on a fluorescence plate reader (Bio-Tek Synergy HT, Winooski, VT) set at 499 nm excitation and 512 nm emission. Caspase-3/7 activity was calculated as [((mean R110 fluorescence performed in triplicate)-(mean background fluorescence)) divided by μg of protein]. In addition, to normalize transfection efficiency of APPc58-myc in caspase assays, cells were co-transfected with 0.25 μg of pCS-βgal construct. The β-galactosidase product was measured at 420 nm. All measurements were performed in triplicate in three independent experiments. Statistically significant differences were determined using Student's t-test.
2.4 Immunoprecipitation and western blot
HEK293T cells stably expressing EGFP or CT-GFP were plated at a density of 2 × 106 cells per 100 mm diameter dish. Cells were transfected with either APP695, APPc58-myc, or APPc58-myc and HA-Fe65 plasmid (Cao and Südhof, 2001) (generously provided by Dr. Thomas Südhof, Southwestern Medical School, Dallas, TX) using FuGene6 reagent according to manufacturer recommendations. At 48 hr post-transfection, cells were washed twice with ice-cold PBS and lysed with lysis buffer (1% triton-X 100, 0.1% SDS, 0.5% deoxycholic acid, 20 mM Tris-Cl pH 7.5, 10% glycerol, 0.5 mM EDTA, 100 mM PMSF, and protease inhibitor cocktail (Roche Applied Science) for 10 min on ice. Cells were scraped, collected, and incubated for 20 min on ice with occasional vortexing. Cell debris was separated from total protein lysates by centrifugation at 13,000g for 20 min at 4°C. Supernatant was stored at -20°C until used. Protein concentrations of the supernatants were determined by either Bradford or bicinchoninic acid protein assay using BSA as a standard. For immunoblot analysis, samples were loaded with 50 μg of protein from total protein lysates and separated by SDS-PAGE. Blots were probed using anti-34 kDa (B2C) mouse monoclonal antibody at a 1:5000 dilution or anti-GFP mouse monoclonal antibody (Molecular Probes-Invitrogen, Carlsbad, CA) at a 1:3000 dilution. The signals were detected by chemiluminescence (Pierce Biotechnology, Rockford, IL). For immunoprecipitation assays, cell lysates of 500 μg of total protein were immunoprecipitated with either anti-APPct (Sigma-Aldrich, St. Louis, MO), anti-GFP (Covance, Emeryville, CA), anti-c-Myc A-14 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Fe65 E-20 (Santa Cruz Biotechnology), or anti-HA.11 (Covance) antibody, and collected with protein G-Agarose (Roche Applied Science, Nutley, NJ). The beads were washed 3 times in lysis buffer. Immunoprecipitated proteins were eluted, resolved by SDS-PAGE, transferred to nitrocellulose (BA-85) for 1 hr or 4 hr (immunoprecipitation with anti-GFP) at 100 V, and analyzed by immunoblotting with chemiluminescent detection.
2.5 Transactivation assays
This assay was performed essentially as described (Cao and Südhof, 2001). For Gal4 transactivation assays, luciferase assays were performed using a Dual-Glo luciferase assay system (Promega, Madison, WI) according to the manufacturer's instructions. Most plasmids for transactivation assays were a generous gift from Dr. T. Südhof (University of Texas Southwestern Medical Center, Dallas, TX); (A) pG5E1B-luc (0.1 μg DNA) which contains five Gal4 binding sites and the E1B minimal promoter in front of the firefly luciferase gene; (B) pMst (Gal4, 0.1 μg DNA), pMst-APP695 (APP-Gal4, 0.1 μg DNA), pMst-APP695* (APP*-Gal4, 0.1 μg DNA), pMst-APPct (APPct-Gal4, 0.1 μg DNA), and pMst-APPct* (APPct*-Gal4, 0.1 μg DNA); Plasmids with * contain NATA mutation instead of NPTY in the C-terminal domain of APP. (C) pRL-TK (kind gift of Dr. J. Lauderdale, University of Georgia, 0.02 μg DNA) which produces Renilla luciferase as a control for transfection efficiency; (D) HA-Fe65 (0.1 μg DNA) which stimulates transcription by binding to the YENPTY sequence of the C-terminal domain of APP. Briefly, HEK293T cells stably expressing EGFP or CT-GFP were co-transfected with three or four plasmids at a density of 1 × 104 cells per well in white, opaque 96-well microtiter plates using FuGene6 reagent. At 48 h after transfection, cells were lysed with Glo lysis buffer, and firefly and Renilla luciferase activities in cell lysates were sequentially measured using the Dual-Glo Luciferase Reporter System and Lmax luminometer (Molecular Devices, Sunnyvale, Calif.). Firefly luciferase activity from reporter plasmid pG5E1B-Luc was produced by adding Dual-Glo luciferase substrate. Renilla luciferase activity from pRL-TK was produced by addition of Dual-Glo Stop&Glo substrate which is a substrate for Renilla luciferase and acts also as the stop solution for firefly luciferase. Firefly luciferase luminescence was normalized by Renilla luciferase luminescence as a control for transfection efficiency and further normalized by beta-galactosidase activity as a control for the transactivation assay. The mean values were obtained from transactivation assays performed in triplicate and repeated in at least three independent experiments. Statistical analyses were performed using Student's t-test.
3. Results
3.1 Localization of the C-terminal domain of amyloid-β precursor protein (AICD) and Fe65 in Hirano bodies
We have developed a cell culture model for the formation of Hirano bodies in Dictyostelium and mammalian cells using altered forms of the 34 kDa actin bundling protein (Davis et al., 2008; Maselli et al., 2002; Maselli et al., 2003). We wanted to verify whether the C-terminal fragment of APP is associated with model Hirano bodies in human H4 cells stably expressing CT-GFP fusion protein as predicted by the original study of Hirano bodies in the CA1 region of the hippocampus (Munoz et al., 1993). Results from immunofluorescence and immunoprecipitation studies of both endogenous and overexpressed AICD, APP, and Fe65 are shown in figures 1-4. Immunofluorescence of H4 stable cells expressing either GFP or CT-GFP was performed utilizing an antibody raised against the C-terminal amino acids 676-695 of APP. This antibody recognizes all endogenous forms of APP that contain the COOH-terminus including APP695, C99, C83, and C57-59. This collection of APP isoforms recognized by the antibody to the COOH terminus will be referred to as APPx. APPx is diffusely distributed through the control H4 cells expressing GFP, and co-localizes with Hirano bodies in CT-GFP H4 cells (Figure 1A).
Figure 1.
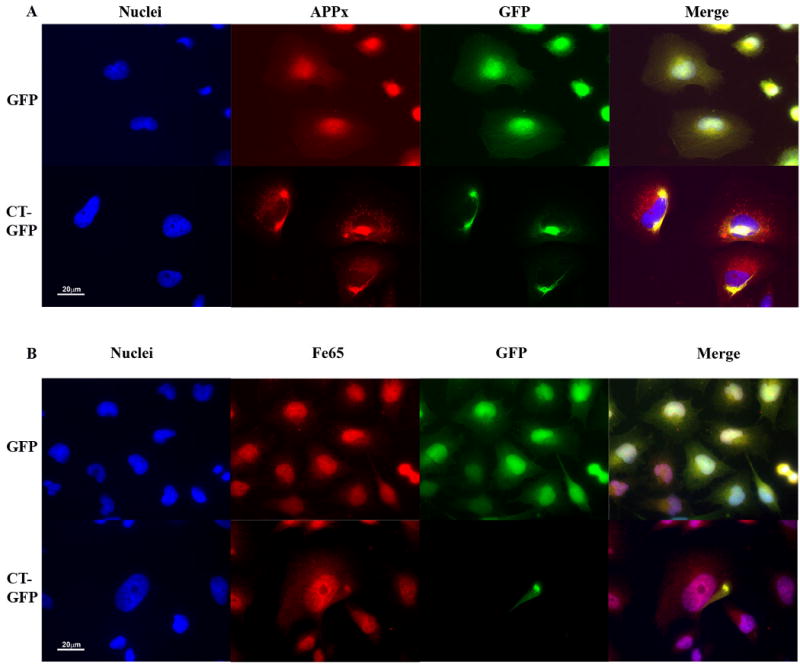
The C-terminal domain of the amyloid-β precursor protein is co-localized with Hirano bodies. H4 cells stably expressing GFP or CT-GFP were stained for the presence of endogenous forms of APP that include the C-terminus (APPx) (A) or Fe65 (B), and visualized by TRITC-conjugated secondary antibodies (red). DNA was stained with Hoechst dye (blue). A. APPx strongly localized with the nucleus and perinuclear region with limited diffuse cytoplasmic staining in GFP stable cells, and showed extensive co-localization with Hirano bodies in the cytoplasm in CT-GFP stable cells. Areas of co-localization appear yellow in the merged image. B. Fe65 co-localized extensively with nuclei in GFP stable cells and is partitioned between the nucleus and Hirano bodies in CT-GFP stable cells. Scale bar = 20 μm.
Figure 4.
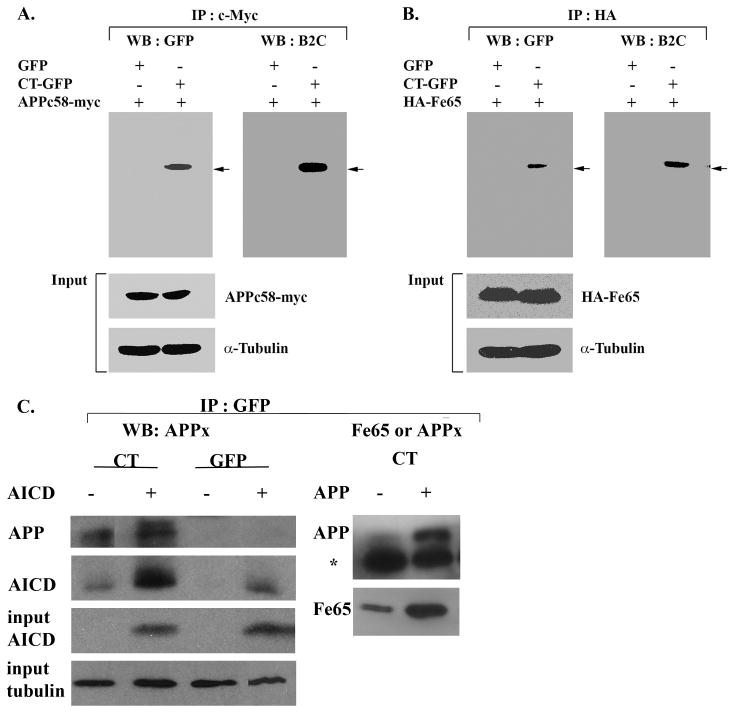
Co-localization of AICD and Fe65 with Hirano bodies. Lysates from cells transfected with APPc58-myc (A, C) or HA-Fe65 (B) or APP695 (C) were immunoprecipitated with anti-myc antibody (A) or anti-HA antibody (B) or anti-GFP (C). The immunoprecipitated proteins were analyzed by western blot with either anti-GFP or anti-34 kDa (B2C) antibody or anti-APPct or anti-Fe65 (B). To verify equal loading of the different cell lysates, α-tubulin was employed as a control as well as APPc58-myc in A, C and HA-Fe65 in B. The results show a specific interaction of APPc58 (A,C), Fe65 (B,C), and APP695 with CT-GFP in model Hirano bodies, but not with GFP in control cells. The * in C indicates the heavy chain of IgG arising from the GFP antibody used for the immunoprecipitation.
Fe65 is a neuronal adaptor protein that interacts with the C-terminal domain of APP through a YENPTY binding motif. The Fe65 is activated by interaction with this YENPTY motif, and the activated form of Fe65 potently activates transcription in the nucleus (Cao and Südhof, 2001, 2004; Kim et al., 2003; Kinoshita et al., 2002). Since APPx is co-localized in model Hirano bodies, we assessed whether Fe65 is also co-localized in model Hirano bodies in H4 stable cells expressing CT-GFP fusion protein. In H4 stable cells expressing GFP, Fe65 was predominantly localized in the nucleus and also diffusely distributed in the cytoplasm (Figure 1B). Fe65 was also localized in the nucleus of CT-GFP stable cells (Figure 1B), but concentrated in model Hirano bodies in the cytoplasm of CT-GFP stable cells. These results show that Fe65 also associates with model Hirano bodies.
Biochemical confirmation of the localization of APPx and Fe65 in Hirano bodies was obtained through immunoprecipitation using antibody recognizing either the C-terminal fragment of APP or anti-Fe65. Western blots were probed with an antibody to GFP that recognizes free GFP and CT-GFP in the control and Hirano body containing cells, respectively (Figure 2A). The immunoprecipitate obtained with antibody to the C-terminal fragment of APP contained full length APP (not shown), and C99/C83 (Figure 2B). The AICD (C57-59) is probably not detectable on this blot due to its low concentration. The 46 kDa CT-GFP fusion protein was detected as a single band in immunoprecipitates prepared with anti-APPx or anti-Fe65, supporting the conclusion that model Hirano bodies expressing CT-GFP fusion protein contain sites for accumulation of endogenous Fe65 (Figure 2C) and APPx including at least the C83 and c99 fragments, and full length APP 695 (Figure 2B). When immunoprecipitates are prepared with anti-GFP and western blots probed with either anti-APPx (Figure 4C), endogenous AICD and a high molecular weight form corresponding to full length APP are present only in the model Hirano bodies. Western blots probed with anti-Fe65 (Figure 4C) reveal the presence of Fe65 in the model Hirano bodies.
Figure 2.
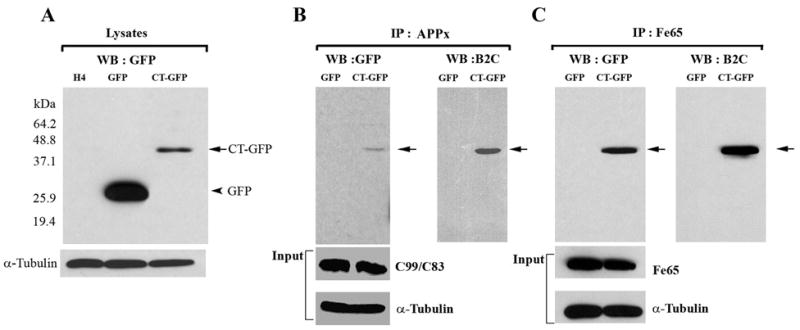
Hirano bodies bind to C-terminal fragments of APP (APP695, APPC99/APPC83 and APPC57-59) and Fe65. A. Cell lysates from wild type, GFP, and CT-GFP stable H4 cells were analyzed by western blot using antibody against GFP. To verify equal loading of the different cell lysates, α-tubulin was employed as a control. The 46 kDa CT-GFP fusion protein (arrow) and 27 kDa GFP protein (arrowhead) are detected as single bands. Marker proteins are 64.2, 48.8, 37.1, 25.9, and 19.4 kDa. B, C. Cell lysates obtained from GFP and CT-GFP stable H4 cells were immunoprecipitated with an antibody against the C-terminal portion of APP (anti-APPx) or with anti-Fe65 antibody, respectively. Products were detected with either anti-GFP or B2C, a monoclonal antibody against 34 kDa protein. To verify equal loading of the different cell lysates, α-tubulin was employed as a control as well as APPx (panel B) and Fe65 (panel C). The immunoprecipitate fraction in panel B contained full length APP (not shown) as well as C99/C83. The results show a specific interaction of APPx (B) and Fe65 (C) with CT-GFP in model Hirano bodies, but not with GFP.
To determine directly whether exogenous AICD (C58) could also associate with Hirano bodies, H4 stable cell lines expressing GFP or CT-GFP were transiently transfected with APPc58-myc (AICD), and the localization of APPc58-myc was determined by immunofluorescence with anti-c-Myc antibody (Figure 3A). The APPc58-myc showed a predominantly nuclear localization with diffuse cytoplasmic staining in GFP stable cells (Figure 3B), in agreement with previous reports of the localization of APPc58 in cells lacking Hirano bodies (Kim et al., 2003; Kinoshita et al., 2002; von Rotz et al., 2004). By contrast, the staining with antibody to myc was also partially nuclear in CT-GFP stable cells, but significantly co-localized with model Hirano bodies in the cytoplasm of these cells (Figure 3A). Similarly, in cells expressing exogenous HA-Fe65 and APPc58-myc, staining with antibody to the HA epitope reveals that Fe65 is co-localized with AICD in the model Hirano bodies (data not shown). Thus, AICD and Fe65 co-localize with Hirano bodies in cells overexpressing APPc58-myc.
Figure 3.
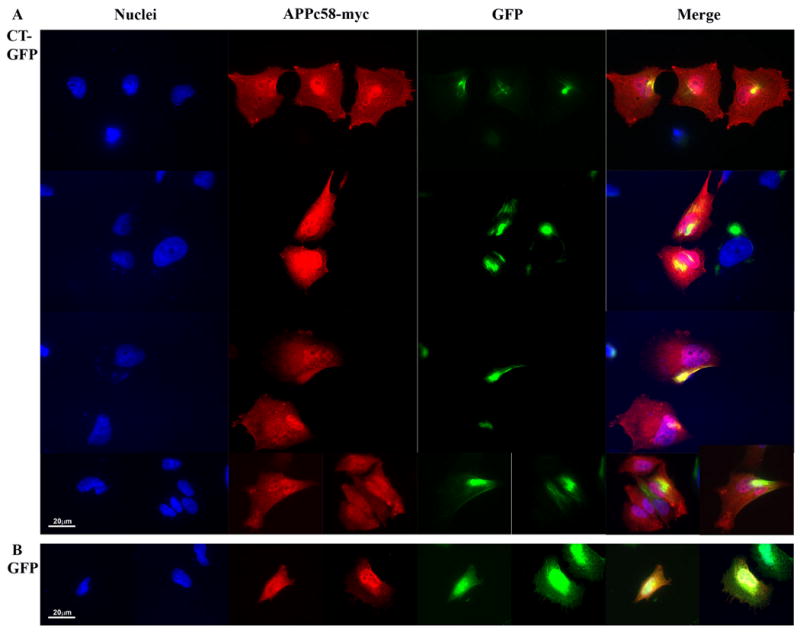
The C-terminal domain of the amyloid-β precursor protein is co-localized with Hirano bodies. H4 stable cells expressing (A) CT-GFP or (B) GFP were transiently transfected with APPc58-myc and visualized by anti-myc, and TRITC-conjugated secondary antibody (red) for APPc58-myc. DNA was stained with Hoechst dye (blue). A. Areas of specific co-localization of APPc58-myc with Hirano bodies in the cytoplasm in CT-GFP stable cells appear yellow in the merged images. B. APPc58-myc is strongly localized in the nucleus in GFP stable cells. Scale bar = 20 μm.
Biochemical confirmation of the molecular interaction of Hirano bodies and AICD was performed by immunoprecipitation from total cell lysates of cells stably expressing GFP or CT-GFP to induce model Hirano bodies, and either APPc58-myc or HA-Fe65 (Figure 4). Cell lysates were immunoprecipitated with anti-c-Myc antibody to APPc58-myc (AICD) or anti-HA antibody to HA-Fe65, and western blot analysis was performed with anti-GFP or anti-34 kDa (B2C) antibody. The 46 kDa CT-GFP fusion protein was detected as a single band in immunoprecipitates prepared from Hirano body containing cells but not from control cells (Figure 4). In addition, total cell lysates of cells stably expressing CT-GFP and transfected with either APPc58-myc (AICD) or APP695 were immunoprecipitated using anti-GFP, and western blot analysis was performed with anti-APPx or anti-Fe65 antibodies (Figure 4c). These results confirm that model Hirano bodies contain sites for accumulation of APP695, and endogenous or exogenously expressed forms of both Fe65 and AICD.
3.2 Inhibition of AICD-induced apoptosis by Hirano bodies
AICD and Fe65 associate with Tip60 and other factors in the nucleus to modify transcription, and to promote apoptosis by inducing GSK-3β and p53 (Alves da Costa et al., 2006; Kim et al., 2003; Kim et al., 2004; Kinoshita et al., 2002; Lu et al., 2003). In this study, we investigated the physiological effect of the presence of Hirano bodies on cell death and caspase-3 activity in GFP and CT-GFP expressing H4 cells transfected with different concentrations of APPc58-myc DNA. Immunoblotting was used to verify increases in expression of APPc58-myc with increasing concentration of plasmid used for transformation (Figure 5). In cells transfected with 0.5 and 1 μg of APPc58-myc, the percentage of dead cells measured by staining with propidium iodide 24 h after transfection was significantly lower in CT-GFP stable cells (10±0.9% and 25.9±2.5%) than in GFP stable cells (13±1.5% and 41.8±3.6%), respectively (means ± S.E.M; p < 0.01) (Figure 5A). Thus, AICD-induced cell death is reduced in CT-GFP expressing H4 cells that contain model Hirano bodies.
Figure 5.
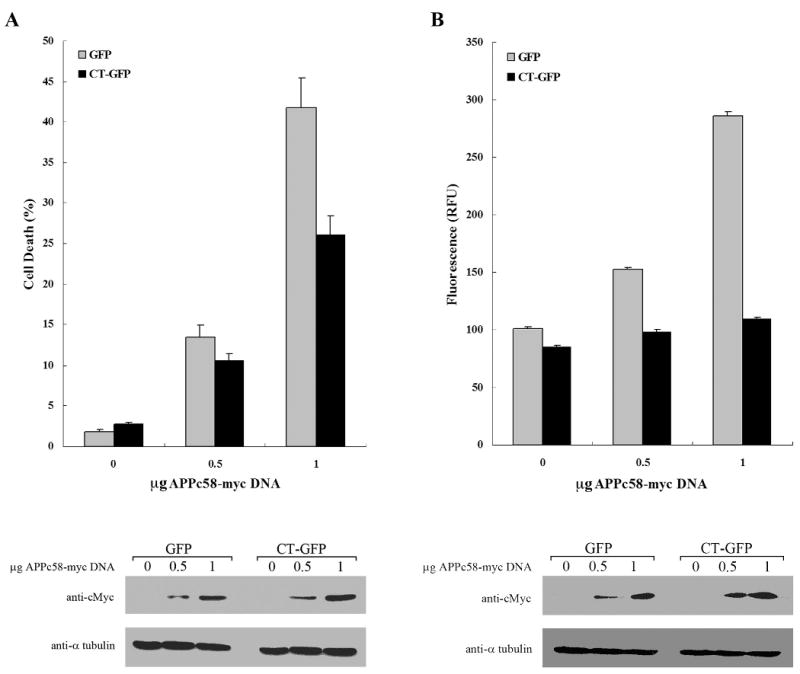
Hirano bodies significantly decrease AICD-induced apoptosis in H4 cells. H4 stable cells stably expressing GFP or CT-GFP were transiently transfected with APPc58-myc DNA (0, 0.5, or 1 μg DNA), and examined at 24 hours for cell viability (A) and caspase 3/7 activation (B). A. Cells were stained with propidium iodide (PI) and Hoechst dye 33342 to assess cell viability. CT-GFP stable cells with Hirano bodies showed a lower percentage of cell death than GFP stable cells (p < 0.001). B. Caspase-3 activity was measured using a fluorescence assay that was normalized to β-galactosidase activity as a control for transfection efficiency of APPc58-myc. Caspase-3 activity represented by relative fluorescence units (RFU) was significantly decreased in CT-GFP stable cells compared to GFP stable cells (p < 0.001). Data shown are mean ± S.E.M. of triplicate measurements, and repeated in three independent experiments. Cell lysates transiently transfected with various concentrations of APPc58-myc DNA were examined by immunoblotting with anti-c-Myc antibodies (bottom panel) to verify that expression of APPc58-myc increased with the amount of DNA transfected.
To confirm the effect of model Hirano bodies on AICD-induced cell death, we assessed the caspase-3 enzyme activity at the same conditions. H4 stable cells were co-transfected with 0, 0.5, and 1 μg of APPc58-myc containing 0.25 μg of βgal DNA to control for transfection efficiency. Caspase-3 activities in GFP stable cells were significantly increased depending on the concentration of APPc58-myc DNA. By contrast, caspase-3 activities in CT-GFP stable cells were not significantly elevated after expression of APPc58-myc DNA (Figure 5B). Further, caspase-3 levels were significantly lower in CT-GFP stable H4 cells as compared to GFP stable H4 cells over the entire range of concentrations of APPc58-myc DNA (Figure 5B; p ≤ 0.001). Taken together, these results show that the presence of model Hirano bodies decreases AICD-induced apoptosis.
3.3 Hirano bodies down-regulate Fe65 dependent transcriptional activity of APP-Gal4 and APPct-Gal4 in HEK293T cells
Since AICD and Fe65 accumulated in model Hirano bodies in H4 cells (Figures 1-4), we hypothesized that Hirano bodies might be able to modulate transcriptional activity. As reported by Cao and Südhof (2001, 2004), Fe65 interacts with the YENPTY binding motif of the C-terminal domain of APP and activates transactivation of APP-Gal4 and APPct-Gal4. We examined whether model Hirano bodies regulate transcriptional activity of AICD in the presence or absence of Fe65. Transcriptional activity was measured in HEK293T stable cells using a dual luciferase reporter method and a Gal4/UAS transactivation assay using 4 different APP and APPct constructs fused to Gal4 (Figure 6C), and further normalized with control Gal4 transcriptional activity. We measured Gal4 transcriptional activity in the absence (Figure 6A) or presence (Figure 6B) of exogenous Fe65 in HEK293T cells stably expressing either GFP or CT-GFP. In the absence of exogenous Fe65, there was a >10 fold enhancement of reporter activity in APPct-Gal4 transfected cells as compared to other constructs, and this enhancement was not observed following transfection with the APPct*-Gal4 mutant bearing a mutation in the YENPTY motif that mediates binding to Fe65 (Figure 6A). The transcriptional activation in the absence of exogenous Fe65 was consistently less in cells with Hirano bodies as compared to control cells, but the fold activation is relatively small and the differences were not statistically significant (Figure 6A). In the presence of exogenous Fe65 expression, transcriptional activation induced by APP and APPct was enhanced by 300- and 700- fold, respectively, in cells expressing GFP (Figure 6B). This dramatic enhancement was not observed following transfection with the APP*-Gal4 and APPct*-Gal4 bearing a mutation in the YENPTY motif. These results are consistent with previous reports that Fe65 is required to increase transcriptional activity of APP-Gal4 and APPct-Gal4 (Cao and Südhof, 2001, 2004; Perkinton et al., 2004; von Rotz et al., 2004; Zhao and Lee, 2003). Most importantly, APP-Gal4 and APPct-Gal4 transcriptional activity in the presence of exogenous Fe65 was significantly decreased in CT-GFP stable cells as compared to GFP stable cells (Figure 6B, p<0.001). These results indicate that Hirano bodies greatly down-regulated APP-Gal4 and APPct-Gal4 transcriptional activity, suggesting a physiological role of Hirano bodies in regulation of AICD- and Fe65-dependent transcriptional regulation.
Figure 6.
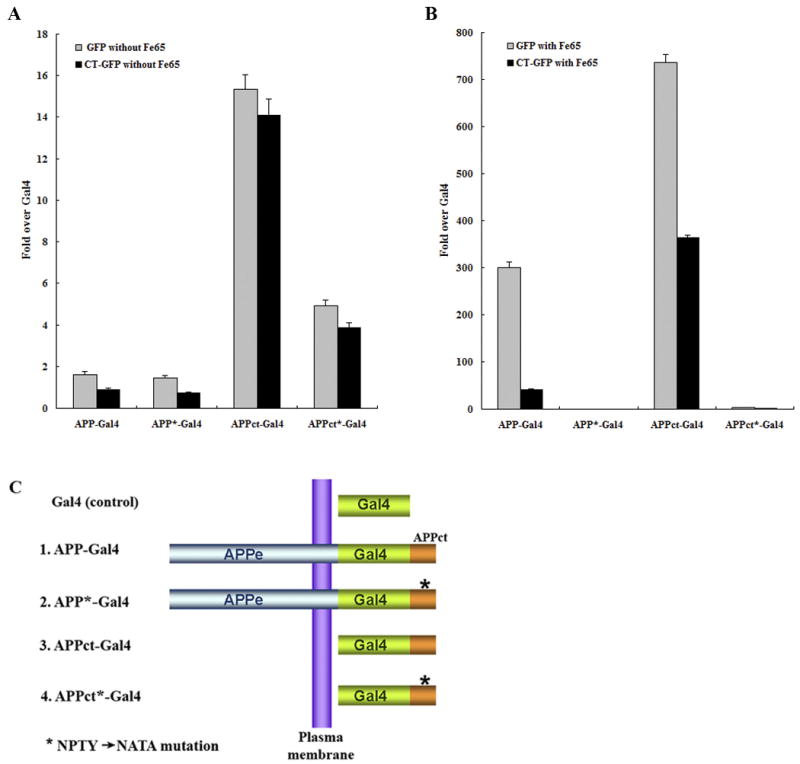
Fe65-dependent transcriptional activity of APP-Gal4 and APPct-Gal4 is down regulated in HEK293T stable cells with Hirano bodies. HEK293T stable cells (1×104 cells) expressing GFP or CT-GFP were co-transfected with 0.1 μg of pG5E1B-luc, 0.02 μg of pRL-TK and one of the following 5 different Gal4 constructs (C): 0.1 μg of pMst (Gal4), 0.1 μg of pMst-APP695 (APP-Gal4), 0.1 μg of pMst-APP695* (APP*-Gal4), 0.1 μg of pMst-APPct (APPct-Gal4), and 0.1 μg of pMst-APPct* (APPct*-Gal4) in the absence (A) or presence (B) of 0.1 μg of HA-Fe65. Plasmids with * contain the NATA mutation instead of NPTY in the C-terminal domain of APP that binds to Fe65. Transactivation activity was normalized to the level of transactivation of control Gal4. APP-Gal4 and APPct-Gal4 induced reporter expression was greatly enhanced by the presence of Fe65 (note the different scale bars in panels A and B). In the presence of Fe65, reporter expression was significantly lower in CT-GFP cells as compared to GFP cells (p < 0.001), and was significantly greater for APP-Gal4 and APPct-Gal4 than for the corresponding constructs with the NATA mutation (marked with *) in the Fe65 binding motif. Data shown are mean ± S.E.M. of triplicate measurements and repeated in three independent experiments.
4. Discussion
The amyloid cascade is the most prominent hypothesis proposed to explain the initiation and progression of Alzheimer's disease (Hardy and Selkoe, 2002), while other hypotheses posit important roles of other factors including oxidative stress (Butterfield et al., 2002), axonal transport (Stokin et al., 2005), presenilin (Tu et al., 2006), tau (Lee et al., 2001; Ballatore et al., 2007), and AICD (Galvan et al., 2006; Saganich et al., 2006). AICD is released from APP by the presenilin-dependent gamma-secretase, and interacts with a number of adaptor proteins that function in signal transduction, to modulate the metabolism and internalization of APP, the secretion of Aβ, and to promote gene transactivation (King and Turner, 2004; Kinoshita et al., 2002; McLoughlin and Miller, 2008; Raychaudhuri and Mukhopadhyay, 2007; Sastre et al., 1998). Further, two of these adaptor proteins, CP2 and Fe65, have genetic polymorphisms that are correlated with Alzheimer's disease (Hu et al., 1998; Lambert et al., 2000). Induction of cell death by AICD (Kinoshita et al., 2002) requires Fe65-dependent gene transactivation (Cao and Südhof, 2001), and is likely mediated by enhanced expression of known neurotoxic agents including GSK3β and p53 (Alves da Costa et al., 2006; Kim et al., 2003; Xu et al., 2007), implying that p53-dependent apoptosis is involved. In addition to its role in initiation of apoptosis, AICD can also lead to changes in calcium signaling, growth factor and NF-kB pathway activation, the production, trafficking, and processing of APP, and neuronal development and differentiation (Raychaudhuri and Mukhopadhyay, 2007).
We initiated this work by revisiting the report (Munoz et al., 1993) that epitopes from the carboxy-terminus, but not the amino-terminus of APP are associated with Hirano bodies. Epitopes from the C-terminus of APP (APPx) and the adapter protein Fe65 have been localized to model Hirano bodies using both immunofluorescence and immunoprecipitation (Figures 1,2). Direct demonstration that AICD itself can associate with model Hirano bodies was obtained in cells transfected with epitope tagged APPc58-myc (Figures 3, 4). Our findings demonstrate conclusively that both endogenous and exogenous AICD and Fe65 can associate with model Hirano bodies. In addition, cells with model Hirano bodies had reduced susceptibility to AICD-induced programmed cell death as revealed by enhanced survival and reduced activation of caspase 3 following expression of AICD (Figure 5). Finally, gene transactivation initiated by expression of APP-Gal4 and APPct-Gal4 is Fe65-dependent, and is significantly reduced in cells bearing model Hirano bodies (Figure 6). These findings have considerable significance with regard to the AICD hypothesis.
First, how could the presence of Hirano bodies modulate AICD-induced apoptosis and gene transactivation? The simplest model is that Hirano bodies sequester AICD and Fe65, and reduce the concentration of these agents available to stimulate transcription. While the mechanism of the regulation of transcription by AICD and Fe65 is controversial, the presence of Fe65 in an active conformation in the nucleus is required for AICD-dependent transcriptional activation (Cao and Südhof, 2004), so the sequestration of Fe65 in Hirano bodies is as significant as the presence of AICD. Fe65 interacts with other nuclear proteins, CP2/LSF/LBP1 and Tip60, and plays a major role in activation of transcription (Cao and Südhof, 2001; Kim et al., 2003; Zambrano et al., 1998). The sequestration of Fe65 in Hirano bodies can also impact the metabolism of APP, and may inhibit production of Aβ and of AICD (King and Turner, 2004), and may also interfere in other cellular functions of Fe65 (McLoughlin and Miller, 2008). In addition, Hirano bodies could impact the course of apoptosis and gene expression by sequestration of other molecules whose presence in Hirano bodies has been reported previously including iNOS (Lee et al., 1999), PKC ι/λ (Shao et al., 2006), and other transcriptional regulators including FAC1 (Jordan-Sciutto et al., 1998), and p130 (Previll et al., 2007). Sequestration of such signaling molecules could impact the results of AICD-induced gene transactivation directly, or indirectly by affecting the metabolism, trafficking, and interactions of AICD at multiple levels. For example, it has been reported that phosphorylation of AICD at threonine 668 is required for its interaction with Fe65, import to the nucleus, and for stimulation of neurotoxicity (Chang et al., 2006). Other transcriptional modulators such as low density lipoprotein receptor related protein intracellular domain (LRPICD) and NF-κB can inhibit or repress AICD dependent gene transactivation (Kinoshita et al., 2003; Zhao and Lee, 2003). Finally, in addition to simple sequestration, the degradation of Hirano bodies would permanently remove AICD, Fe65, and other signaling molecules that traffic to these cytoskeletal inclusions. Consistent with this proposal, we have recently found that autophagy contributes to the degradation of model Hirano bodies (Kim et al., 2009).
A second major question concerns the mechanism of association of AICD and Fe65 with Hirano bodies. The immunofluorescence and immunoprecipitation assays revealed an association of AICD and Fe65 with CT-GFP (Figures 3 and 4), and our working model is that this interaction is indirect. It has been known for some time that Fe65 binds to the actin associated protein mena (Ermekova et al., 1997). Studies of the localization of Fe65 show nuclear accumulation, or cytoplasmic staining that is frequently punctate rather than diffuse (Cao and Südhof, 2001). It is consistent to propose that these punctate foci arise from Fe65 that is anchored to the actin cytoskeleton through mena. In addition, interactions of APP with Fe65 and mena in lamellipodia have been proposed to modulate cell movement (Sabo et al., 2003). One explanation of this effect is that components of the actin cytoskeleton are major targets of AICD- and Fe65-dependent gene transcription (Muller et al., 2007). It will be exciting to test the proposal that mena is present in Hirano bodies and could mediate the association of Fe65 and AICD with Hirano bodies.
The reported association of tau with Hirano bodies (Davis et al., 2008; Galloway et al., 1987; Peterson et al., 1988) suggests another possible mechanism by which the presence of Hirano bodies in cells could promote cell survival, and impact the progression of disease. AICD and Fe65 also induce expression of glycogen synthase kinase-3β that is involved in the phosphorylation of tau (Kim et al., 2003; Xu et al., 2007). Thus, their sequestration in Hirano bodies might modulate these interactions. In addition, Fe65 is reported to be co-localized with tau proteins in neurofibrillary tangles (Delatour et al., 2001). Finally, a recent study reveals interactions of tau with filamentous actin, and implicates rearrangement of the actin cytoskeleton in tau-induced neurodegeneration (Fulga et al., 2007). These studies focus additional attention on the fact that the actin cytoskeleton can modulate pathological processes that induce neurodegeneration. The molecular basis for such modulation presents an exciting prospect for future study.
It is particularly noteworthy that studies using both mouse models and AD patients have generated evidence that directly implicates the COOH-terminal regions of APP in the pathogenesis of AD. AICD can be cleaved at aspartate 664 to generate a small cytotoxic fragment called APPc31. Expression in mice of a human familial Alzheimer's APP with alanine inserted in place of aspartate (D664A) at position 664 suppresses loss of synaptic function in mouse models of AD, even though Aβ42 levels increase and plaque deposits at normal levels (Galvan et al., 2006; Saganich et al., 2006; Galvan et al., 2008). These results highlight the potential significance in the progression of AD of the generation of an APPc31 fragment by cleavage of AICD at aspartate 664. Since the APPc31 fragment contains the YENPTY motif required for interaction with adapter proteins, and induces cell death and promotes gene transactivation with a potency similar to or slightly less than that of APPc59 (Cao and Südhof, 2004; Kim et al., 2003; Kim et al., 2004), it is possible that Hirano bodies could affect either this cleavage event, or the localization and biological activity of APPc31. Further, mice expressing AICD show cognitive decline with no elevation of Aβ or deposition of plaque (Ghosal et al., 2009). Finally, studies of brain from Alzheimer's patients do reveal elevated levels of both AICD and of APP cleaved at aspartate 664 offering support to the conclusions from the mouse models that intracellular regions of APP do promote disease progression (Banwait et al., 2008; Ghosal et al., 2009).
In summary, studies of cells with model Hirano bodies in cell culture provide direct evidence that model Hirano bodies do not inhibit cell growth, and are not necessarily involved in a stage of cell death (Davis et al., 2008; Maselli et al., 2002; Maselli et al., 2003). Further, we have shown that model Hirano bodies contain sites for accumulation of AICD as well as Fe65, and that model Hirano bodies suppress apoptosis induced by AICD and Fe65. Finally, Gal4-transactivation assays confirm that Hirano bodies reduce AICD- and Fe65-dependent transcription. Thus, our current results strongly support the hypothesis that the formation of Hirano bodies is an adaptive response involving the actin cytoskeleton that may play a protective role in the progression of neurodegenerative disease like Alzheimer's disease. Further studies will be required to explore and to critically test this hypothesis.
Acknowledgments
We thank Drs. Thomas Südhof, Bradley T. Hyman, and James Lauderdale for kindly providing plasmids. This work was supported by awards to RF and MF from NSF (MCB 98-08748), the Alzheimer's Association (IIRG-00-2436), and NIH (1R01-NS04645101).
The abbreviations used are
- APP
amyloid-β precursor protein
- AICD
the C-terminal fragment of amyloid-β precursor protein
- APPx
all truncated forms of APP that include the C-terminal region
- β-gal
β-galactosidase
- PI
propidium iodide
- MAP
microtubule-associated protein
- LRP
Low density lipoprotein-related protein
Footnotes
Disclosure Statement: there are no conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sangdeuk Ha, Email: sha@bidmc.harvard.edu.
Ruth Furukawa, Email: furukawa@cb.uga.edu.
Marcus Fechheimer, Email: fechheim@cb.uga.edu.
References
- Alves da Costa C, Sunyach C, Pardossi-Piquard R, Sevalle J, Vincent B, Boyer N, Kawarai T, Girardot N, St George-Hyslop P, Checler F. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer's disease. J Neurosci. 2006;26:6377. doi: 10.1523/JNEUROSCI.0651-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Banwait S, Galvan V, Zhang J, Gorostiza OF, Ataie M, Huang W, Crippen D, Koo EH, Bredesen DE. C-terminal cleavage of the amyloid-beta protein precursor at Asp664: a switch associated with Alzheimer's disease. J Alzheimer's Disease. 2008;13:1–16. doi: 10.3233/jad-2008-13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Griffin S, Munch G, Pasinetti GM. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer's disease brain exists. J Alzheimer's Dis. 2002;4:193. doi: 10.3233/jad-2002-4309. [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC. A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC. Dissection of amyloid-β precursor protein-dependent transcriptional transactivation. J Biol Chem. 2004;279:24601. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- Chang KA, Kim HS, Ha TY, Ha JW, Shin KY, Jeong YH, Lee JP, Park CH, Kim S, Baik TK, Suh YH. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol Biol Cell. 2006;26:4327. doi: 10.1128/MCB.02393-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupers P, Orlans I, Craessarts K, Annaert W, De Strooper B. The amyloid precursor protein (APP)-cytoplasmic fragment generated by gamma-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurons in culture. J Neurochem. 2001;78:1168. doi: 10.1046/j.1471-4159.2001.00516.x. [DOI] [PubMed] [Google Scholar]
- Davis RC, Furukawa R, Fechheimer M. A cell culture model for investigation of Hirano bodies. Acta Neuropathol (Berl) 2008;115:205. doi: 10.1007/s00401-007-0275-9. [DOI] [PubMed] [Google Scholar]
- Delatour B, Mercken L, Hachimi KH, Colle MA, Padier L, Duyckaerts C. FE65 in Alzheimer's disease: neuronal distribution and association with neurofibrillary tangles. Am J Clin Pathol. 2001;158:1585. doi: 10.1016/S0002-9440(10)64113-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermekova KS, Zambrano N, Linn H, Minopoli G, Gertler F, Russo T, Sudol M. The WW domain of neural protein FE65 interacts with proline-rich motifs in mena, the mammalian homolog of Drosophila enabled. J Biol Chem. 1997;272:32869. doi: 10.1074/jbc.272.52.32869. [DOI] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhib ML, Spires TL, T HB, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Galloway PG, Perry G, Gambetti P. Hirano body filaments contain actin and actin-associated proteins. J Neuropathol Exp Neurol. 1987;46:185. doi: 10.1097/00005072-198703000-00006. [DOI] [PubMed] [Google Scholar]
- Galloway PG, Perry G, Kosik KS, Gambetti P. Hirano bodies contain tau protein. Brain Res. 1987;403:337. doi: 10.1016/0006-8993(87)90071-0. [DOI] [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci U S A. 2006;103:7130. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Zhang J, Gorostiza OF, Sanwait S, Huang W, Ataie M, Tang H, Br edesen DE. Long-term prevention of Alzheimer's disease-like behavioral deficits in PDAPP mice carrying a mutation in Asp664. Behav Brain Res. 2008;191:246. doi: 10.1016/j.bbr.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009;106:18367–72. doi: 10.1073/pnas.0907652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson PH, Tomlinson BE. Numbers of Hirano bodies in the hippocampus of normal and demented people with Alzheimer's disease. J Neurol Sci. 1977;33:199. doi: 10.1016/0022-510x(77)90193-9. [DOI] [PubMed] [Google Scholar]
- Goldman JE. The association of actin with Hirano bodies. J Neuropathol Exp Neurol. 1983;42:146. doi: 10.1097/00005072-198303000-00004. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002;297:353. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hirano A. Hirano bodies and related neuronal inclusions. Neuropathol Appl Neurobiol. 1994;20:3. doi: 10.1111/j.1365-2990.1994.tb00951.x. [DOI] [PubMed] [Google Scholar]
- Hu Q, Kukull WA, Bressler SL, Gray MD, Cam JA, Larson EB, Martin GM, Deeb SS. The human FE65 gene: genomic structure and an intronic biallelic polymorphism associated with sporadic dementia of the Alzheimer type. Human Mol Genet. 1998;103:295. doi: 10.1007/s004390050820. [DOI] [PubMed] [Google Scholar]
- Jordan-Sciutto K, Dragich J, Walcott D, Bowser R. The presence of FAC1 protein in Hirano bodies. Neuropathol Appl Neurobiol. 1998;24:359. doi: 10.1046/j.1365-2990.1998.00140.x. [DOI] [PubMed] [Google Scholar]
- Kim DH, Davis RC, Furukawa R, Fechheimer M. Autophagy Contributes to Degradation of Hirano Bodies. Autophagy. 2009;5:44. doi: 10.4161/auto.5.1.7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Kim EM, Lee JP, Park CH, Kim S, Seo JH, Chang KA, Yu E, Jeong SJ, Chong YH, Suh AYH. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3β expression. FASEB J. 2003;17:1951. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- Kim HS, Kim EM, Kim NJ, Chang KA, Choi Y, Ahn KW, Lee JH, Kim S, Park CH, Suh YH. Inhibition of histone deacetylation enhances the neurotoxicity induced by the C-terminal fragments of amyloid precursor protein. J Neurosci Res. 2004;75:117. doi: 10.1002/jnr.10845. [DOI] [PubMed] [Google Scholar]
- King GD, Turner RS. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol. 2004;185:208. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Shah T, Tangredi MM, Strickland DK, Hyman BT. The intracellular domain of the low density lipoprotein receptor-related protein modulates transactivation mediated by amyloid precursor protein and Fe65. J Biol Chem. 2003;278:41182. doi: 10.1074/jbc.M306403200. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Whelan CM, Berezovska O, Hyman BT. The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J Biol Chem. 2002;277:28530. doi: 10.1074/jbc.M203372200. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Whelan CM, Smith CJ, Berezovska O, Hyman BT. Direct visualization of the gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein: association with Fe65 and translocation to the nucleus. J Neurochem. 2002;82:839. doi: 10.1046/j.1471-4159.2002.01016.x. [DOI] [PubMed] [Google Scholar]
- Laas R, Hagel C. Hirano bodies and chronic alcoholism. Neuropathol Appl Neurobiol. 1994;20:12. doi: 10.1111/j.1365-2990.1994.tb00952.x. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Goumidi L, Vrieze FW, Frigard B, Harris JM, Cummings A, Coates J, Pasquier F, Cottel D, Gaillac M, St Clair D, Mann DM, Hardy J, Lendon CL, Amouyel P, Chartier-Harlin MC. The transcription factor LBP-1c/CP2//LSF gene on chromosome 12 is a genetic determinant of Alzheimer's disease. Human Mol Genet. 2000;9:2275. doi: 10.1093/oxfordjournals.hmg.a018918. [DOI] [PubMed] [Google Scholar]
- Lee SC, Zhao ML, Hirano A, Dickson DW. Inducible nitric oxide synthase immunoreactivity in the Alzheimer disease hippocampus: association with Hirano bodies, neurofibrillary tangles, and senile plaques. J Neuropathol Exp Neurol. 1999;58:1163. doi: 10.1097/00005072-199911000-00006. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Ann Rev Neurosci. 2001;24:1121. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lim RWL, Furukawa R, Fechheimer M. Evidence of intramolecular regulation of the Dictyostelium discoideum 34,000 dalton F-actin bundling protein. Biochemistry. 1999;38:16323. doi: 10.1021/bi991100o. [DOI] [PubMed] [Google Scholar]
- Lu DC, Soriano S, Bredesen DE, Koo EH. Caspase cleavage of the amyloid precursor protein modulates amyloid beta-protein toxicity. J Neurochem. 2003;87:733. doi: 10.1046/j.1471-4159.2003.02059.x. [DOI] [PubMed] [Google Scholar]
- Maciver SK, Harrington CR. Two actin binding proteins, actin depolymerizing factor and cofilin, are associated with Hirano bodies. Neuroreport. 1995;6:1985. doi: 10.1097/00001756-199510010-00008. [DOI] [PubMed] [Google Scholar]
- Maselli AG, Davis R, Furukawa R, Fechheimer M. Formation of Hirano bodies in Dictyostelium and mammalian cells induced by expression of a modified form of an actin cross-linking protein. J Cell Sci. 2002;115:1939. doi: 10.1242/jcs.115.9.1939. [DOI] [PubMed] [Google Scholar]
- Maselli AG, Furukawa R, Thomson SAM, Davis RC, Fechheimer M. Formation of Hirano bodies induced by expression of an actin cross-linking protein with a gain of function mutation. Eucaryot Cell. 2003;2:778. doi: 10.1128/EC.2.4.778-787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoughlin DM, Miller CCJ. The Fe65 proteins and Alzheimer's disease. J Neurosci Res. 2008;86:744. doi: 10.1002/jnr.21532. [DOI] [PubMed] [Google Scholar]
- Mitake S, Ojika K, Hirano A. Hirano bodies and Alzheimer's disease. Kao Hsiung I Hsueh Ko Hsueh Tsa Chih. 1997;13:10. [PubMed] [Google Scholar]
- Mori H, Tomonaga M, Baba N, Kanaya K. The structure analysis of Hirano bodies by digital processing on electron micrographs. Acta Neuropathol. 1986;71:32. doi: 10.1007/BF00687959. [DOI] [PubMed] [Google Scholar]
- Muller T, Concannon CG, Ward MW, Walsh CM, Tirniceriu AL, Tribl F, Kogel D, Prehn JHM, Egensperger R. Modulation of gene expression and cytoskeletal dynamics of the amyloid precursor protein intracellular domain (AICD) Mol Biol Cell. 2007;18:201. doi: 10.1091/mbc.E06-04-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DG, Wang D, Greenberg BD. Hirano bodies accumulate C-terminal sequences of beta-amyloid precursor protein (beta-APP) epitopes. J Neuropathol Exp Neurol. 1993;52:14. doi: 10.1097/00005072-199301000-00003. [DOI] [PubMed] [Google Scholar]
- Ogata J, Budzilovich GN, Cravioto H. A study of rod-like structures (Hirano bodies) in 240 normal and pathological brains. Acta Neuropathol. 1972;21:61. doi: 10.1007/BF00688000. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Hirai S, Hirano A. Hirano bodies in myelinated fibers of hepatic encephalopathy. Acta Neuropathol. 1982;58:307. doi: 10.1007/BF00688615. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Standen CL, Lau KF, Kesavapany S, Byers HL, Ward M, McLoughlin DM, Miller CCJ. The c-Abl tyrosine kinase phosphorylates the Fe65 adaptor protein to stimulate Fe65/amyloid precursor protein nuclear signaling. J Biol Chem. 2004;279:22084. doi: 10.1074/jbc.M311479200. [DOI] [PubMed] [Google Scholar]
- Peterson C, Kress Y, Vallee R, Goldman JE. High molecular weight microtubule-associated proteins bind to actin lattices (Hirano bodies) Acta Neuropathol. 1988;77:168. doi: 10.1007/BF00687427. [DOI] [PubMed] [Google Scholar]
- Previll LA, Crosby ME, Castellani RJ, Bowser R, Perry G, Smith MA, Zhu X. Increased expression of p130 in Alzheimer's disease. Neurochem Res. 2007;32:639. doi: 10.1007/s11064-006-9146-3. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri M, Mukhopadhyay D. AICD and its adapters-In search of new players. J Alzheimer's Dis. 2007;11:343. doi: 10.3233/jad-2007-11311. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The amyloid precursor protein and its regulatory protein FE65, in growth cones and synapses in vitro and in vivo. J Neurosci. 2003;23:5407. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require c-terminal cleavage of APP. J Neurosci. 2006;26:13428. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Scott Turner R, Levy E. X11 Interaction with β-amyloid precursor protein modulates its cellular stabilization and reduced amyloid β-protein secretion. J Biol Chem. 1998;273:22351. doi: 10.1074/jbc.273.35.22351. [DOI] [PubMed] [Google Scholar]
- Schmidt ML, Lee VM, Trojanowski JQ. Analysis of epitopes shared by Hirano bodies and neurofilament proteins in normal and Alzheimer's disease hippocampus. Lab Invest. 1989;60:513. [PubMed] [Google Scholar]
- Schochet SS, Jr, McCormick WF. Ultrastructure of Hirano bodies. Acta Neuropathol. 1972;21:50. doi: 10.1007/BF00687999. [DOI] [PubMed] [Google Scholar]
- Shao CY, Crary JF, Rao C, Sacktor TC, Mirra SS. Atypical protein kinase C in neurodegenerative disease II: PKC ι/λ in tauopathies and α-synucleinopathies. J Neuropathol Exp Neurol. 2006;65:327. doi: 10.1097/01.jnen.0000218441.00040.82. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LSB. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci. 2004;117:4435. doi: 10.1242/jcs.01323. [DOI] [PubMed] [Google Scholar]
- Xu Y, Kim HS, Joo Y, Choi Y, Chang KA, Park CH, Shin KY, Kim S, Cheon YH, Baik TK, Kim JH, Suh YH. Intracellular domains of amyloid precursor-like protein 2 interact with CP2 transcription factor in the nucleus and induce glycogen synthase kinase-3beta expression. Cell Death Diff. 2007;14:79. doi: 10.1038/sj.cdd.4401928. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Hirano A. Hirano bodies in the perikaryon of the Purkinje cell in a case of Alzheimer's disease. Acta Neuropathol. 1985;67:167. doi: 10.1007/BF00688139. [DOI] [PubMed] [Google Scholar]
- Zambrano N, Minopoli G, de Candia P, Russo T. The Fe65 adaptor protein interacts through its PID1 domain with the transcription factor CP2/FSF/LBP1. J Biol Chem. 1998;273:20128. doi: 10.1074/jbc.273.32.20128. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Lee FS. The transcriptional activity of the APP intracellular domain-Fe65 complex is inhibited by activation of the NF-kappaB pathway. Biochemistry. 2003;42:3627. doi: 10.1021/bi027117f. [DOI] [PubMed] [Google Scholar]