

Abstract
In refractory temporal lobe epilepsy, seizures often arise from a shrunken hippocampus exhibiting a pattern of selective neuron loss called “classic hippocampal sclerosis.” No single experimental injury has reproduced this specific pathology, suggesting that hippocampal atrophy might be a progressive “endstage” pathology resulting from years of spontaneous seizures. We posed the alternate hypothesis that classic hippocampal sclerosis results from a single excitatory event that has never been successfully modeled experimentally because convulsive status epilepticus, the insult most commonly used to produce epileptogenic brain injury, is too severe and necessarily terminated before the hippocampus receives the needed duration of excitation. We tested this hypothesis by producing prolonged hippocampal excitation in awake rats without causing convulsive status epilepticus. Two daily 30-minute episodes of perforant pathway stimulation in Sprague-Dawley rats increased granule cell paired-pulse inhibition, decreased epileptiform afterdischarge durations during 8 hours of subsequent stimulation, and prevented convulsive status epilepticus. Similarly, one 8-hour episode of reduced-intensity stimulation in Long-Evans rats, which are relatively resistant to developing status epilepticus, produced hippocampal discharges without causing status epilepticus. Both paradigms immediately produced the extensive neuronal injury that defines classic hippocampal sclerosis, without giving any clinical indication during the insult that an injury was being inflicted. Spontaneous hippocampal-onset seizures began 16–25 days post-injury, before hippocampal atrophy developed, as demonstrated by sequential magnetic resonance imaging. These results indicate that classic hippocampal sclerosis is uniquely produced by a single episode of clinically “cryptic” excitation. Epileptogenic insults may often involve prolonged excitation that goes undetected at the time of injury.
Keywords: hippocampus, epileptogenesis, rat, dentate gyrus
Introduction
Acquired mesial temporal lobe epilepsy (MTLE) with hippocampal sclerosis is a common, often medically refractory neurological disorder thought to be initiated by a brain insult (Chang and Lowenstein, 2003). Refractory MTLE is associated with a characteristic pattern of selective and extensive hippocampal neuron loss that results in hippocampal atrophy (Bruton, 1988; Meldrum and Bruton, 1992). The atrophic hippocampus is one apparent source of the electrical events that cause spontaneous epileptic seizures (Spencer and Spencer, 1994; Spencer, 1998), and its surgical removal produces clinical improvement (Falconer and Taylor, 1968), a result consistent with the view that hippocampal sclerosis is an epileptogenic pathological entity (Falconer, 1974).
Suspected causes of acquired temporal lobe epilepsy with hippocampal sclerosis include prolonged febrile seizures in childhood, and seizures, head trauma, or infection at any age (Marks et al., 1992; Maher and McLachlan, 1995; Mathern et al., 1995; Jackson et al., 1999; Sokol et al., 2003; Cendes, 2004; Lewis, 2005). However, the precise cause of the extreme hippocampal atrophy often associated with refractory MTLE has remained a mystery for more than a century (Sommer, 1880; Bratz, 1899; Margerison and Corsellis, 1966; Meldrum and Bruton, 1992) because no single experimental brain insult replicates classic hippocampal sclerosis in animals. Paradoxically, many patients with classic hippocampal sclerosis on initial imaging (Lehericy et al., 1997; Van Paesschen et al., 1997) report no antecedent febrile seizures, or any identified brain insult, which raises the question in these cases: if no known injury caused their hippocampal atrophy, did a more subtle, “cryptic” injury occur, and if so, what was its nature?
Efforts to model epileptogenic brain injury have most frequently employed systemic chemoconvulsants (Purpura and Gonzalez-Monteagudo 1960; Meldrum et al., 1974; Turski et al., 1983; Tauck and Nadler, 1985). Although chemoconvulsant-induced status epilepticus (SE) produces a permanent epileptic state in rats (Pisa et al., 1980; Nadler, 1981; Cavalheiro et al., 1982), the available data indicate that prolonged convulsive SE in animals is a severe, often lethal insult that causes more extra-hippocampal injury than hippocampal injury (Schwob et al., 1980; Clifford et al., 1987). In contrast to the extensive hippocampal atrophy and limited extra-hippocampal pathology of human mesial temporal lobe epilepsy with classic hippocampal sclerosis, convulsive SE in rodents, whether initiated by kainate, pilocarpine, or electrical stimulation, causes variable hippocampal damage and extensive extra-hippocampal damage (Roch et al., 2002; Tongiorgi et al., 2004; Nairismägi et al., 2004; Fabene et al., 2003; 2007; Harvey and Sloviter, 2005; Bumanglag and Sloviter, 2008; Navarro Mora et al., 2009). Even a much longer duration (24 hrs) of hippocampal excitation in urethane-anesthetized rats only causes limited hippocampal injury (Sloviter and Damiano, 1981; Sloviter, 1987; 1991b), which resembles human “endfolium sclerosis” more closely than classic hippocampal sclerosis (Margerison and Corsellis, 1966; Bruton, 1988). In cases of chemoconvulsant-induced SE in which extensive hippocampal injury does follow hours of seizures, granule cell and pyramidal cell loss often involves a vascular abnormality that causes ischemic, rather than excitotoxic injury (Sloviter, 2005; Biagini et al., 2008), and the resulting pattern of pathology is not classic hippocampal sclerosis.
The failure of any single experimental brain injury to produce classic hippocampal sclerosis, and the presence of classic hippocampal sclerosis in many patients with no history of febrile seizures, brain infection, or traumatic head injury (Hauser and Hesdorffer, 1990; Meldrum and Bruton, 1992), has led to the hypothesis that classic hippocampal sclerosis may be a progressive “end-stage” pathology, rather than the result of a single initial insult (Mathern et al., 1995; Briellmann et al., 2002). According to this view, epileptogenesis might be initiated by an injury or a pre-existing structural hippocampal abnormality (Fernandez et al., 1998), but classic hippocampal sclerosis is ultimately produced only by incremental and cumulative neuron loss that occurs with each spontaneous seizure (Cavazos and Sutula, 1990; Mathern et al., 1995; Kälviäinen et al., 1998; O’Brien et al., 1999; Tasch et al., 1999; Briellmann et al., 2002). However, the recent finding that spontaneous epileptic seizures in animals do not produce any detectable hippocampal neuron loss (Pitkänen et al., 2002; Gorter et al., 2003), suggests that classic hippocampal sclerosis may be caused by an as yet unidentified single initial insult, a possibility with significant implications for understanding the etiology of hippocampal sclerosis, identifying epileptogenic mechanisms, and knowing when to attempt therapeutic strategies to prevent pathology and interfere with the epileptogenic process.
We formulated a “cryptic injury” hypothesis, which holds that the extensive hippocampal neuron loss and limited extra-hippocampal pathology often associated with refractory MTLE is caused by a single excitatory event of long duration, but limited intensity, that stays sequestered within the temporal lobe. We hypothesized that this focal activity is insufficiently intense to spread to the motor pathways that need to be activated to cause convulsive SE and to produce the widespread brain damage that convulsive SE can cause (Schwob et al., 1980; Clifford et al., 1987; Fujikawa et al., 2000; Mikaeloff et al., 2006). This “cryptic injury” hypothesis is consistent with the observations that few patients report episodes of convulsive SE prior to the development of MTLE (Hauser and Hesdorffer, 1990; French et al., 1993), and that prolonged convulsive status epilepticus in humans, which can result in severe cognitive impairment and widespread brain damage (Fujikawa et al., 2000; Mikaeloff et al., 2006), is distinct from the limited pathology found in otherwise neurologically normal patients with refractory MTLE (French et al., 1993; Engel, 1996).
If classic hippocampal sclerosis is caused by a single episode of prolonged excitation that is less intense than the excitation that causes convulsive SE, and the duration of excitation is a critical factor, then why does perforant pathway stimulation for 24 hours in urethane-anesthetized rats produce endfolium sclerosis (mainly hilar cell loss), but not classic hippocampal sclerosis (Sloviter, 1991b)? We observed that direct unilateral stimulation of the CA3 pyramidal cell layer in urethane-anesthetized rats produced extensive bilateral damage to CA3 and CA1 pyramidal cells (Sloviter, 1991b), a finding that confirmed the vulnerability of nearly all pyramidal cells, and the injuriousness of forced CA3 discharge under urethane anesthesia. Thus, we posited that during perforant pathway stimulation, which involves excitation one synapse removed from CA3 activation, urethane anesthesia suppresses CA3 pyramidal cell discharge in response to granule cell excitation, thereby preventing CA3 pyramidal cells from destroying their area CA3 and CA1 target cells and causing classic hippocampal sclerosis. Therefore, we sought a way to produce simultaneous dentate granule cell and CA3 pyramidal cell excitation in awake animals for a prolonged period (8 hours) without producing the seizure spread and lethal convulsive SE that causes widespread brain damage and requires premature termination of the experiment.
To find a way to evoke hippocampal excitation in awake animals without causing convulsive SE, we utilized an approach developed in other laboratories to decrease the seizure intensity and neuronal injury caused by prolonged convulsive SE. Kelly and McIntyre (1994) and Penner and colleagues (2001) reported that kindling stimuli decreased the brain damage caused by a subsequent episode of convulsive SE. In addition, other studies have shown that a brief episode of chemoconvulsant-induced seizures decreased the seizure intensity and the extent of damage evoked by a subsequent chemoconvulsant treatment (Sasahira et al., 1995; El Bahh et al., 1997; Najm et al., 1998; Kondratyev et al., 2001; Hatazaki et al., 2007), even if behavioral SE still develops (Zhang et al., 2002). Because our primary purpose was to evoke prolonged, focal hippocampal excitation in awake rats without producing convulsive SE, we determined whether two relatively brief daily episodes of hippocampal excitation in Sprague-Dawley rats effectively reduced the intensity of granule cell afterdischarges evoked by stimulation on the third test day, and permitted 8 hours of stimulation to be delivered to awake animals without causing lethal convulsive SE. A second approach involved simply using a less intense stimulation paradigm in rat strains inherently more resistant than Sprague-Dawley rats to developing convulsive SE (Long-Evans and Brown-Norway; Xu et al., 2004; Young et al., 2009). Using these methods, we determined whether a single focal excitatory episode can immediately cause the selective pattern of temporal lobe injury that: 1) defines classic hippocampal sclerosis; 2) involves the limited extra-hippocampal injury that defines mesial temporal sclerosis (Meldrum and Bruton, 1992), and; 3) produces a permanent epileptic state.
Materials and Methods
Animal treatment
Sixty-four male Sprague-Dawley rats, forty-nine male Long-Evans rats, and four male Brown-Norway rats (300–400g; Harlan Sprague Dawley; Indianapolis, IN) were treated in accordance with the guidelines of the National Institutes of Health for the humane treatment of animals. The University of Arizona Institutional Animal Care and Use Committee approved all methods used.
Perforant pathway stimulation paradigms
Rats were implanted bilaterally with unipolar recording electrodes in the dentate granule cell layer of the dorsal hippocampus, and with bipolar stimulating electrodes in the angular bundles of the perforant pathway, as previously described (Harvey and Sloviter, 2006; Bumanglag and Sloviter, 2008). Bipolar stainless-steel stimulating electrodes (Rhodes Medical Instruments, Summerland, CA) were placed bilaterally in the angular bundles of the perforant pathway (~4.5 mm lateral from the midline suture and immediately rostral to the lambdoid suture). Unipolar recording electrodes, fabricated from Teflon-coated 0.003-inch-diameter stainless-steel microwires (7910; A-M Systems, Inc., Carlsborg, WA) were lowered into the brain bilaterally (~2 mm lateral from the midline, ~3 mm caudal to bregma, and 3.5 mm below the brain surface). Final tip locations in the granule cell layer were reached by optimizing the potentials evoked by perforant pathway stimulation (Andersen et al., 1966). A layer of dental acrylic cement attached the electrodes to the screws and skull. Plastic connectors (Ginder Scientific, Ottawa, Ontario, Canada) were fitted to the electrodes and then embedded in acrylic cement to form a mechanically stable cap. The scalp was restored around the cap using a single staple at the caudal end of the incision.
All stimulation protocols in Sprague-Dawley rats utilized a paradigm designed to evoke and maintain hippocampal granule cell afterdischarges throughout the period of stimulation (Sloviter, 1983). Stimulation consisted of continuous, bilateral 2 Hz paired-pulse stimuli, with a 40 msec interpulse interval, plus a 10 sec train of 20 Hz single-pulse stimuli delivered once per minute. All pulses (0.1 msec duration) were delivered at 15 V, as this voltage evoked granule cell epileptiform discharges in all animals without producing the tissue-damaging hydrolysis at the electrode tips that is caused by higher stimulus voltages.
After 3 hours of continuous stimulation, which reliably produced convulsive status epilepticus in naive animals, electrographic seizure activity and behavioral seizures were terminated abruptly by isoflurane inhalation, and their re-occurrence was suppressed by a sub-anesthetic dose of urethane (0.8 g/kg sc). Animals stimulated for 30 minutes on two consecutive days required only isoflurane to terminate status epilepticus. Eight hour stimulation on the third day did not cause convulsive SE, or epileptiform electrographic activity that outlasted the stimulation, and therefore did not require pharmacological termination. Stimulations in Long-Evans and Brown Norway rats utilized a different stimulation paradigm, which was designed to cause focal hippocampal discharges over a period of 8 hours. In this case, stimulation involved only 10 sec-long trains of 20 Hz, single-pulse stimuli delivered once per minute. This paradigm did not cause convulsive SE in Long-Evans or Brown Norway rats, or epileptiform electrographic activity that outlasted the stimulation, and therefore, these animals also required no treatment to terminate seizure activity.
Electrophysiological and video monitoring methods
Electrical stimulation utilized stimuli (0.1 msec duration) generated by a Grass S88 stimulator, which was used in conjunction with a stimulus isolation unit (Grass Instruments, West Warwick, RI). Granule cell layer activity during and after perforant pathway stimulation was amplified and recorded digitally at 10 kHz using Chart 5 software (AD Instruments, Mountain View, CA). Immediately following perforant pathway stimulation, spontaneous granule cell layer activity was recorded continuously (24/7), and stored digitally and automatically in 3-hour epochs. Each day, the preceding 24 hours of recordings were assessed visually, and all events with amplitudes obviously larger than baseline were analyzed. Each confirmed seizure was related to behavior on the timestamped video recordings, as described below. Continuous (24/7) video monitoring began immediately following stimulation utilizing Panasonic model 9622 color CCD day/night infrared cameras. Video files were captured at 30 frames/sec and time-stamped for integration with the electrophysiological data using automated surveillance software (Ben Software, London, United Kingdom), and stored digitally. Spontaneous behavioral seizures were scored according to the Racine scale (Racine, 1972).
Perfusion-fixation and tissue treatment
Rats were anesthetized with urethane (1.25 g/kg ip) and perfused through the aorta by gravity feed with normal saline for 2 minutes, followed by either 4% paraformaldehyde or 1.5% glutaraldehyde/1% paraformaldehyde, both in 0.1 M phosphate buffer, pH 7.4, for 10 minutes. Other rats, intended for Timm staining (Sloviter, 1982), were perfused with saline and 0.1% sodium sulphide in 0.1M phosphate buffer, pH 7.4, for one min each prior to the saline and aldehyde solutions described above. After overnight fixation in situ at 4° C, brains were removed from the skull and 40-μm-thick sections were cut in 0.1 M hydroxymethylaminomethane buffer, pH 7.6 (Tris), using a Vibratome. Brains were first cut in the horizontal plane from the base of the brain, and then the remaining tissue was cut coronally. All histological and immunocytochemical procedures were performed as previously described (Sloviter et al., 2006).
Immunocytochemistry protocol
Sections were mounted on Superfrost Plus slides, air dried, and placed in 0.1 M Tris buffer. Slides were then immersed in 85°C Tris for 1 minute, washed in room temperature Tris, and placed in Tris containing 0.25% bovine serum albumin (BSA; fraction V; Sigma) and 0.1% Triton X-100 (Sigma), pH 7.6. Slides were incubated overnight in primary mouse anti-NeuN (diluted 1:10,000; MAB377; lot No. 22021251; Chemicon). Anti-NeuN was raised against purified cell nuclei from mouse brain, and recognizes 2–3 bands in Western blots in the 46–48 kDa range, and possibly another band at approximately 66 kDa, which constitutes unknown nuclear proteins (Mullen et al., 1992). NeuN was used solely as a neuronal marker. Although NeuN is constitutively expressed in all temporal cortical neurons, to our knowledge, it is not expressed by all neurons of all species (Sarnat et al., 1998; Weyer and Schilling, 2003; Kumar and Buckmaster, 2007), and its expression can become undetectable after brain insults that do not result in neuronal death (McPhail et al., 2004; Unal-Cevik et al., 2004; Collombet et al., 2006). Therefore, Nissl staining was used to verify that loss of NeuN expression was due to cell death.
After primary antibody incubation, slides were washed in Tris-BSA-Triton buffer (2 × 5 minutes minimum). Slides were then incubated in biotinylated secondary antibody solution (1:2,000 dilution of goat anti-mouse or goat anti-rabbit; Vector, Burlingame, CA) in Tris-BSA-Triton buffer for 2 hours, washed in the same buffer, and then incubated for 2 hours in avidin-biotin-HRP complex (Vector Elite Kit diluted 1:1,000 in Tris-BSA-Triton buffer). Slides were then washed in Tris (3 × 5 minutes minimum) and incubated in a hydrogen peroxide-generating diaminobenzidine (DAB) solution (100 ml Tris containing 50 mg DAB, 40 mg ammonium chloride, 0.3 mg glucose oxidase, and 200 mg β-D-glucose). After incubation in DAB solution for 20–30 minutes, slides were rinsed in Tris, dehydrated in graded ethanols and xylene, and coverslipped with Permount. Sets of matched sections were Nissl stained with 1% cresyl violet or stained with the neurodegeneration marker Fluoro-Jade B (Schmued and Hopkins, 2000).
Light microscopy imaging methods
Brightfield images were acquired digitally on a Nikon E800M microscope with a Hamamatsu C5180 camera. Adobe Photoshop CS3 was used to acquire images and optimize contrast and brightness, but not to change the image content.
Quantitative assessment of neuron loss
To assess the approximate extent of acute injury caused by 8 hours of perforant pathway stimulation in Sprague-Dawley rats, we analyzed five non-adjacent sections from the dorsal hippocampus of naïve and experimental animals (30,30,8) that were immunostained for NeuN, a neuronal marker protein (Mullen et al., 1992). Ventral sections involved so much hippocampal shrinkage that horizontal sections often contained no hippocampal tissue. Therefore, quantitative analysis was limited to the dorsal hippocampus. We used matching coronal sections from the dorsal hippocampus of control and experimental animals, which were based on the identification of extra-hippocampal brain regions, e.g. the dorsal thalamic nuclei and the shape of the third ventricle. Cell counts of immunopositive neurons included all neuronal somata that contained a visible nucleus. In the hilus of the dentate gyrus, counted neurons included somata not in contact with the granule cell layers, and those that were outside the somal and dendritic regions of area CA3c. In the naïve sections, immunoreactive neurons counted in the pyramidal cell layers included somata in apparent contact with at least one other neuron. In this way, only neurons that were part of the pyramidal cell layer were counted. In stimulated animals with hippocampal atrophy, in which strata boundaries were eliminated, all immunoreactive neurons within the remaining hippocampus, but located outside the dentate gyrus, were counted. Quantitative neuron counts should be regarded as approximations of the extent of neuronal loss and survival. Neurons were not counted in Nissl-stained sections because neurons could not be reliably differentiated from the many glial cells that proliferate after seizure-induced damage.
We measured the nuclear diameter of each cell population (hilus, CA3, CA1) in both naïve and stimulated animals using the CS3 Adobe Photoshop measuring tool because over-counting of neurons can occur if a significant difference in cell size exists within one class of cells between control and experimental groups. We applied Abercrombie’s correction method (Abercrombie, 1946; Saper, 1996) to the cell counts per unit area. The number of counted cells with visible nuclei per unit area in sections 40um thick was divided by the section thickness plus the diameter of the CA3 pyramidal cell nucleus, for example, according to the formula: N=n*t/t+d, where N is the true value (cells per unit area), “n” is the observed cell count per unit area, t is the section thickness, and d is the diameter of the object counted.
Hippocampal volume measurement
To estimate the extent of hippocampal atrophy produced by 8 hours of stimulation in Sprague-Dawley rats, we calculated whole hippocampal volumes. This was done by collecting the entire series of 40μm-thick coronal sections that contained any hippocampal tissue, and then measuring the areas of the hippocampus in every other section (e.g. sections 1,3,5,7, etc). Measurements were made in 4 sham control rats, 4 rats stimulated for 3 hr, 4 rats stimulated 2 × 30 min (30,30,0-group), and 4 rats stimulated 2 × 30 min plus 8 hours on the third day (30,30,8-group). All rats used for area measurements were perfusion-fixed with 4% paraformaldehyde at least 9 weeks post-stimulation. Areas were measured using the Adobe Photoshop CS3 Extended Measurement feature to calculate the area bounded by an irregular border. Whole hippocampal area included the dentate gyrus and the hippocampus “proper,” excluding the fimbria. The total hippocampal volume was calculated by multiplying the area of each section by the thickness of two sections (80 μm), as every other section was measured. The accuracy of area measurement was verified by measuring the area of a 1 mm square on a micrometer, which yielded an area of 1.003 mm2 by the Adobe Photoshop CS3 Extended Measure program. Group means were compared using Student’s t-test.
Magnetic resonance imaging (MRI)
MRI experiments were performed on animals in which all metal electrodes had been removed within 24 hours of the end of stimulation or sham stimulation. Images were obtained using a Bruker Biospec Tomograph (Bruker Medical, Ettlingen, Germany) equipped with an Oxford, 33 cm bore, horizontal magnet operating at 4.7 T and a Bruker gradient insert (maximum intensity 20 G/cm), as previously described in detail (Fabene et al., 2006). Rats were anesthetized by inhalation of a mixture of oxygen and air (1 l/min) containing 1% halothane (initial dose 4% halothane); rectal temperature and heart rate were monitored by a Small Animal Monitoring and Gating System (SA Instrument, Stony Brook, NY) and were similar in control and experimental animals. The rats were placed into a 7.2 cm inside diameter bird cage transmitter coil. The signal was received through a helmet coil, optimized for the rat brain, actively decoupled from the transmitter coil and placed directly on the animal’s head. Three mutually perpendicular slices were acquired through the brain as scout images. A T1W-SpinEcho image was acquired in the sagittal plane in order to localize the olfactory bulbs. Twenty contiguous, transversal, T2W, 1-mm-thick slices were imaged starting 1 mm posterior to the olfactory bulbs. T2W images were acquired using a RARE sequence with the following parameters: repetition time (TR) = 5117 ms; echo time (TE) = 65 ms, RARE factor = 8; field of view (FOV) = 3.5×3.5m2, matrix size 256 × 256 corresponding to an in-plane resolution of 137×137 μm2.
RESULTS
Three hours of confirmed hippocampal status epilepticus plus months of spontaneous epileptic seizures did not cause classic hippocampal sclerosis
A single, prolonged episode of chemoconvulsant-induced status epilepticus (SE) fails to cause classic hippocampal sclerosis (Schwob et al., 1980; Harvey and Sloviter, 2005), in part, because chemoconvulsant treatment rarely produces continuous hippocampal seizure activity (Sloviter et al., 2003). Therefore, we determined whether 3 hour of verified, continuous hippocampal electrographic status epilepticus ever produces classic hippocampal sclerosis. Bilateral electrical stimulation of the perforant pathways in awake, chronically implanted rats at 2 Hz continuously, with additional 10 sec-long 20 Hz trains delivered once per minute, evoked granule cell population spikes during the 20 Hz trains and epileptiform discharges during the 2 Hz stimulation between successive 20 Hz trains (Fig. 1, A1). This stimulation paradigm reliably evoked behavioral status epilepticus, which was lethal in all animals tested (n=6) when stimulation was continued for 4 hours or more. When convulsive status epilepticus was terminated pharmacologically after 3 hours of continuous dentate granule cell seizure discharges, all animals survived. Fluoro-Jade B staining (Schmued and Hopkins, 2000) 4 days later revealed acute injury to hilar neurons and scattered CA1 and CA3 pyramidal cells, but no evidence of extensive hippocampal pyramidal cell injury (n=9; Fig. 1, D). These results confirm that a single episode of continuous hippocampal seizure activity for 3 hours in awake rats that also caused convulsive SE does not produce the extensive or nearly complete pyramidal cell loss that defines human classic hippocampal sclerosis (Meldrum and Bruton, 1992).
Figure 1.

Hippocampal granule cell epileptiform activity during 3 hours of perforant pathway stimulation-induced status epilepticus (SE) in awake, freely moving Sprague-Dawley rats, and pathology following stimulation. A1: 125 sec of electrographic activity recorded directly from the granule cell layer during stimulation. Note that stimulation consisted of continuous 2 Hz paired-pulse stimuli (marked “2 Hz”) plus 10 sec-long 20 Hz trains delivered once per minute (marked “20 Hz”). A1a: 500 msec of evoked granule cell layer activity (responses to one pair of pulses at 2 Hz plus epileptiform granule cell discharges). The expanded trace in A1a is the area marked “a” in panel A1. Note that perforant pathway stimulation forced the granule cells to discharge and cause convulsive SE throughout the 3 hour stimulation period. B: Nissl-stained section of the normal dorsal hippocampus. C: NeuN-immunoreactivity in a section adjacent to that shown in panel B. D: FluoroJade-B (FJB) staining in the dorsal hippocampus 4 days after 3 hours of perforant pathway stimulation-induced convulsive SE in an awake rat. Note that FJB-positive neurons are evident in the dentate hilus (h) and sporadically in areas CA3 and CA1. Arrows denote degeneration in the inner molecular layer, indicating degeneration of hilar mossy cells and their axon terminals in the inner molecular layer. Note that despite confirmed granule cell epileptiform discharges for 3 hours, extensive pyramidal cell injury was not produced. Green FJB fluorescence was photographed, converted to grayscale, and then inverted to produce grayscale images of FJB-positive-, acutely degenerating neurons on a white background. E: NeuN immunoreactivity in the dorsal hippocampus 196 days after 3 hours of perforant pathway stimulation in an awake rat. Note that despite a long survival period during which many spontaneous epileptic seizures occurred, obvious cell loss was apparent only in the hilus (h), and extensive pyramidal cell loss did not occur. Scale bar in A1a: 5 sec in A1 and 50 msec in A1a; 10 mV in A1 and A1a; 200 μm in B–D.
Analysis of identically treated animals (n=5) that survived for 2–14 months after 3 hours of stimulation-induced SE, and had exhibited months of spontaneous epileptic seizures, revealed that there was no detectable evidence of acute neuronal death as reflected by Fluoro-Jade B staining (data not shown), and no significant hippocampal atrophy characteristic of classic hippocampal sclerosis in any rat (Fig. 1, E). The absence of Fluoro-Jade B-positive cells in all analyzed brain sections from animals that were actively exhibiting spontaneous epileptic seizures in the days preceding perfusion-fixation is consistent with previous studies indicating that spontaneous epileptic seizures do not cause any detectable neuron death (Pitkanen et al., 2002; Gorter et al., 2003). This lack of acute seizure-induced damage to vulnerable hippocampal neurons following a succession of individual spontaneous seizures is distinct from the report that individual seizures increase the rate of dentate granule cell turnover (Bengzon et al., 1997), which is a naturally-occurring granule cell death process distinct from seizure-induced, excitotoxic cell death of vulnerable hilar and pyramidal layer neurons (Sloviter et al., 1996). That the lack of FJB staining after individual seizures was not due to an inability of FJB to detect injury in chronically epileptic rats was demonstrated in one previously stimulated animal that developed spontaneous convulsive SE ~8 months after stimulation and months of spontaneous epileptic seizures. In this animal, spontaneous convulsive SE that lasted for more than 8 hours caused widespread brain damage that included extensive hippocampal injury to areas CA1 and CA3 (data not shown).
Two brief daily episodes of perforant pathway stimulation prevent convulsive status epilepticus on the third day of stimulation
The failure of 3 hours of confirmed hippocampal electrographic SE and convulsive SE to produce extensive hippocampal pyramidal cell loss led us to develop a pre-stimulation paradigm designed to attenuate the duration of hippocampal epileptiform discharges evoked by perforant pathway stimulation, thereby decreasing the spread of hippocampal focal seizure activity to motor pathways. In this way, we hypothesized that less intense hippocampal excitation might remain sequestered within the temporal cortical region and not cause lethal behavioral status epilepticus, thereby allowing a longer period of hippocampal excitation to be delivered. We found in pilot experiments that 30 minutes of stimulation on two consecutive days (continuous 2 Hz stimulation with paired pulses 40 msec apart plus 10 sec long 20 Hz trains of single stimuli added once per minute) prevented behavioral status epilepticus from developing on the third day in response to the identical stimulation paradigm.
Granule cell paired-pulse inhibition, as defined and assessed previously (Sloviter, 1991a) was markedly and reliably increased on the third day by the two 30 min-long stimulations on the two preceding days in all 9 rats tested. Evoked responses (an average of 5 evoked potentials) on the third day were compared to the responses evoked in the same animals on the first day (Fig. 2, A). Prior to the first 30 min-long stimulation, the average amplitude of the second of two paired pulses evoked by perforant pathway stimulation at 0.1 Hz was 3.7 ± 2.5 mV (n=9). One day after the second of two daily 30 min stimulations, the average amplitude of the second of two paired pulses in the same nine animals was 0.7 ± 0.8 mV. This 81% decrease in second population spike amplitude with minimal change in the amplitude of the first population spike (Fig. 2, C) was significant (n=9; p<0.001; paired t-test). Consistent with the observed increase in granule cell paired-pulse inhibition, the durations of granule cell epileptiform discharges evoked during the 30 min episodes of perforant pathway stimulation were decreased by 74% on the day following the two 30 min-long stimulation episodes (Fig. 2, B). On the first day of the 30 min pre-stimulation, granule cell epileptiform discharges during the 50 sec intervals between each 20 Hz stimulus train were an average of 43.4 ± 4.6 sec in duration (n= 266 afterdischarges from 9 rats). On the third stimulation day, during the first 30 min of stimulation, the duration of granule cell epileptiform discharges between each 20 Hz stimulus train in the same animals was 11.4 ± 3.9 sec (26% of the discharge duration on the first test day; Fig. 2,D), which was a significant mean difference (n=8; p<0.001; paired t-test). These two daily 30 min stimulations prevented the development of lethal behavioral status epilepticus, and permitted a prolonged, 8 hour-long stimulation to be delivered on the third day. This 8 hour-long stimulation caused “wet-dog” shakes and a few brief seizures, but mostly what appeared to be normal eating, drinking, and exploratory behaviors.
Figure 2.

Decreased granule cell excitability after two daily 30 min-long episodes of non-injurious perforant pathway stimulation in freely moving Sprague-Dawley rats. A: Granule cell layer responses to low frequency (0.1 Hz) perforant pathway stimulation before (Day 1) and after (Day 3) two daily 30 min-long stimulations. Arrows denote the second spike responses to identical afferent stimulation in the same rat. Note that the amplitudes of the second population spike were always smaller on Day 3, indicating increased paired-pulse inhibition as a result of prior stimulation. B1, B2: Representative granule cell layer activity (120 sec long) during stimulation on Day 1 (B1) and Day 3 (B2), consisting of continuous 2 Hz paired-pulse stimulation plus 10 sec-long 20 Hz trains of single stimuli delivered once per minute. Arrows show the decrease in large amplitude, negative-going spikes on Day 3 compared to Day 1, and (“a”) marks the locations expanded in B1a and B2a, which show the presence of shorter granule cell epileptiform discharges on Day 3 compared to Day 1. B1a, B2a: 500 msec-long extracts (one paired-pulse at 2 Hz and its associated evoked activity) occurring shortly after the 20 Hz train. Note that stimulation on Day 3 (B2a) evoked attenuated granule cell responses compared to the responses to identical stimuli in the same rat on Day 1. C: Quantitative analysis of the second spike amplitudes before and after two 30 minute stimulations in 10 rats. The decreased spike amplitude was found to be statistically significant (p<0.0001) by a paired t-test. D: Quantitative analysis of total afterdischarge duration during the 30 min of stimulation on Day 1 and the first 30 min of stimulation on Day 3 (n=9). Note that the afterdischarge durations on Day 3 were paired and compared with responses on Day 1 in each animal. The reduction was statistically significant (p<0.0001) by a paired t-test. Calibration bar: 5 ms and 10 mV in A, 10 mV; 5 sec and 10 mV in B1 and B2; 20 msec and 10 mV in B1a and B2a.
Perhaps the most reliable observation made during live monitoring of 49 rats during stimulation on 3 consecutive days was the obvious causal relationship between the prolonged granule cell epileptiform discharges that occurred between stimulus trains, and the occasional occurrence of behavioral seizures. In naïve rats, the 20 Hz stimulus trains delivered once per minute were reliably followed by prolonged epileptiform discharges that nearly filled most or all of the 50 sec periods between the 20 Hz stimulus trains (Fig. 2, B1a; expanded trace), and these epileptiform discharges were accompanied by the continuous behavioral seizures that define convulsive status epilepticus. Conversely, after the two 30 min stimulations on days 1 and 2 that decreased the inter-train epileptiform discharge duration (Fig. 2,D), stimulation on the third day of stimulation caused only 15–20 brief behavioral seizures in response to the 480 stimulus trains delivered during 8 hours of stimulation. In every case (216 behavioral seizures observed in 12 rats during 8 hour stimulation on day 3), the brief behavioral seizures were always preceded by a prolonged granule cell epileptiform discharge between the two 20 Hz trains. That is, most 20 Hz stimulus trains (>95%) evoked granule cell discharges during each stimulus train, but only brief afterdischarges during the 50 sec periods between the trains (Fig. 2, B2a; expanded trace). These truncated afterdischarges were never associated with behavioral seizures. Conversely, whenever a 20 Hz stimulus train was followed by a rare prolonged epileptiform afterdischarge during the 50 sec inter-train interval (as shown in Fig. 2, B1a), a behavioral seizure lasting approximately 1 min invariably occurred. Thus, the appearance of a prolonged granule cell epileptiform discharge during the 50 sec-long period between 20 Hz trains was perfectly predictive of an immediately impending behavioral seizure that started within seconds of the prolonged granule cell population discharges. In no case did a behavioral seizure occur without an immediately preceding prolonged granule cell epileptiform discharge. This same apparently causal relationship between granule cell epileptiform discharges and behavioral seizures was evident when spontaneous epileptic seizures developed; all seizures were preceded by spontaneous granule cell epileptiform discharges (see below).
Hippocampal pathology after conditioning and test stimulation in awake Sprague-Dawley rats
Acute pathology
Male Sprague-Dawley rats stimulated bilaterally for two 30-minute periods on two consecutive days, with- (30,30,8) or without (30,30,0) a subsequent 8 hour-long stimulation on day 3, were perfusion-fixed 4 and 7 days after the end of stimulation to assess acute neuronal injury. FJB-positive neurons were not detected anywhere in the brain in any 30,30,0-group animals (n=6). In addition, the two 30 min stimulation episodes prevented the hippocampal damage produced by 3 hours of stimulation on day 3, a duration of stimulation that always involved extensive hilar neuron injury in naive (not pre-stimulated) rats (Fig. 1,D). Thus, the “neuroprotective” effect of relatively short seizures before a longer seizure episode (Sasahira et al., 1995; El Bahh et al., 1997; Najm et al., 1998; Kondratyev et al., 2001; Zhang et al., 2002; Hatazaki et al., 2007), was the apparent result of a seizure-induced increase in inhibition that decreased the intensity of the main insult, i.e. fewer epileptiform granule cell afterdischarges and therefore less glutamate released onto monosynaptic target cells (hilar neurons and CA3 pyramidal cells).
Although 3 hours of stimulation on day 3 caused no detectable hippocampal injury (n=4), indicating apparent “neuroprotection” (3 hours of normally injurious stimulation produced no detectable damage), continuation of the same stimulation for 5 additional hours on day 3 immediately produced the extensive hippocampal neuron loss that results in classic hippocampal sclerosis. Staining of brain sections with the degeneration marker FJB was not necessary to detect the acute seizure-induced hippocampal injury produced by 8 hours of stimulation on day 3 (30,30,8-group). Microscopic examination of freshly cut, unstained brain sections under brightfield illumination revealed the full pattern of hippocampal brain injury (Fig. 3,A) because degenerating neurons apparently change shape, and their membranes reflect light out of the path of the objective lens (in the manner of a darkfield condenser), making dying cells appear darker than adjacent undamaged tissue that faithfully transmits light into the objective lens. Similarly, dying neurons in unstained sections appear autofluorescent in the green wavelength, making degenerating neurons in unstained sections appear as though they have been weakly FJB-stained (data not shown). FJB staining of the same sections facilitated the visual assessment of degeneration, but the pattern of degeneration was the same. In animals stimulated for 8 hours on day 3 (30,30,8-group), FJB staining was mainly restricted to the hippocampus, thalamic nuclei, the lateral septum, and the entorhinal cortex (Figs 3 and 4). Granule cells of the dentate gyrus, and neurons of the subiculum, presubiculum, and parasubiculum, were reliably and conspicuously spared (Fig. 3). Despite this extensive temporal cortical injury, prolonged stimulation did not produce the widespread brain damage caused by chemoconvulsant-induced SE (Schwob et al., 1980; Clifford et al., 1987; Chen and Buckmaster, 2005; Harvey and Sloviter, 2005). Furthermore, there was no evidence in stimulated rats of the hippocampal vascular abnormalities that cause significant ischemic injury and pyramidal neuron loss following kainate- or pilocarpine-induced status epilepticus (Fabene, 2003; Sloviter, 2005; Biagini et al., 2008). Another notable difference between stimulated animals and rats subjected to chemoconvulsant-induced SE was that stimulated rats appeared behaviorally normal after stimulation, rather than aggressive, hyper-reflexic, and sickly, as they often do after chemoconvulsant-induced SE.
Figure 3.

Hippocampal injury four days after perforant pathway stimulation in awake Sprague-Dawley rats. Rats were subjected to 30 min of stimulation on Days 1 and 2, and then 8 hours of identical stimulation on Day 3, followed by perfusion-fixation 4 days after the Day 3 stimulation. A: Unstained, freshly-cut, wet section of dorsal hippocampus under brightfield illumination. Note that injured neurons are visible in unstained sections, and are darker than undamaged regions (the dentate gyrus molecular layer; DG), apparently because degenerating neurons change shape, and their membranes reflect light out of the path of the objective lens (in the manner of a darkfield condenser), making dying cells appear darker than adjacent undamaged tissue that faithfully transmits light into the objective lens. B: FluoroJade-B (FJB)-stained coronal section of the dorsal hippocampus, showing FJB-positive cells in the hilus (h), and in areas CA3 and CA1. Note also the FJB-positive plexus in the inner molecular layer (arrows), which represents the degenerating axon terminals of the associational/commissural projection from damaged hilar mossy cells. C: FJB-stained horizontal section from the ventral hippocampus, demonstrating apparent neurodegeneration in the dentate hilus (h), in areas CA1 and CA3, and in layers 3 (III) and 5 (V) of the entorhinal cortex (EC). Note also the conspicuous sparing of the subiculum (S), presubiculum (Pr), and parasubiculum (Pa). Scale bar: 220 μm in (A) and (B); 450 μm in (C).
Figure 4.

Limited ventral hippocampal- and extra-hippocampal injury after perforant pathway stimulation in awake Sprague-Dawley rats. Rats were subjected to 30 min of stimulation on Days 1 and 2, and then 8 hours of identical stimulation on Day 3, followed by perfusion-fixation 4 days after the Day 3 stimulation. A: FluoroJade-B (FJB) staining of a coronal brain section at low power, demonstrating acutely degenerating neurons in the hippocampus and in thalamic nuclei (box) and the cortex (arrow). A1: Magnification of the box in (A) showing FJB-positive cells in central lateral thalamic nucleus (CL), mediodorsal thalamic nucleus central (MDC) and lateral (MDL), and the paraventricular thalamic nucleus (PV). B: FJB staining of a horizontal section of a rat treated identically to the rat shown in Figure 3 above, but exhibiting little or no apparent injury to ventral dentate hilar neurons (h). Despite this variability in the extent of hilar cell loss throughout the longitudinal hippocampal axis, injury in the hippocampus “proper” (areas CA1-CA3) and in the entorhinal cortex (box expanded in B1) was highly consistent, as was the sparing of subiculum (S), presubiculum (Pr), and parasubiculum (Pa). B1: Magnification of the box in (B1). Note that FJB-positive cells are primarily in layers 3 (III) and 5 (V) of the entorhinal cortex (EC). Green FJB fluorescence was photographed, converted to grayscale, and then inverted to produce the images of black FJB-positive-, acutely degenerating neurons on a white background. Scale bar in B: 1 mm in (A) and (B); 200 μm in (A1) and (B1).
Comparison of coronal and horizontal sections from most animals 4 days after stimulation revealed more extensive hilar neuron injury in the dorsal- than in the ventral hippocampus, but similarly extensive pyramidal layer injury along the hippocampal longitudinal axis (Fig. 4). However, other identically stimulated animals exhibited hilar neuron injury in both the dorsal and ventral hippocampus (Fig. 3). This minor variability among identically treated rats was the only obvious variability noted, as no animals died and none failed to exhibit the pattern of pathology shown in Figures 3 and 4 (n>40). Ten days post-stimulation, Nissl-stained sections from stimulated brains (30,30,8-group) exhibited extensive neuron loss and glial proliferation (Fig. 5, D1 and E1).
Figure 5.

Acute and chronic pathology after perforant pathway stimulation in awake Sprague-Dawley rats. Rats were subjected to 30 min of stimulation on Days 1 and 2, and then 8 hours of identical stimulation on Day 3, followed by perfusion-fixation 4-117 days after the Day 3 stimulation. A: NeuN immunostaining in the dorsal hippocampus 77 days after being stimulated for 30 minutes on each of two consecutive days. Note no obvious damage after only the first two brief stimulations (30,30,0-group). B: FluoroJade-B (FJB) staining 4 days after the two 30 min-long stimulations plus 8 hours of identical stimulation on Day 3. Note extensive injury to CA1 and CA3 pyramidal cells, as well as hilar mossy cells, the origin of the degenerating axon plexus in the dentate inner molecular layer (arrows). Note also the lack of FJB staining of the dentate gryus (DG) and the unstained mossy fiber pathway in the stratum lucidum of area CA3 (asterisk). Green FJB fluorescence was photographed, converted to grayscale, and then inverted to produce the images of black FJB-positive-, acutely degenerating neurons on a white background. C: NeuN immunoreactivity in the dorsal hippocampus 117 days after 30,30,8-group stimulation, showing hippocampal atrophy and survival of dentate granule cells and “resistant zone” pyramidal cells (arrow). D: Nissl-stained section (1% cresyl violet) of dorsal hippocampus from a sham control rat, showing normal anatomy. E: Nissl-stained section from a 30,30,8-group rat 10 days after the third, 8 hr-long stimulation, showing the nearly complete loss of CA3 and CA1 neurons and proliferation of smaller cells throughout the area CA1 and area CA3 neuropil. D1 and E1: Magnification of the area CA1 boxes in panels D and E, showing CA1 pyramidal cell loss and apparent gliosis. F: NeuN immunostaining in a horizontal section from a control rat, showing normal anatomy. G: NeuN immunostaining in a horizontal section from the rat shown in panel C. Note the virtually total loss of CA3 and CA1 pyramidal cells and the survival of the dentate gyrus (DG) and subiculum (S). Note also the loss of neurons in the entorhinal cortex (EC). F1 and G1: Magnification of entorhinal cortex boxes in panels F and G, showing the prominent loss of NeuN-positive cells in layer III of the entorhinal cortex. Scale bar: 200 μm in A–E; D1,E1 = 65 μm in D1,E1; 300 μm in F,G; 98 μm in F1,G1.
Additional rats were stimulated for 12 hours (n=3) and 16 hours (n=3) on day 3, and these animals additionally exhibited significant granule cell degeneration (data not shown). Thus, even relatively resistant dentate granule cells are vulnerable, and prolonged stimulation results in total hippocampal sclerosis (Meldrum and Bruton, 1992).
Long-term neuropathology
Chronic neuron loss and hippocampal atrophy were assessed in rats that survived for 2–14 months following 8 hours of perforant pathway stimulation on day 3. Figure 5,A shows a NeuN-immunostained coronal section from a chronically-implanted control rat subjected to two 30 min stimulations on days 1 and 2, but no stimulation on day 3 (30,30,0 group), which exhibited normal hippocampal morphology. In contrast, rats subjected to the same two 30 min stimulations plus 8 hours of stimulation on day 3 (30,30,8 group) and perfusion-fixed months later exhibited highly reproducible and significant hippocampal atrophy (Fig. 5, C and G) compared with sham control- (n=6) or 30,30,0-group (n=6) rats (Fig. 5, A and F). The hippocampal atrophy evident in both coronal (Fig. 5, C) and horizontal (Fig. 5, G) sections of each brain was the result of widespread loss of both hilar neurons and pyramidal cells in areas CA1 and CA3.
Quantitative analysis confirmed the qualitative data. Rats subjected to two 30 min stimulations plus 8 hours of stimulation on day 3 (30,30,8 group) exhibited a 62% reduction of total hippocampal volume (Fig. 6). Conversely, rats subjected to 3 hours of stimulation-induced convulsive SE (n=4), which mainly affected hilar neurons, appeared slightly shrunken qualitatively and exhibited a calculated 10% reduction in total hippocampal volume that was not significantly different from control (Fig. 6). Rats subjected to two episodes of stimulation-induced convulsive SE for 30 min on days 1 and 2 (30,30,0 group; n=6) also did not exhibit a statistically significant decrease in total hippocampal volume compared to naïve rats (Fig. 6), which is consistent with their lack of FJB-positive cells during the first week after two 30-min that evoked convulsive SE. Cell counts gave similar results. At least 9 weeks after 30,30,8 stimulation, rats exhibited ~74% neuron loss in the dorsal hilus, ~80% neuron loss in area CA3, and ~93% neuron loss in area CA1 of the dorsal hippocampus (Table 1).
Figure 6.
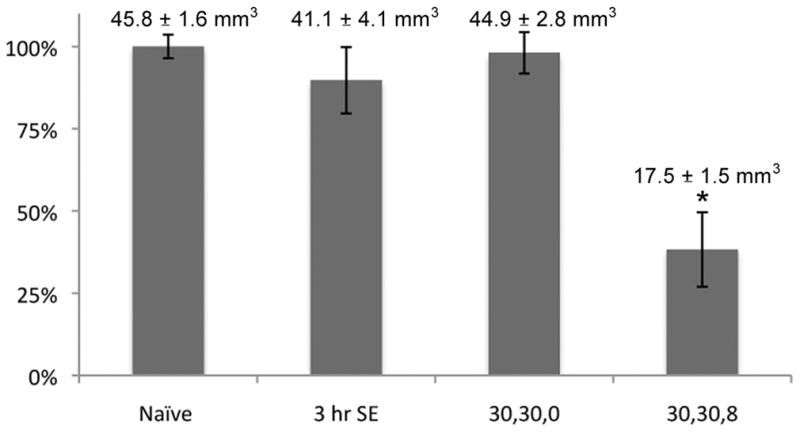
Total hippocampal volume two months after perforant pathway stimulation in freely moving Sprague-Dawley rats. The area of the hippocampus in every second section throughout the entire rostrocaudal extent of the hippocampus was measured using the Adobe Photoshop CS3 Extended Measurement program, which calculates the area bounded by an irregular border. The numerical data are presented above each graph bar. The asterisk denotes that the 30,30,8 group mean was significantly different from naïve control (p < 0.001 by Student’s t-test). Total hippocampal volumes >2 months after 3 hours of convulsive status epilepticus or 30,30,0 stimulation were not significantly different from naïve control (p > 0.05).
Table 1.
Hippocampal neuron loss in the dorsal hippocampus 9 weeks after perforant path stimulation that evoked hippocampal excitation for 8 hrs, but not convulsive status epilepticus. Five 40 μm-thick, non-adjacent sections throughout the dorsal hippocampus of naive control rats and of rats stimulated for 30 min on two consecutive days, followed by 8 hr stimulation on the third day (30,30,8 group), were immunostained for the neuron marker NeuN. Immunoreactive neurons with visible nuclei in each population were counted (n = 4 animals per group). Hilar neurons (hilus) include the number of somata not in contact with the granule cell layers, and outside the somal and dendritic regions of CA3c. In naïve sections, immunoreactive neurons in the CA3 and CA1 pyramidal cell layers included somata that appeared to contact at least one other neuron. This ensured that only neurons that were part of stratum pyramidale were counted. To circumvent the issue that the number of cells per section varies throughout the longitudinal axis of the hippocampus, we used matching coronal sections from the dorsal hippocampus of control and experimental animals, which were selected based on the identification of extra-hippocampal brain regions, e.g. the dorsal thalamic nuclei and the shape of the third ventricle. Note that all cell populations were significantly reduced, with area CA1 showing the greatest loss. Group means are the average number of cells per 40 μm-thick section
| control(naïve) | 30,30,8 | % of control | |
|---|---|---|---|
| Hilus | 66.6±6.9 | 17.2±6.4* | 25.8 ±9.6 |
| CA3 | 577.9±44.3 | 113.3±54.2* | 19.6 ±9.4 |
| CA1 | 1238.2±174.1 | 88.1±37.0* | 7.1 ±2.9 |
p < 0.001 by Student’s t-test.
The ability of a single prolonged episode of hippocampal excitation to replicate all of the defining features of human classic hippocampal sclerosis was evident from the populations of surviving neurons in 30,30,8-group animals. Following degeneration of the initially Fluoro-Jade B-positive hilar neurons, CA3 and CA1 pyramidal cells (Fig. 5,B), and a virtually complete collapse and virtual disappearance of the CA3 and CA1 pyramidal layers (Fig. 5,C), the only surviving hippocampal principal cell populations were the dentate granule cells (Fig. 5,C), an island of pyramidal cells corresponding approximately to area CA2 (arrow in Fig. 5,C), and the subiculum, which survived next to an abruptly decimated and collapsed area CA1 (Fig. 5, F and G). Thus, the single 8 hr-long period of focal hippocampal excitation decimated the previously identified three most vulnerable hippocampal neuron populations, i.e. hilar neurons, CA3 and CA1 pyramidal cells (Nadler et al., 1978; Sloviter, 1991b), and did not cause similar injury to the three principal neuron populations that characteristically survive in human classic hippocampal sclerosis, i.e. dentate granule cells, CA2 pyramidal cells (the “resistant zone”), and subicular pyramidal cells (Meldrum and Bruton, 1992).
Magnetic resonance imaging of convulsive- and non-convulsive status epilepticus
The difference in the extent of extra-hippocampal damage produced by convulsive- vs. non-convulsive status epilepticus was dramatically illustrated by a serendipitous observation made during imaging experiments in Long-Evans rats stimulated for 8 hours (single 8 hour stimulation paradigm described below). On several unusually warm days on which the laboratory temperature was elevated, all stimulated rats developed mild convulsive SE (“mild” because “normal” convulsive SE is always lethal in our experience) for the 8 hours of stimulation, and survived. These rats were compared 11 months later to identically treated rats that did not develop convulsive status epilepticus on cooler days. Magnetic resonance imaging revealed that even mild convulsive SE was associated with obvious changes in the extra-hippocampal piriform/perirhinal cortical regions (n=3; Fig. 7, B1 and B2), whereas the non-convulsive excitation normally produced by the 8 hours of stimulation produced dramatic changes that were limited to the hippocampus (n=4; Fig. 7, C1 and C2). Histological analysis confirmed that the convulsive SE group exhibited a pan-necrosis of the amygdala and piriform area (Fig. 7, B3), whereas animals that did not develop convulsive SE exhibited severe hippocampal atrophy, but no obvious macroscopic damage in those same extra-hippocampal brain regions (Fig. 7, C3).
Figure 7.

Coronal slices from T2-weighted magnetic resonance imaging 11 months after 8 hours of perforant pathway stimulation in Long-Evans rats that exhibited convulsive status epilepticus (CSE) (B), or non-convulsive satatus epilepticus (NCSE) (C). A1,A2: Naïve images acquired from the same animal seen in row B, prior to electrode implantation. B1,B2: Images acquired 11 months after 8 hours of perforant path stimulation (10 sec- long-20 Hz stimulus trains delivered once per minute for 8 hours) that caused mild (non-lethal) status epilepticus for the duration of the 8 hours of perforant pathway stimulation as a result of increased ambient temperature (see text). Arrows denote an elevated signal in extra-hippocampal areas. C1,C2: Images acquired 11 months after identical stimulation at normal ambient temperature, which did not cause convulsive status epilepticus. Note the lack of apparent injury in extra-hippocampal areas compared to the section in (B1). A3-C3: Nissl-stained coronal sections from rats subjected to convulsive vs. non-convulsive SE. A3: Nissl-stained section from a sham-stimulated rat. B3: Nissl-stained coronal section of the rat shown in B1 and B2. Note that the enhanced T2 signal reflected a pan-necrosis and loss of brain tissue presumably filled in vivo with cerebrospinal fluid. C3: Nissl-stained section from a stimulated rat that did not exhibit convulsive status epilepticus. Note the corresponding lack of extra-hippocampal damage in (A3) compared to panel (A2), but also greater pyramidal cell loss and hippocampal atrophy after non-convulsive status epilepticus (C3) than after convulsive status epilepticus (C3). Images were obtained in animals in which all metal electrodes had been removed within 24 hours of the end of stimulation or sham stimulation. Scale bar = 1 mm.
Mossy fiber sprouting after prolonged hippocampal excitation
Timm staining in rats that survived for 3–7 months after 30,30,8-group perforant pathway stimulation and developed classic hippocampal sclerosis (Fig. 8,D) revealed the presence of dense mossy fiber sprouting in the inner molecular layer of the dorsal hippocampus (Fig. 8,E), an expected consequence of hilar mossy cell loss (Jiao and Nadler, 2007). These animals also exhibited an apparent innervation by mossy fibers of the surviving island of pyramidal cells that presumably represent area CA2 neurons (arrow in Fig. 8, D). We say “presumably” because it is not possible to identify surviving pyramidal cells as CA2 pyramidal cells simply because they are in the location previously occupied by CA2 pyramidal cells. CA2 pyramidal cells can only be identified in the normal hippocampus with some degree of certainty by a lack of, or minimal, mossy fiber innervation. That is, CA2 pyramidal cells are specifically defined as large pyramidal cells (like CA3 pyramidal cells) at the end of the CA3 pyramidal cell layer that do not receive mossy fiber input (Lorente de Nó, 1934). In all stimulated rats assessed with Timm staining, the Timm-positive mossy fibers innervated the surviving pyramidal cells (Fig. 8,F). This observation suggests that the CA2 pyramidal cells survived, presumably because they did not receive lethal mossy fiber input during stimulation, and were then aberrantly innervated by surviving mossy fibers, a scenario previously described in the human epileptic hippocampus (Williamson and Spencer, 1994).
Figure 8.

Hippocampal atrophy and synaptic reorganization (mossy fiber sprouting) in the dorsal hippocampus after perforant pathway stimulation in awake Sprague-Dawley rats. Rats were subjected to 30 min of stimulation on Days 1 and 2, and then 8 hours of identical stimulation on Day 3 (30,30,8-group). A: Nissl-stained section of the dorsal hippocampus from a naïve rat, illustrating normal anatomy. B: NeuN immunostaining in a dorsal hippocampal section from a naïve rat, showing the normal location of hippocampal neurons. C: NeuN immunostaining 56 days after 30,30,8-group stimulation, demonstrating classic hippocampal sclerosis from a single episode of stimulation on Day 3. Arrow points to surviving “resistant zone” neurons. D: Timm staining in a dorsal hippocampal section of a rat 181 days after two daily 30 minute episodes of perforant pathway stimulation (30,30,0 group). Note the normal pattern of Timm staining derived from the axons of dentate granule cells (mossy fibers; MF) and the axon terminals of CA3 pyramidal cells in areas CA3 and CA1 (asterisks). E: Timm-stained and Nissl- counterstained section of the dorsal hippocampus 193 days after 30,30,8-group stimulation, demonstrating classic hippocampal sclerosis, with survival of granule cells that form an aberrant axon plexus in the inner molecular layer (arrows). F: Magnification of box in panel E, showing the apparent innervation of surviving hippocampal neurons by the terminal portion of the mossy fiber pathway (arrows). Scale bar: 250 μm in A–E; 55 μm in F.
Lower intensity perforant pathway stimulation for 8 hours in Long-Evans and Brown-Norway rats
The observation that two brief daily stimulations decreased the electrographic and behavioral severity of seizure activity evoked by the same stimulation paradigm on the third day raised the issue of whether the prevention of convulsive status epilepticus by the two daily stimulations was due simply to the increased granule cell inhibition and reduced granule cell excitability that we observed 24 hours after the second stimulation, or might be a consequence of other changes triggered over the two-day pre-stimulation period, such as changes in GABA receptor expression (Brooks-Kayal et al., 1998), or other gene-dependent processes. Therefore, we determined whether a single 8 hour episode of less intense perforant pathway stimulation, without any pre-stimulation, was capable of producing the same pattern of classic hippocampal sclerosis. We accomplished this by using a rat strain (Long-Evans) that reportedly has a decreased tendency to develop convulsive status epilepticus in response to kainate treatment (Xu et al., 2004; Young et al., 2009). Because Long-Evans rats are a cross between an albino rat and a Brown Norway rat, we also tested the single stimulation paradigm in four Brown Norway rats, predicting that they might also be relatively resistant to developing behavioral status epilepticus.
In the studies described above in Sprague-Dawley rats, we used a stimulation paradigm that involved continuous 2 Hz stimulation plus 10 sec-long 20 Hz stimulus trains, and this paradigm also caused convulsive SE in Long-Evans rats. Therefore, we omitted the continuous 2 Hz stimulation and simply delivered 10 sec-long 20 Hz stimulus trains once per minute for 8 hrs. Although this paradigm still produced convulsive SE in Sprague-Dawley rats, stimulus trains alone did not produce convulsive SE in Long-Evans or Brown-Norway rats. During 8 hours of this reduced-intensity stimulation, Long-Evans and Brown-Norway rats exhibited motionless periods, occasional wet-dog shakes, but otherwise little abnormal behavior. Occasional behavioral seizures, occurring once or twice per hour, were always accompanied by prolonged granule cell epileptiform discharges between stimulus trains, as described above in Sprague-Dawley rats. Long-Evans and Brown-Norway rats stimulated for one 8 hour episode exhibited acute pathology (FJB-positive neurons) 3 or 4 days later that was indistinguishable from that seen in Sprague-Dawley rats after the three day stimulation paradigm (as shown in Figures 3 and 4). Figure 9 shows the extensive hippocampal atrophy in both the dorsal and ventral hippocampus 261 days after a single 8 hour episode of afferent stimulation in a Long-Evans rat. Thus, the 2-day pre-treatment stimulation was not necessary for extensive pathology to occur, and apparently served simply to increase granule cell inhibition, reduce the duration of granule cell afterdischarge, limit seizure spread, prevent the development of convulsive status epilepticus, and decrease the intensity of the 8 hour-long insult.
Figure 9.

Hippocampal pathology after perforant pathway stimulation in awake, freely moving Long-Evans rats. A1 and A2: Nissl-stained coronal and horizontal sections of the dorsal (A1) and ventral (A2) hippocampus, respectively, of a naïve Long-Evans rat, showing normal hippocampal anatomy. B1 and B2: Nissl-stained sections of the dorsal (B1) and ventral (B2) hippocampus of a Long-Evans rat 261 days after a single 8 hour- long episode of perforant pathway stimulation that did not cause convulsive status epilepticus. Note classic hippocampal sclerosis throughout the hippocampal longitudinal axis. Scale bar: 450 μm.
Spontaneous hippocampal-onset seizures following 8 hours of perforant pathway stimulation
Sprague-Dawley rats subjected to two 30 min-long pre-stimulations and an 8 hour episode of perforant pathway stimulation on the third stimulation day (30,30,8-group) were monitored after stimulation by continuous electrical and video recording to determine the latency to spontaneous electrographic and behavioral seizures. However, given the severe atrophy that followed hippocampal injury, it is doubtful that the recording electrodes tips remained within the granule cell layers for very long. Thus, in most animals, the electrodes were removed at the end of stimulation, and new electrodes were re-implanted months later, after most of the hippocampal shrinkage had occurred. As a result, the activity we recorded from original electrodes after this initial stimulation must be regarded as EEG activity rather than granule cell layer activity, except in cases where the responses to perforant pathway stimulation showed that the electrode tip was in the granule cell layer. Conversely, the later recordings with newly implanted recording electrodes were verified to be within the granule cell layer of the surviving and relocated dentate gyrus by assessing the evoked responses.
In the first series of recordings, made shortly after the initial stimulation using the original electrodes, continuous video and electrographic recordings revealed the average latency to spontaneous electrographic and clinical epilepsy to be 21.5 days ± 3.7 (range 16–25 days; n=6). All six animals exhibited “hippocampal-onset” seizures, i.e. high amplitude electrographic activity was recorded before behavioral onset, which was defined as the first unequivocal signs (forepaw clonus) of an ensuing generalized seizure. Behavioral arrest routinely preceded forepaw clonus, but behavioral immobilization was not regarded as behavioral onset. None of the spontaneous behavioral seizures, all of which were judged to be stage 5 seizures (Racine, 1972), occurred without concurrent electrographic seizure activity, and vice versa. However, uncertainty regarding the electrode tip location in the acute post-injury period (stimuli were not delivered out of concern that any stimulation could affect the latency to clinical epilepsy) prevents us from stating unequivocally whether undetected subclinical hippocampal-onset seizures preceded the clinical seizures. Thus, the only reliable latency information we obtained was that spontaneous behavioral seizures started ~3 weeks post-stimulation, with a range of 16–25 days post-stimulation in the 6 rats monitored for initial behavioral seizure occurrence.
Spontaneous behavioral seizures were also recorded from chronically epileptic animals after electrode re-implantation to determine whether or not spontaneous seizures were granule cell-onset seizures, as only re-implanted electrodes could be known to be recording directly from the granule cell layer. A total of 65 spontaneous behavioral seizures were recorded in 6 chronically epileptic rats, and these recordings revealed that all involved spontaneous negative-going granule cell population spikes (Fig. 10), which occurred as early as day 16 post-stimulation and as late as 154 days post-stimulation, the latest period recorded. Of the 65 clinical seizures video-recorded, large-amplitude granule cell activity preceded the first obvious seizure sign (forepaw clonus) in 64 of 65 spontaneous seizures analyzed. In the remaining seizure, electrographic seizure activity was coincident with the initial immobilization that preceded forepaw clonus.
Figure 10.

Spontaneous granule cell layer activity recorded in an awake Sprague-Dawley rat 129 days after perforant pathway stimulation that evoked hippocampal excitation for 8 hrs, but not convulsive status epilepticus. Treatment was 30 min of stimulation on Days 1 and 2, and then 8 hours of identical stimulation on Day 3, followed by continuous electrographic and video monitoring. A: 60 seconds of activity recorded directly from the granule cell layer. Asterisk marks the occurrence of the first sign of a spontaneous behavioral seizure (forepaw clonus leading to rearing). Note that high amplitude activity (boxes 1, 2, and 3) preceded the behavioral seizure onset. Expanded trace 1: Expanded view of box 1 in (A) above. Note that large amplitude activity began as positive-going potentials with a small superimposed negative-going population spike, activity consistent with seizure onset in the entorhinal cortex and propagation to the granule cell layer via the perforant pathway. Expanded trace 2: Expanded view of box 2 in (A) above. Note that the spontaneous granule cell layer events increase in amplitude and frequency, and appear virtually identical to the granule cell evoked potentials recorded in response to perforant pathway stimulation (compare the spontaneous potentials in (B2) with the evoked potential in (B1). Expanded trace 3: Spontaneous granule cell layer events increase in amplitude and frequency just prior to behavioral seizure onset (asterisk). Expanded trace 4: Spontaneous granule cell layer events continue after seizure onset. Calibration bars: 5 sec in (A); 25 msec in panels 1–4; 5 msec in B1 and B2; 5 mV in all panels.
Electrographic seizure activity during these 65 spontaneous behavioral seizures had an average duration of 49.4 ± 15.7 seconds (Table 2). Early seizures (those recorded on or before day 33, the latest period monitored with original electrodes) had a shorter average duration (34.9 ± 13.1 sec) than those seen at later time points (on or after day 79, the earliest time of electrode re-implantation), which were 53.3 ± 13.9 sec in duration. A two-sample t-test revealed a significant difference between these two group means (p<0.005), suggesting a correlation between lengthening seizure duration with increasing time post-stimulation. In addition, it may be significant that the nine longest electrographic- and the seven longest behavioral seizures were recorded at least 81 days after stimulation (Table 2). The absence of data points between 39 and 81 days post-stimulation was due to the practical issue of the changing recording electrode position described above. All late seizures were recorded on or after day 79 post-stimulation, and these were recorded from animals that had their electrodes removed within 24 hours after stimulation. Thus, the early and late seizures were recorded from different groups of animals and therefore the differences in the durations of the early and late electrographic and behavioral seizures may not be comparable or reliable indicators of differences with respect to changes in seizure severity or duration as a function of the post-stimulation period.
Table 2.
Electrographic and behavioral seizure duration after perforant path stimulation that evoked hippocampal excitation for 8 hrs, but not convulsive status epilepticus. Continuous electrographic recording of spontaneous activity from the granule cell layer, and continuous video recording of behavioral activity were analyzed for 65 spontaneous seizures, all of which were rated stage 5 on the Racine scale (Racine, 1972). Electrographic seizure duration was always longer than that of seizure behavior, and preceded behavioral onset in 64 of 65 spontaneous seizures. Early seizures (those recorded on or before day 33) had a slightly shorter mean duration than those seen at later time points (on or after day 79), and a two sample t-test revealed a statistically significant difference between group means (p < 0.005), suggesting a correlation between increasing time post-stimulation and lengthening seizure duration. All late seizures, i.e. those that occurred on or after day 79, were recorded from animals that had their electrodes removed immediately following stimulation and then had new electrodes re-implanted after day 70. This was done to ensure that electrodes did not interfere with developing hippocampal atrophy, and to obtain granule cell layer recordings from atrophied hippocampi after maximal shrinkage had occurred.
| All seizures | Earlyseizures | Lateseizures | |
|---|---|---|---|
| Electrographic duration | 49.4±15.7 sec (range 21 to 85) | 34.9±13.1 (21 to 61) | 53.3±13.9 (27 to 85) |
| Behavioral duration | 43.6±15.9 sec (range 12 to 82) | 29.1±15.7 (14 to 58) | 47.4±13.8 (12 to 82) |
The clearest information gleaned from recordings made directly from the granule cell layers, with the electrode tip locations verified by assessing the morphology of the evoked potentials, was that following the 8 hours of stimulation that caused extensive hilar neuron loss and the death of CA1 and CA3 pyramidal cells, granule cells became spontaneously active, generating synchronous positive-going potentials with superimposed negative-going population spikes, as we previously demonstrated after 3 hours of convulsive SE-induced hippocampal injury (Bumanglag and Sloviter, 2008). Spontaneous granule cell layer potentials invariably began as large amplitude, positive-going potentials with small negative-going spikes (Fig. 10, A1) that were virtually identical to the compound potentials evoked by stimulation of the efferent entorhinal cortical input to the dentate gyrus (Andersen et al., 1966; Sloviter, 1991a). That is, the first spontaneous potentials closely resembled what would be expected from the granule cell layer if the seizures originated in the entorhinal cortex. These early spontaneous potentials were quickly followed by the appearance of larger-amplitude, negative-going granule cell population spikes that were virtually identical in amplitude, duration, and population spike morphology to potentials evoked minutes later by perforant pathway stimulation (Fig. 10, B1 and B2). Thus, the available evidence is consistent with spontaneous “hippocampal-onset” granule cell seizure discharges that may have been preceded and driven by spontaneous and synchronized entorhinal cortex discharges.
Sham-control rats (n=4) and rats subjected to two 30 min-long stimulations (n=4) were observed intermittently for 4 months. Neither spontaneous granule cell layer population spikes or epileptiform discharges, nor spontaneous behavioral seizures, were observed in any control animals.
Progressive hippocampal atrophy after extensive hippocampal injury
Sequential magnetic resonance imaging was utilized as a qualitative measure of hippocampal atrophy in animals stimulated using the 30,30,8 paradigm to identify the approximate time course of maximal hippocampal atrophy and to determine, in relation to the onset of spontaneous epilepsy, whether epileptogenesis preceded or followed hippocampal atrophy. This addressed the question of whether epileptogenesis occurs approximately coincident with initial neuron loss, or if epileptogenesis requires hippocampal atrophy before epilepsy begins. T2-weighted images were obtained in naïve rats prior to electrode implantation and at various time points up to 7 months after stimulation and immediate removal of all electrodes. Images obtained 10 days post-stimulation revealed an elevated T2 signal in both hippocampi, which was presumably indicative of acute hippocampal edema and gliosis, rather than later hippocampal atrophy and ventricular expansion, because histology at 10 days post-stimulation revealed an unshrunken hippocampus containing dead neurons and glial proliferation (Fig. 5,E). Imaging at successive time points (8 and 28 weeks) demonstrated progressive hippocampal atrophy that was remarkably selective in that the rest of the brain appeared relatively unaffected. Notably, however, progressive shrinkage was evident in both the hippocampus and the ventral cortex (Fig. 11, B3 and B4; arrows) between 8 and 28 weeks post-stimulation, indicating a progressive, long-lasting process of tissue removal and atrophy that follows both a single initial insult and the early development of clinical epilepsy. Thus, continuing tissue shrinkage may not be synonymous with ongoing neuronal death (Van Paesschen et al., 1998).
Figure 11.

Coronal slices from T2-weighted magnetic resonance images at various time points after sham stimulation or perforant pathway stimulation in awake Sprague-Dawley rats. Sham stimulation involved no delivery of stimuli to chronically implanted rats, followed by removal of the implanted electrodes in preparation for imaging. Stimulated rats were subjected to 30 min of stimulation on Days 1 and 2, and then 8 hours of identical stimulation on Day 3, followed by removal of the electrodes and repeated magnetic resonance imaging 10, 56, and 196 days later. Note that all images in each column were acquired in the same rat. A1,B1: Images from each naïve rat prior to electrode implantation. A2,B2: 10 days after sham (A2) or 8 hour stimulation (B2). Note elevated signal in the hippocampi of the 8 hr-stimulated animal. A3,B3: 8 weeks after stimulation, images of the same rats show hippocampal atrophy (apparent expansion of ventricular volume), as well as cortical shrinkage (arrow). A4,B4: 28 weeks after stimulation, images show continuing hippocampal atrophy and cortical shrinkage (arrow). Scale bar: 1 mm.
Discussion
The results of this study clarify a variety of issues surrounding the etiology of acquired temporal lobe epilepsy with hippocampal sclerosis, the nature of insults that most likely cause hippocampal sclerosis and initiate the epileptogenic process, and the features of animal models that use electrical stimulation or chemoconvulsants to produce a permanent epileptic state. The main original findings are as follows.
First, one prolonged episode of moderate-intensity hippocampal excitation, which involved no clinical indication during the insult that an injury was being inflicted, nonetheless produced classic hippocampal sclerosis with minimal variability. The pathology was characterized by the death of dentate hilar neurons and virtually all hippocampal pyramidal cells, as well as neurons of the entorhinal cortex and thalamus, with consistent sparing of dentate granule cells, a “resistant zone” of pyramidal cells, and subicular neurons.
Second, the cryptic injury that caused classic hippocampal sclerosis reliably initiated a clinical state in which spontaneous hippocampal granule cell epileptiform discharges preceded or accompanied each spontaneous behavioral seizure. Thus, we believe this to be the first animal model that reliably reproduces the pathology and pathophysiology of human mesial temporal lobe epilepsy with classic hippocampal sclerosis (Meldrum and Bruton, 1992).
Third, the first 3 hours of the ultimately highly injurious 8 hour-long excitatory insult produced no detectable hippocampal injury, indicating that although vulnerable, the rat hippocampus is not fragile and can endure prolonged, potentially injurious excitation without suffering any detectable neuron loss.
Fourth, the unexpectedly cryptic nature of the insult that causes classic hippocampal sclerosis and hippocampal-onset epilepsy in rats may explain why some patients with hippocampal atrophy report no known brain injury prior to developing epilepsy. That is, subclinical signs of focal excitation during prolonged febrile seizures, or after a head injury, which perhaps involves only confusion or an amnestic period at the time of the initial insult, may be frequently missed.
Fifth, spontaneous “hippocampal-onset” seizures, defined as events in which spontaneous dentate granule cell epileptiform discharges precede each behavioral seizure onset, began shortly after insult, indicating that epileptogenesis is a rapid process that precedes the extended tissue shrinkage process that ultimately results in hippocampal atrophy.
Finally, the similarity of hippocampal pathology between patients that suffered prolonged febrile seizures or head trauma (Swartz et al., 2006) suggests that classic hippocampal sclerosis following head trauma may involve undetected subclinical hippocampal excitation, or glutamate release in the immediate post-injury period that may occur (Eid et al., 2004; Cavus et al., 2008) without producing detectable electrographic activity. If so, the treatment of head trauma-induced epileptogenesis might benefit from more aggressive EEG monitoring and pharmacological suppression of excitation and/or calcium influx (Evans et al., 1984) during the immediate post-injury period, when cell death is actively occurring.
On the etiology of classic hippocampal sclerosis
The discovery that a single episode of hippocampal excitation of moderate intensity and long duration reliably and uniquely replicates classic hippocampal sclerosis in awake rats, taken together with current knowledge of hippocampal circuitry, explains why no other experimental insults produce this characteristic hippocampal pathology. In urethane-anesthetized rats, even prolonged perforant pathway stimulation for 24 hours causes only the death of some of the neurons directly innervated by granule cells (Sloviter, 1983; 1987), including most hilar neurons and a minority of CA3 pyramidal cells, but spares most pyramidal cells (Sloviter, 1991b). This pattern of limited cell loss results in “endfolium sclerosis,” rather than classic hippocampal sclerosis (Margerison and Corsellis, 1966; Bruton, 1988). Conversely, a virtually complete loss of CA3 and CA1 pyramidal cells results from the identical stimulation paradigm under urethane anesthesia when the CA3 pyramidal cell layer is stimulated directly (Sloviter, 1991b). Thus, we conclude that urethane anesthesia inhibits CA3 pyramidal cell discharge during perforant pathway stimulation, and prevents the extensive CA3 and CA1 pyramidal cell loss that CA3 discharge produces in the awake state.
The different patterns of hippocampal neuron loss caused by different stimulation patterns in awake or anesthetized rats can be closely correlated with the four distinct patterns of human hippocampal pathology associated with MTLE (Blümcke et al., 2007). The four patterns include: 1) “Mesial Temporal Sclerosis (MTS)-1a,” defined as classic hippocampal sclerosis (loss of hilar neurons, CA3 and CA1 pyramidal cells, with survival of granule cells and “resistant zone” neurons); 2) “MTS-1b,” defined as extensive cell loss in all hippocampal neuron subpopulations (total hippocampal sclerosis); 3) “MTS-2,” defined as a loss of CA1 pyramidal cells, with survival of hilar neurons, and; 4) “MTS-3,” defined as mainly hilar cell loss (endfolium sclerosis) (Blümcke et al., 2007). Perforant pathway stimulation for 24 hours under urethane anesthesia (Sloviter and Damiano, 1981; Sloviter, 1983; 1987; 1991b) produces “endfolium sclerosis” (human pattern MTS-3), whereas CA3 pyramidal layer stimulation for 24 hours under urethane anesthesia (Sloviter, 1991b) causes pyramidal cell loss without significant dentate hilar loss (human pattern MTS-2; Blümcke classification). These two patterns of cell loss (mainly hilar cell loss or mainly pyramidal cell loss) are combined in classic hippocampal sclerosis (“MTS type 1a”; Blümcke et al., 2007), which apparently requires simultaneous granule cell- and CA3 pyramidal cell discharging that doesn’t spread to cause convulsive SE. Thus, the results of this study indicate that, in the awake state, and with a less-than-maximally-intense hippocampal excitation, both granule cells and CA3 pyramidal cells discharge continuously, causing: 1) death of cells monosynaptically driven by granule cells (hilar neurons and a minority of CA3 pyramidal cells), and; 2) the death of neurons driven monosynaptically by the CA3 pyramidal cells (all CA3 and CA1 pyramidal cells), while avoiding the extensive brain damage caused by convulsive SE.
We attribute the failure of perforant pathway stimulation under urethane anesthesia, and all chemoconvulsant treatments in awake rats, to cause extensive pyramidal cell loss (and therefore classic hippocampal sclerosis) to their failure to produce sustained CA3 pyramidal cell discharge, and therefore the loss of pyramidal cells, which is the cell loss that causes maximal hippocampal atrophy. Conversely, the consistently extensive loss of hilar neurons after perforant pathway stimulation under urethane anesthesia (Sloviter, 1991b; Zappone and Sloviter, 2004), and the variable loss of hilar neurons after pilocarpine- or kainate-induced status epilepticus (Sloviter, 1992; Harvey and Sloviter, 2005), apparently reflects consistent or variable granule cell discharging during these treatments, respectively, as we demonstrated in kainate-treated rats using awake recording methods (Sloviter et al., 2003). However, all of these treatments fail to cause extensive loss of CA3 and CA1 pyramidal cells, an effect that can be obtained only by direct excitation of the CA3 pyramidal cell layer (Sloviter, 1991b), or by the new stimulation paradigm presented in this study. In the case of human patients who mainly exhibit CA1 pyramidal cell loss, with preservation of all other hippocampal neuron populations (pattern MTS-2; Blümcke classification), we attribute this pattern of cell loss to CA3 pyramidal cell excitation of an intensity and duration that is sufficient to destroy CA1 pyramidal cells, but insufficient to cause the death of most CA3 pyramidal cells, via their recurrent excitatory associational axonal projections (Miles and Wong, 1987; Ishizuka et al., 1990). Whether this CA3 pyramidal cell layer excitation is produced by an intensity of granule cell discharge sufficient to activate CA3, but insufficient to kill most hilar neurons, or by CA3 discharge not involving granule cell discharge, is unknown.
The existence of a fourth pattern of hippocampal sclerosis (pattern MTS-1b), called “total hippocampal sclerosis,” in which the hippocampus is virtually depleted of all neuronal populations in some part of the hippocampus (Meldrum and Bruton, 1992), indicates that all hippocampal neurons are vulnerable, if not always killed. We attribute total hippocampal sclerosis to a particularly long duration of hippocampal excitation. We produced this pattern of damage, which includes extensive dentate granule cell loss as well as hilar and pyramidal cell loss, by simply continuing the stimulation for 12–16 hrs. Thus, we view all hippocampal neurons to be potentially vulnerable to excitotoxic injury (Olney, 1981), and suggest that the different cell loss patterns associated with MTLE are the result of differences in the intensity and duration of granule cell and CA3 pyramidal cell discharges, rather than being the result of seizure activity propagating to hippocampal neurons via different pathways, although this is a speculative point.
Neuronal vulnerability is not synonymous with neuronal fragility
Although we reported originally that hilar mossy cells and hilar interneurons appeared to be the most vulnerable neurons in the urethane-anesthetized rat after 24 hours of perforant pathway stimulation (Sloviter, 1987), all hippocampal principal neurons are clearly vulnerable, as we subsequently demonstrated in rats (Sloviter, 1991b; Sloviter et al., 1996), and as clearly indicated by the existence of total hippocampal sclerosis in some human patients (Meldrum and Bruton, 1992). We have recently confirmed the relatively extreme vulnerability of dentate hilar mossy cells in mice, which exhibit extensive mossy cell loss in response to a brief duration of perforant pathway stimulation (2 hrs) that spares adjacent hilar inhibitory interneurons (Kienzler et al., 2009). Despite the clear vulnerability of hippocampal neurons, many animals subjected to convulsive SE (Milgram et al., 1991; Scharfman et al., 2001; Sloviter et al., 2003; Zappone and Sloviter, 2004), and a subset of MTLE patients (Margerison and Corsellis, 1966; de Lanerolle et al., 2003; Thom et al., 2005; Blümcke et al., 2007; Seress et al., 2009), exhibit minor or no detectable hippocampal cell loss. However, we do not regard the survival of hippocampal neurons in some cases as evidence of their lack of vulnerability, any more than the survival of a home located just outside the direct path of a tornado is evidence of its invulnerability to tornadoes. In our view, the survival of some neurons after an insult indicates only that the insult was insufficient to kill the surviving neurons under the conditions experienced, or that a hierarchy of relative vulnerability may exist (Nadler et al., 1978; Sloviter, 1987; 1991b). Regardless, the present results indicate that hippocampal neurons, although vulnerable, are not fragile, because they survive 30 minutes of continuous convulsive hippocampal SE, or 3 hours of less intense, but nonetheless eventually injurious, excitation. Our results clearly indicate the parameters of the insult that destroys virtually all of the most vulnerable hippocampal neuron populations (hilar neurons and pyramidal cells), and spares the more resistant populations (dentate granule cells, CA2 pyramidal cells, and subicular neurons), thus replicating classic hippocampal sclerosis.
Presumably, a threshold of significant duration must be exceeded to produce irreversible neuron loss, which may involve reaching a threshold at which glia lose their ability to maintain ionic homeostasis, and calcium entry becomes irreversible and deleterious (Evans et al., 1984). The apparent requirement for an excitatory event of a particular intensity and duration to produce any detectable neuron loss (and presumably epilepsy) probably explains why only ~10% of MTLE patients exhibit classic hippocampal sclerosis, and why most patients exhibit less extensive neuron loss (Lehericy et al., 1997; Van Paesschen et al., 1997; Salmenperä et al., 2005; Blümcke et al., 2007). That is, there may be a narrow “window” for excitation-induced injury: too little excitation, and no damage results; too much excitation and status epilepticus results, causing widespread brain damage and a different clinical state (Mikaeloff et al., 2006). Thus, only the rare injury event is of the intensity and duration needed to both activate all hippocampal principal cells and to activate them long enough to irreversibly injure their vulnerable target cells. The apparent natural resistance of all neurons to prolonged excitation may also explain why most children who experience febrile seizures do not develop epilepsy, and why some patients who experience years of spontaneous and relatively brief epileptic seizures often exhibit no evidence of hippocampal injury or atrophy (Margerison and Corsellis, 1966; Bruton, 1988; de Lanerolle et al., 2003; Thom et al., 2005; Blümcke et al., 2007). Although most febrile seizures and mild head traumas do not cause hippocampal sclerosis and epilepsy, the hypothesized causal relationship between hippocampal neuron loss and hippocampal epileptogenesis (Sloviter, 1991b; 1994; Bumanglag and Sloviter, 2008), is consistent with the observation that a large proportion of those patients who do exhibit refractory MTLE and classic hippocampal sclerosis report a history of prolonged febrile seizures (Maher and McLachlan, 1995; Cendes, 2004; Lewis, 2005), infection (Marks et al., 1992), or head trauma (Bruton, 1988; Marks et al., 1995; Mathern et al., 1995).
Implications for epileptogenesis and the latent period
Given the fact that spontaneous granule cell-onset seizures are generated directly from the granule cell layers of awake epileptic rats whether or not extensive pyramidal cell loss occurs (Bumanglag and Sloviter, 2008 and this study), and given the observation that there is a close correlation between the extent of hilar neuron loss and immediate granule cell hyperexcitability (Sloviter, 1991b; Sloviter, 1992; Zappone and Sloviter; 2004), we suggest that granule cell hyperexcitability and spontaneous granule cell seizure discharges are primarily caused by seizure- or head injury-induced hilar neuron loss (Sloviter, 1991b; 1994; Lowenstein et al., 1992), whereas extensive pyramidal cell loss, when it occurs, may primarily explain some of the memory defects associated with epilepsy (Sass et al., 1991; 1995; Cohen-Gadol et al., 2004).
The observation that widespread brain damage caused by convulsive status epilepticus results in the appearance of clinical epilepsy without delay (Harvey and Sloviter, 2005; Raol et al., 2006; Goffin et al., 2007; Jung et al., 2007; Bumanglag and Sloviter, 2008), whereas the less extensive extra-hippocampal brain damage observed in this study was associated with a latent period lasting ~ 3 weeks, suggests that the time needed for focal hippocampal discharges to become clinically obvious events may be related to the location and extent of extra-hippocampal brain damage (Bumanglag and Sloviter, 2008), rather than the maturation of a time-dependent secondary process triggered by neuronal injury (Sloviter, 2008). That is, initially focal (subclinical) discharges in minimally damaged brains may face barriers to seizure spread that delay the appearance of generalized seizures, resulting in a detectable “latent period.” Conversely, initial damage to all constituent nuclei in a more severely damaged seizure circuit may cause even the earliest focal discharges to become clinically obvious events without delay (Bumanglag and Sloviter, 2008). Thus, acquired epileptogenesis may primarily involve neuron loss plus a time-requiring “kindling” process during which initially subclinical focal epileptiform events become clinically detectable disturbances of motor pathways. From this network perspective, epilepsy, defined at minimum as a state of abnormal focal discharges, develops at the time of the initial injury, with the latent period involving a progressive process that gradually lowers the seizure threshold (“kindling”), rather than a “quiet” pre-epileptic period after injury that awaits the development of a distinct secondary process (Sloviter, 2008).
The primary value of the electrical stimulation models used in this and previous studies lies in their similarity to the human neurological condition. That is, the human patterns of endfolium sclerosis or classic hippocampal sclerosis with limited temporal cortical damage are produced in rats with negligible variability and no lethality, and every animal develops hippocampal-onset epilepsy, features that are needed in any model of hippocampal epileptogenesis. The available data indicate that after damage to the entorhinal cortex and hippocampus, spontaneous granule cell discharges develop without delay and closely resemble the potentials evoked from granule cells by perforant path stimulation (Bumanglag and Sloviter, 2008). We hypothesize that the seizures in this model begin outside the hippocampus, possibly within the entorhinal cortex (Du et al., 1993; 1995; Schwarcz et al., 2000) or in the relatively undamaged subiculum (Cohen et al., 2002), presubiculum or parasubiculum (Eid et al., 1996), and that only when the originating seizure activity recruits epileptiform discharges from disinhibited dentate granule cells (Sloviter, 1991b; 1994) do clinical seizures occur.
Finally, the availability of animal models that involve no chemoconvulsant drugs, minimal variability and no lethality, and that exhibit the human patterns of limited pathology with verified hippocampal-onset seizures, should make it possible to begin to understand the relationship between neuron loss, subclinical focal events, and the network process that causes those events to either quickly or eventually become clinical seizures. Although true anti-epileptogenesis may be an unrealistic goal if injury-induced neuron loss is epileptogenic and cannot be entirely prevented, it should nonetheless be possible to identify the mechanisms that convert focal discharges into life-disrupting clinical seizures, and to develop strategies to prolong the latent period indefinitely.
Acknowledgments
This work was supported by grant NS18201 from the National Institute of Neurological Disorders and Stroke, National Institutes of Health (RSS), by the Fondazione Cariverona, and by a European Community grant (LSH-CT-2006-037315; EPICURE, thematic priority LIFESCIHEALTH (PFF).
Literature Cited
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Andersen P, Holmqvist B, Voorhoeve PE. Entorhinal activation of dentate granule cells. Acta Physiol Scand. 1966;66:448–460. doi: 10.1111/j.1748-1716.1966.tb03223.x. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Elmér E, Nanobashvili A, Kokaia M, Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci USA. 1997;94:10432–10437. doi: 10.1073/pnas.94.19.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagini G, Baldelli E, Longo D, Contri MB, Guerrini U, Sironi L, Gelosa P, Zini I, Ragsdale DS, Avoli M. Proepileptic influence of a focal vascular lesion affecting entorhinal cortex-CA3 connections after status epilepticus. J Neuropathol Exp Neurol. 2008;67:687–701. doi: 10.1097/NEN.0b013e318181b8ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümcke I, Pauli E, Clusmann H, Schramm J, Becker A, Elger C, Merschhemke M, Meencke HJ, Lehmann T, von Deimling A, Scheiwe C, Zentner J, Volk B, Romstöck J, Stefan H, Hildebrandt M. A new clinico-pathological classification system for mesial temporal sclerosis. Acta Neuropathol. 2007;113:235–244. doi: 10.1007/s00401-006-0187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratz E. Ammonshornbefunde der Epileptischen. Arch Psychiat Nervenkr. 1899;31:820–836. [Google Scholar]
- Briellmann R, Berkovic S, Syngeniotis, King M, Jackson GD. Seizure associated hippocampal volume loss: a longitudinal magnetic resonance study of temporal lobe epilepsy. Ann Neurol. 2002;51:641–644. doi: 10.1002/ana.10171. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Bruton CJ. The neuropathology of temporal lobe epilepsy. Oxford: Oxford University Press; 1988. [Google Scholar]
- Bumanglag AV, Sloviter RS. Minimal latency to hippocampal epileptogenesis and clinical epilepsy after perforant pathway stimulation-induced status epilepticus in awake rats. J Comp Neurol. 2008;510:561–580. doi: 10.1002/cne.21801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalheiro EA, Riche DA, Le Gal La Salle G. Long-term effects of intrahippocampal kainic acid injection in rats: a method for inducing spontaneous recurrent seizures. Electroencephalogr Clin Neurophysiol. 1982;53:581–589. doi: 10.1016/0013-4694(82)90134-1. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Sutula TP. Progressive neuronal loss induced by kindling: a possible mechanism for mossy fiber synaptic reorganization and hippocampal sclerosis. Brain Res. 1990;527:1–6. doi: 10.1016/0006-8993(90)91054-k. [DOI] [PubMed] [Google Scholar]
- Cavus I, Pan JW, Hetherington HP, Abi-Saab W, Zaveri HP, Vives KP, Krystal JH, Spencer SS, Spencer DD. Decreased hippocampal volume on MRI is associated with increased extracellular glutamate in epilepsy patients. Epilepsia. 2008;49:1358–1366. doi: 10.1111/j.1528-1167.2008.01603.x. [DOI] [PubMed] [Google Scholar]
- Chen S, Buckmaster PS. Stereological analysis of forebrain regions in kainate-treated epileptic rats. Brain Res. 2005;1057:141–152. doi: 10.1016/j.brainres.2005.07.058. [DOI] [PubMed] [Google Scholar]
- Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol. 2004;17:161–164. doi: 10.1097/00019052-200404000-00013. [DOI] [PubMed] [Google Scholar]
- Chang BS, Lowenstein DH. Mechanisms of disease; epilepsy. N Engl J Med. 2003;349:1257–1266. doi: 10.1056/NEJMra022308. [DOI] [PubMed] [Google Scholar]
- Clifford DB, Olney JW, Maniotis A, Collins RC, Zorumski CF. The functional anatomy and pathology of lithium-pilocarpine and high-dose pilocarpine seizures. Neuroscience. 1987;23:953–968. doi: 10.1016/0306-4522(87)90171-0. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cohen-Gadol AA, Westerveld M, Alvarez-Carilles J, Spencer DD. Intracarotid Amytal memory test and hippocampal magnetic resonance imaging volumetry: validity of the Wada test as an indicator of hippocampal integrity among candidates for epilepsy surgery. J Neurosurg. 2004;101:926–931. doi: 10.3171/jns.2004.101.6.0926. [DOI] [PubMed] [Google Scholar]
- Collombet JM, Masqueliez C, Four E, Burckhart MF, Bernabé D, Baubichon D, Lallement G. Early reduction of NeuN antigenicity induced by soman poisoning in mice can be used to predict delayed neuronal degeneration in the hippocampus. Neurosci Lett. 2006;398:337–342. doi: 10.1016/j.neulet.2006.01.029. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, Spencer DD. A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia. 2003;44:677–687. doi: 10.1046/j.1528-1157.2003.32701.x. [DOI] [PubMed] [Google Scholar]
- Du F, Whetsell WO, Jr, Abou-Khalil B, Blumenkopf B, Lothman EW, Schwarcz R. Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 1993;16:223–233. doi: 10.1016/0920-1211(93)90083-j. [DOI] [PubMed] [Google Scholar]
- Du F, Eid T, Lothman EW, Kohler C, Schwarcz R. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J Neurosci. 1995;15:6301–6313. doi: 10.1523/JNEUROSCI.15-10-06301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Jorritsma-Byham B, Schwarcz R, Witter MP. Afferents to the seizure-sensitive neurons in layer III of the medial entorhinal area: a tracing study in the rat. Exp Brain Res. 1996;109:209–218. doi: 10.1007/BF00231782. [DOI] [PubMed] [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, Rundén-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, de Lanerolle NC. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- El Bahh B, Lurton D, Sundstrom LE, Rougier A. Induction of tolerance and mossy fibre neuropeptide-Y expression in the contralateral hippocampus following a unilateral intrahippocampal kainic acid injection in the rat. Neurosci Lett. 1997;227:135–139. doi: 10.1016/s0304-3940(97)00261-9. [DOI] [PubMed] [Google Scholar]
- Engel J., Jr Introduction to temporal lobe epilepsy. Epilepsy Res. 1996;26:141–150. doi: 10.1016/s0920-1211(96)00043-5. [DOI] [PubMed] [Google Scholar]
- Evans MC, Griffiths T, Meldrum BS. Kainic acid seizures and the reversibility of calcium loading in vulnerable neurons in the hippocampus. Neuropathol Appl Neurobiol. 1984;10:285–302. doi: 10.1111/j.1365-2990.1984.tb00360.x. [DOI] [PubMed] [Google Scholar]
- Fabene PF, Marzola P, Sbarbati A, Bentivoglio M. Magnetic resonance imaging of changes elicited by status epilepticus in the rat brain: diffusion-weighted and T2-weighted images, regional blood volume maps, and direct correlation with tissue and cell damage. Neuroimage. 2003;18:375–389. doi: 10.1016/s1053-8119(02)00025-3. [DOI] [PubMed] [Google Scholar]
- Fabene PF, Merigo F, Galiè M, Benati D, Bernardi P, Farace P, Nicolato E, Marzola P, Sbarbati A. Pilocarpine-induced status epilepticus in rats involves ischemic and excitotoxic mechanisms. PLoS One. 2007;2:e1105. doi: 10.1371/journal.pone.0001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabene PF, Weiczner R, Marzola P, Nicolato E, Calderan L, Andrioli A, Farkas E, Süle Z, Mihaly A, Sbarbati A. Structural and functional MRI following 4-aminopyridine-induced seizures: a comparative imaging and anatomical study. Neurobiol Dis. 2006;21:80–89. doi: 10.1016/j.nbd.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Falconer MA. Mesial temporal (Ammon’s horn) sclerosis as a common cause of epilepsy. Aetiology, treatment, and prevention. Lancet. 1974;2:767–770. doi: 10.1016/s0140-6736(74)90956-8. [DOI] [PubMed] [Google Scholar]
- Falconer MA, Taylor DC. Surgical treatment of drug-resistant epilepsy due to mesial temporal lobe sclerosis; etiology and significance. Arch Neurol. 1968;19:353–361. doi: 10.1001/archneur.1968.00480040019001. [DOI] [PubMed] [Google Scholar]
- Fernández G, Effenberger O, Vinz B, Steinlein O, Elger CE, Döhring W, Heinze HJ. Hippocampal malformation as a cause of familial febrile convulsions and subsequent hippocampal sclerosis. Neurology. 1998;50:909–917. doi: 10.1212/wnl.50.4.909. [DOI] [PubMed] [Google Scholar]
- French JA, Williamson PD, Thadani VM, Darcey TM, Mattson RH, Spencer SS, Spencer DD. Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol. 1993;34:774–780. doi: 10.1002/ana.410340604. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Itabashi HH, Wu A, Shinmei SS. Status epilepticus-induced neuronal loss in humans without systemic complications or epilepsy. Epilepsia. 2000;41:981–991. doi: 10.1111/j.1528-1157.2000.tb00283.x. [DOI] [PubMed] [Google Scholar]
- Goffin K, Nissinen J, Van Laere K, Pitkanen A. Cyclicity of spontaneous recurrent seizures in pilocarpine model of temporal lobe epilepsy in rat. Exp Neurol. 2007;205:501–505. doi: 10.1016/j.expneurol.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Gorter JA, Gonçalves Pereira PM, van Vliet EA, Aronica E, Lopes da Silva FH, Lucassen PJ. Neuronal cell death in a rat model for mesial temporal lobe epilepsy is induced by the initial status epilepticus and not by later repeated spontaneous seizures. Epilepsia. 2003;44:647–658. doi: 10.1046/j.1528-1157.2003.53902.x. [DOI] [PubMed] [Google Scholar]
- Harvey BD, Sloviter RS. Hippocampal granule cell activity and c-Fos expression during spontaneous seizures in awake, chronically epileptic, pilocarpine-treated rats; implications for hippocampal epileptogenesis. J Comp Neurol. 2005;488:441–462. doi: 10.1002/cne.20594. [DOI] [PubMed] [Google Scholar]
- Hatazaki S, Bellver-Estelles C, Jimenez-Mateos EM, Meller R, Bonner C, Murphy N, Matsushima S, Taki W, Prehn JH, Simon RP, Henshall DC. Microarray profile of seizure damage-refractory hippocampal CA3 in a mouse model of epileptic preconditioning. Neuroscience. 2007;150:467–77. doi: 10.1016/j.neuroscience.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A, Hesdorffer D. Prognosis. In: Hauser WA, Hesdorffer DC, editors. Epilepsy: frequency, causes and consequences. Demos; New York: 1990. pp. 197–243. [Google Scholar]
- Ishizuka N, Weber J, Amaral DG. Organization of intrahippocampal projections originating from CA3 pyramidal cells in the rat. J Comp Neurol. 1990;295:580–623. doi: 10.1002/cne.902950407. [DOI] [PubMed] [Google Scholar]
- Jackson GD, Chambers BR, Berkovic SF. Hippocampal sclerosis: development in adult life. Dev Neurosci. 1999;21:207–214. doi: 10.1159/000017400. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Nadler JV. Stereological analysis of GluR2-immunoreactive hilar neurons in the pilocarpine model of temporal lobe epilepsy: correlation of cell loss with mossy fiber sprouting. Exp Neurol. 2007;205:569–582. doi: 10.1016/j.expneurol.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo JN, Sheerin JH, Miller JW, D’Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kälviäinen R, Salmenperä T, Partanen K, Vainio P, Riekkinen P, Pitkänen A. Recurrent seizures may cause hippocampal damage in temporal lobe epilepsy. Neurology. 1998;50:1377–1382. doi: 10.1212/wnl.50.5.1377. [DOI] [PubMed] [Google Scholar]
- Kelly ME, McIntyre DC. Hippocampal kindling protects several structures from the neuronal damage resulting from kainic acid-induced status epilepticus. Brain Res. 1994;634:245–256. doi: 10.1016/0006-8993(94)91927-5. [DOI] [PubMed] [Google Scholar]
- Kienzler F, Norwood BA, Sloviter RS. Hippocampal injury, atrophy, synaptic reorganization, and epileptogenesis after perforant pathway stimulation-induced status epilepticus in the mouse. J Comp Neurol. 2009;515:181–196. doi: 10.1002/cne.22059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratyev A, Sahibzada N, Gale K. Electroconvulsive shock exposure prevents neuronal apoptosis after kainic acid-evoked status epilepticus. Brain Res Mol Brain Res. 2001;91:1–13. doi: 10.1016/s0169-328x(01)00099-7. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Buckmaster PS. Neuron-specific nuclear antigen NeuN is not detectable in gerbil subtantia nigra pars reticulata. Brain Res. 2007;1142:54–60. doi: 10.1016/j.brainres.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehericy S, Semah F, Hasboun D, Dormont D, Clemenceau S, Granat O, Marsault C, Baulac M. Temporal lobe epilepsy with varying severity: MRI study of 222 patients. Neuroradiology. 1997;39:788–796. doi: 10.1007/s002340050507. [DOI] [PubMed] [Google Scholar]
- Lewis DV. Losing neurons: selective vulnerability and mesial temporal sclerosis. Epilepsia. 2005;46(Suppl 7):39–44. doi: 10.1111/j.1528-1167.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- Lorente de Nó R. Studies on the structure of the cerebral cortex. II. Continuation of the study of the ammonic system. J Psychol Neurol. 1934;46:113–177. [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher J, McLachlan RS. Febrile convulsions. Is seizure duration the most important predictor of temporal lobe epilepsy? Brain. 1995;118:1521–1528. doi: 10.1093/brain/118.6.1521. [DOI] [PubMed] [Google Scholar]
- Margerison JH, Corsellis JA. Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- Marks DA, Kim J, Spencer DD, Spencer SS. Characteristics of intractable seizures following meningitis and encephalitis. Neurology. 1992;42:1513–1518. doi: 10.1212/wnl.42.8.1513. [DOI] [PubMed] [Google Scholar]
- Marks DA, Kim J, Spencer DD, Spencer SS. Seizure localization and pathology following head injury in patients with uncontrolled epilepsy. Neurology. 1995;45:2051–2057. doi: 10.1212/wnl.45.11.2051. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Vickrey BG, Melendez M, Pretorius JK. The clinical-pathogenic mechanisms of hippocampal neuron loss and surgical outcomes in temporal lobe epilepsy. Brain. 1995;118:105–118. doi: 10.1093/brain/118.1.105. [DOI] [PubMed] [Google Scholar]
- McPhail LT, McBride CB, McGraw J, Steeves JD, Tetzlaff W. Axotomy abolishes NeuN expression in facial but not rubrospinal neurons. Exp Neurol. 2004;185:182–190. doi: 10.1016/j.expneurol.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Meldrum BS, Bruton CJ. Epilepsy. In: Adams JH, Duchen LW, editors. Greenfield’s Neuropathology. New York: Oxford University Press; 1992. pp. 1246–1283. [Google Scholar]
- Meldrum BS, Horton RW, Brierley JB. Epileptic brain damage in adolescent baboons following seizures induced by allylgycine. Brain. 1974;97:407–418. doi: 10.1093/brain/97.1.407. [DOI] [PubMed] [Google Scholar]
- Mikaeloff Y, Jambaque I, Hertz-Pannier L, Zamfirescu A, Adamsbaum C, Plouin P, Dulac O, Chiron C. Devastating epileptic encephalopathy in school-aged children (DESC): a pseudo encephalitis. Epilepsy Res. 2006;69:67–79. doi: 10.1016/j.eplepsyres.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RK. Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J Physiol. 1987;388:611–629. doi: 10.1113/jphysiol.1987.sp016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgram NW, Yearwood T, Khurgel M, Ivy GO, Racine R. Changes in inhibitory processes in the hippocampus following recurrent seizures induced by systemic administration of kainic acid. Brain Res. 1991;551:236–246. doi: 10.1016/0006-8993(91)90938-r. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nadler JV. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981;29:2031–2042. doi: 10.1016/0024-3205(81)90659-7. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- Nairismägi J, Gröhn OH, Kettunen MI, Nissinen J, Kauppinen RA, Pitkänen A. Progression of brain damage after status epilepticus and its association with epileptogenesis: a quantitative MRI study in a rat model of temporal lobe epilepsy. Epilepsia. 2004;45:1024–1034. doi: 10.1111/j.0013-9580.2004.08904.x. [DOI] [PubMed] [Google Scholar]
- Najm IM, Hadam J, Ckakraverty D, Mikuni N, Penrod C, Sopa C, Markarian G, Lüders HO, Babb T, Baudry M. A short episode of seizure activity protects from status epilepticus-induced neuronal damage in rat brain. Brain Res. 1998;810:72–75. doi: 10.1016/s0006-8993(98)00886-5. [DOI] [PubMed] [Google Scholar]
- Navarro Mora G, Bramanti P, Osculati F, Chakir A, Nicolato E, Marzola P, Sbarbati A, Fabene PF. Does pilocarpine-induced epilepsy in adult rats require status epilepticus? PLoS One. 2009;4:e5759. doi: 10.1371/journal.pone.0005759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien TJ, So EL, Meyer FB, Parisi JE, Jack CR. Progressive hippocampal atrophy in chronic intractable temporal lobe epilepsy. Ann Neurol. 1999;45:526–529. [PubMed] [Google Scholar]
- Olney JW. Kainic acid and other excitotoxins: a comparative analysis. Adv Biochem Psychopharmacol. 1981;27:375–384. [PubMed] [Google Scholar]
- Penner MR, Pinaud R, Robertson HA. Rapid kindling of the hippocampus protects against neural damage resulting from status epilepticus. Neuroreport. 2001;12:453–457. doi: 10.1097/00001756-200103050-00007. [DOI] [PubMed] [Google Scholar]
- Pisa M, Sandberg PR, Corcoran ME, Fibiger HC. Spontaneous recurrent seizures after intracerebral injections of kainic acid in rats: a possible model of human temporal lobe epilepsy. Brain Res. 1980;200:481–487. doi: 10.1016/0006-8993(80)90938-5. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Nissinen J, Nairismägi J, Lukasiuk K, Gröhn OH, Miettinen R, Kauppinen R. Progression of neuronal damage after status epilepticus and during spontaneous seizures in a rat model of temporal lobe epilepsy. Prog Brain Res. 2002;135:67–83. doi: 10.1016/S0079-6123(02)35008-8. [DOI] [PubMed] [Google Scholar]
- Purpura DP, Gonzalez-Monteagudo O. Acute effects of methoxypyridoxine on hippocampal end-blade neurons: an experimental study of “special pathoclisis” in the cerebral cortex. J Neuropathol Exp Neurol. 1960;19:421–432. doi: 10.1097/00005072-196007000-00009. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Raol YH, Lund IV, Bandyopadhyay S, Zhang G, Roberts DS, Wolfe JH, Russek SJ, Brooks-Kayal AR. Enhancing GABA(A) receptor alpha 1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J Neurosci. 2006;26:11342–11346. doi: 10.1523/JNEUROSCI.3329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch C, Leroy C, Nehlig A, Namer IJ. Magnetic resonance imaging in the study of the lithium-pilocarpine model of temporal lobe epilepsy in adult rats. Epilepsia. 2002;43:325–335. doi: 10.1046/j.1528-1157.2002.11301.x. [DOI] [PubMed] [Google Scholar]
- Salmenperä T, Könönen M, Roberts N, Vanninen R, Pitkänen A, Kälviäinen R. Hippocampal damage in newly diagnosed focal epilepsy: a prospective MRI study. Neurology. 2005;64:62–68. doi: 10.1212/01.WNL.0000148643.36513.2A. [DOI] [PubMed] [Google Scholar]
- Saper CB. Any way you cut it: a new journal policy for the use of unbiased counting methods. J Comp Neurol. 1996;364:5. doi: 10.1002/(SICI)1096-9861(19960101)364:1<5::AID-CNE1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Sarnat HB, Nochlin D, Born DE. Neuronal nuclear antigen (NeuN): a marker of neuronal maturation in early human fetal nervous system. Brain Dev. 1998;20:88–94. doi: 10.1016/s0387-7604(97)00111-3. [DOI] [PubMed] [Google Scholar]
- Sasahira M, Lowry T, Simon RP, Greenberg DA. Epileptic tolerance: prior seizures protect against seizure-induced neuronal injury. Neurosci Lett. 1995;185:95–98. doi: 10.1016/0304-3940(94)11233-9. [DOI] [PubMed] [Google Scholar]
- Sass KJ, Lencz T, Westerveld M, Novelly RA, Spencer DD, Kim JH. The neural substrate of memory impairment demonstrated by the intracarotid amobarbital procedure. Arch Neurol. 1991;48:48–52. doi: 10.1001/archneur.1991.00530130056020. [DOI] [PubMed] [Google Scholar]
- Sass KJ, Buchanan CP, Kraemer S, Westerveld M, Kim JH, Spencer DD. Verbal memory impairment resulting from hippocampal neuron loss among epileptic patients with structural lesions. Neurology. 1995;45:2154–2158. doi: 10.1212/wnl.45.12.2154. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Smith KL, Goodman JH, Sollas AL. Survival of dentate hilar mossy cells after pilocarpine-induced seizures and their synchronized burst discharges with area CA3 pyramidal cells. Neuroscience. 2001;104:741–759. doi: 10.1016/s0306-4522(01)00132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Eid T, Du F. Neurons in layer III of the entorhinal cortex. A role in epileptogenesis and epilepsy? Ann NY Acad Sci. 2000;911:328–342. doi: 10.1111/j.1749-6632.2000.tb06735.x. [DOI] [PubMed] [Google Scholar]
- Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;5:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- Seress L, Abrahám H, Horváth Z, Dóczi T, Janszky J, Klemm J, Byrne R, Bakay RA. Survival of mossy cells of the hippocampal dentate gyrus in humans with mesial temporal lobe epilepsy. J Neurosurg. 2009;111:1237–1247. doi: 10.3171/2008.11.JNS08779. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. A simplified Timm stain procedure compatible with formaldehyde fixation and routine paraffin embedding of rat brain. Brain Res Bull. 1982;8:771–774. doi: 10.1016/0361-9230(82)90104-6. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. “Epileptic” brain damage in rats induced by sustained electrical stimulation of the perforant path. I. Acute electrophysiological and light microscopic studies. Brain Res Bull. 1983;10:675–697. doi: 10.1016/0361-9230(83)90037-0. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Feedforward and feedback inhibition of hippocampal principal cell activity evoked by perforant path stimulation: GABA-mediated mechanisms that regulate excitability in vivo. Hippocampus. 1991a;1:31–40. doi: 10.1002/hipo.450010105. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the dormant basket cell hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991b;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Possible functional consequences of synaptic reorganization in the dentate gyrus of kainate-treated rats. Neurosci Lett. 1992;137:91–96. doi: 10.1016/0304-3940(92)90306-r. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann Neurol. 1994;35:640–654. doi: 10.1002/ana.410350604. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. The neurobiology of temporal lobe epilepsy; too much information, not enough knowledge. CR Biologies. 2005;328:143–153. doi: 10.1016/j.crvi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis; the importance of the “latent period” and other concepts. Epilepsia. 2008;49(Suppl 9):85–92. doi: 10.1111/j.1528-1167.2008.01931.x. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Damiano BP. Sustained electrical stimulation of the perforant path duplicates kainate-induced electrophysiological effects and hippocampal damage in rats. Neurosci Lett. 1981;24:279–284. doi: 10.1016/0304-3940(81)90171-3. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Dean E, Sollas AL, Goodman JH. Apoptosis and necrosis induced in different hippocampal neuron populations by repetitive perforant path stimulation in the rat. J Comp Neurol. 1996;366:516–533. doi: 10.1002/(SICI)1096-9861(19960311)366:3<516::AID-CNE10>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA, Frotscher M. “Dormant basket cell” hypothesis revisited; relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J Comp Neurol. 2003;459:44–76. doi: 10.1002/cne.10630. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Frotscher M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyperinhibition in chronically epileptic rats. J Comp Neurol. 2006;494:944–960. doi: 10.1002/cne.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol DK, Demyer WE, Edwards-Brown M, Sanders S, Garg B. Swelling to sclerosis: acute change in mesial hippocampus after prolonged febrile seizure. Seizure. 2003;12:237–240. doi: 10.1016/s1059-1311(02)00195-4. [DOI] [PubMed] [Google Scholar]
- Sommer W. Erkrankung des Ammonshorns als aetiologisches Moment der Epilepsien. Arch Psychiat Nervenkr. 1880;10:631–675. [Google Scholar]
- Spencer SS. Substrates of localization-related epilepsies: biological implications of localizing findings in humans. Epilepsia. 1998;39:114–123. doi: 10.1111/j.1528-1157.1998.tb01349.x. [DOI] [PubMed] [Google Scholar]
- Spencer SS, Spencer DD. Entorhinal-hippocampal interactions in medial temporal lobe epilepsy. Epilepsia. 1994;35:721–727. doi: 10.1111/j.1528-1157.1994.tb02502.x. [DOI] [PubMed] [Google Scholar]
- Swartz BE, Houser CR, Tomiyasu U, Walsh GO, DeSalles A, Rich JR, Delgado-Escueta A. Hippocampal cell loss in posttraumatic human epilepsy. Epilepsia. 2006;47:1373–1382. doi: 10.1111/j.1528-1167.2006.00602.x. [DOI] [PubMed] [Google Scholar]
- Tasch E, Cendes F, Li LM, Dubeau F, Andermann F, Arnold DL. Neuroimaging evidence of progressive neuronal loss and dysfunction in temporal lobe epilepsy. Ann Neurol. 1999;45:568–576. doi: 10.1002/1531-8249(199905)45:5<568::aid-ana4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, Zhou J, Martinian L, Sisodiya S. Quantitative post-mortem study of the hippocampus in chronic epilepsy: seizures do not inevitably cause neuronal loss. Brain. 2005;128:1344–1357. doi: 10.1093/brain/awh475. [DOI] [PubMed] [Google Scholar]
- Tongiorgi E, Armellin M, Giulianini PG, Bregola G, Zucchini S, Paradiso B, Steward O, Cattaneo A, Simonato M. Brain-derived neurotrophic factor mRNA and protein are targeted to discrete dendritic laminas by events that trigger epileptogenesis. J Neurosci. 2004;24:6842–6852. doi: 10.1523/JNEUROSCI.5471-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- Unal-Cevik I, Kilinc M, Gürsoy-Ozdemir Y, Gurer G, Dalkara T. Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res. 2004;1015:169–174. doi: 10.1016/j.brainres.2004.04.032. [DOI] [PubMed] [Google Scholar]
- Van Paesschen W, Duncan JS, Stevens JM, Connelly A. Etiology and early prognosis of newly diagnosed partial seizures in adults: a quantitative hippocampal MRI study. Neurology. 1997;49:753–757. doi: 10.1212/wnl.49.3.753. [DOI] [PubMed] [Google Scholar]
- Van Paesschen W, Duncan JS, Stevens JM, Connelly A. Longitudinal quantitative hippocampal magnetic resonance imaging study of adults with newly diagnosed partial seizures: one-year follow-up results. Epilepsia. 1998;39:633–639. doi: 10.1111/j.1528-1157.1998.tb01432.x. [DOI] [PubMed] [Google Scholar]
- Weyer A, Schilling K. Developmental and cell type-specific expression of the neuronal marker NeuN in the murine cerebellum. J Neurosci Res. 2003;73:400–409. doi: 10.1002/jnr.10655. [DOI] [PubMed] [Google Scholar]
- Williamson A, Spencer DD. Electrophysiological characterization of CA2 pyramidal cells from epileptic humans. Hippocampus. 1994;4:226–237. doi: 10.1002/hipo.450040213. [DOI] [PubMed] [Google Scholar]
- Xu B, McIntyre DC, Fahnestock M, Racine RJ. Strain differences affect the induction of status epilepticus and seizure-induced morphological changes. Eur J Neurosci. 2004;20:403–418. doi: 10.1111/j.1460-9568.2004.03489.x. [DOI] [PubMed] [Google Scholar]
- Young NA, Vuong J, Ozen LJ, Flynn C, Teskey GC. Motor map expansion in the pilocarpine model of temporal lobe epilepsy is dependent on seizure severity and rat strain. Exp Neurol. 2009;217:421–428. doi: 10.1016/j.expneurol.2009.03.043. [DOI] [PubMed] [Google Scholar]
- Zappone CA, Sloviter RS. Translamellar disinhibition in the rat hippocampal dentate gyrus after seizure-induced degeneration of vulnerable hilar neurons. J Neurosci. 2004;24:853–864. doi: 10.1523/JNEUROSCI.1619-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cui SS, Wallace AE, Hannesson DK, Schmued LC, Saucier DM, Honer WG, Corcoran ME. Relations between brain pathology and temporal lobe epilepsy. J Neurosci. 2002;22:6052–6061. doi: 10.1523/JNEUROSCI.22-14-06052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]