

SUMMARY
Age-related thymic involution may be triggered by gene expression changes in lymphohematopoietic and/or non-hematopoietic thymic epithelial cells (TECs). The role of epithelial cell-autonomous gene FoxN1 may be involved in the process, but it is still a puzzle due to shortage of evidence from gradual loss-of-function and exogenous gain-of-function studies. Using our recently generated loxP-floxed-FoxN1(fx) mouse carrying the ubiquitous CreERT (uCreERT) transgene with a low dose of spontaneous activation, which causes gradual FoxN1 deletion with age, we found that the uCreERT-fx/fx mice showed an accelerated age-related thymic involution due to progressive loss of FoxN1+ TECs. The thymic aging phenotypes were clearly observable as early as at 3–6 months of age, resembling the naturally aged (18–22-month-old) murine thymus. By intrathymically supplying aged wild-type mice with exogenous FoxN1-cDNA, thymic involution and defective peripheral CD4+ T-cell function could be partially rescued. The results support the notion that decline of a single epithelial cell-autonomous gene FoxN1 levels with age causes primary deterioration in TECs followed by impairment of the total postnatal thymic microenvironment, and potentially triggers age-related thymic involution in mice.
Keywords: Thymic aging, thymic epithelium, loxP/CreERT system, spontaneous FoxN1 gene recombination
INTRODUCTION
Aging process is gradual and complex involved in degeneration in many physiological systems and organs. The rate of degeneration varies among individual organs, tissue types, and cells. The primary organ responsible for acquired immunity, thymus gland, ages much earlier and more rapidly than most other organs. In human beings the onset of degeneration begins at puberty. Typical phenotypes of thymic aging are the thymic involution (Lynch et al. 2009), thymic adiposity (Youm et al. 2009), and decline of T-lymphopoiesis, i.e. reduction of thymic niches and gradual loss of newly produced naïve T cells (Bodey et al. 1997; Petrie 2002; Gruver & Sempowski 2008), which results in a restricted T cell receptor repertoire and an expansion of pre-existing memory T cell pool. It therefore compromises T-cell-mediated immunity due to inability to generate new immune responses. The aged thymus also shows a disproportionate loss of thymic epithelial cells (TECs) (Gray et al. 2006) and disrupted thymic architecture (Gui et al. 2007), which results in an increased risk of autoimmunity through the escape of potentially self-reactive T cells from the disrupted thymic microenvironment. Therefore, age-related immune deficiency impacts elderly individuals encountering new and chronic infections, as well as those undergoing radio-chemo-therapeutics for cancer or other ailments.
Age-related thymic involution is generally believed to result from deterioration of the interactions between lymphohematopoietic progenitor cells (LPCs) and non-hematopoietic thymic stromal cells (TSCs), primarily composed of thymic epithelial cells (TECs), in the thymus (Taub & Longo 2005; Ladi et al. 2006). These interactions encompass many genetic pathways or programs in either LPCs of hematopoietic origin or TSCs of non-hematopoietic origin. We (Gui et al. 2007; Zhu et al. 2007) and others (Aspinall & Andrew 2000) have hypothesized that age-related thymic involution is triggered primarily by deterioration of the thymic microenvironment, due to TEC dysfunction. In support of this concept, improving TEC function by infusing keratinocyte growth factor (Min et al. 2007; Rossi et al. 2007), or directly supplying TEC-derived interlukin-7 (Henson et al. 2005), which is required for early T-cell development in the thymus, ameliorated deficient thymopoiesis due to aging. Therefore, changes in gene expression related to TEC function may primarily regulate age-related thymic involution.
FoxN1 is an epithelial cell-autonomous gene, which is expressed in epithelium of the thymus, skin, and mammary gland, and regulates thymic organogenesis in the thymus (Nehls et al. 1994; Nehls et al. 1996) and is involved in successful generation of the T-cell immune system (Frank et al. 1999; Adriani et al. 2004; Coffer & Burgering 2004; Cunningham-Rundles & Ponda 2005; Amorosi et al. 2008). The mechanism by which FoxN1 regulates thymic organogenesis and T-cell development in the fetal murine thymus has been extensively studied (Nehls et al. 1996; Su et al. 2003; Bleul et al. 2006). However, the function of FoxN1 in the postnatal thymus and during thymic aging has not been fully addressed, and remains uncertain, because of the lack of a temporally-controlled and tissue-specific loss-of-function model. Although FoxN1 mRNA expression in the thymus declines with age in wild-type (WT) mice (Ortman et al. 2002), changes in FoxN1 protein levels have not been detected. Furthermore, it is unclear if reduced FoxN1 mRNA expression is the cause or the effect of thymic aging, because aging-induced TEC deterioration may affect gene expression in TECs. To address this question, we developed a loxP-floxed-FoxN1 mouse model, denoted as “fx” (Cheng et al. 2010). Crossbreeding these mice with ubiquitously-expressed inducible CreERT (uCreERT) transgenic mice, we attained a temporally-controlled FoxN1 knockout mouse allele (denoted as “uCreERT-fx” mouse). Conditional ubiquitous deletion of FoxN1 in the young adult thymus of uCreERT-fx/fx mouse by tamoxifen (TM)-induction caused acute thymic atrophy within 5 days, whereas gradual deletion of FoxN1 in these mice through spontaneous uCreERT activation accelerated thymic involution. The thymic aging phenotype in the uCreERT-fx/fx mice could be observed at a much earlier age (~3–6 months old), which mimicked the naturally aged (~18–22 months old) thymus. Thymic involution was accompanied with changes in thymic architecture, and a sharp decrease in the numbers of CD4+8+ double positive (DP) and CD4+ single positive (SP) thymocytes, and MHC-IIhiUEA-1hi medullary TECs (mTECs). By intrathymic administration of exogenous FoxN1-cDNA in middle-aged and aged WT mice, a rapid test of gain-of-function approach, thymic involution and declining thymic function could be partially rescued, resulting in increased thymic size, thymocyte numbers, and functional CD4+ T cells. Thus, we for the first time provided the empirical evidence supporting that the role of FoxN1 is a potential cause to be related to age-related thymic involution.
RESULTS
Spontaneous FoxN1 deletion and decline of FoxN1+ TECs with age in uCreERT-fx/fx mice
Homozygous FoxN1fx (fx/fx) mice carrying the uCreERT transgene (Hayashi & McMahon 2002) were intraperitoneally injected with ≥3-doses of TM, which caused deletion of exons 5 and 6 (denoted as “ΔE5&6”) of the FoxN1 locus (Fig. 1A) mediated by Cre-recombinase, and resulted in significant thymic atrophic phenotype, as described in our parallel paper (Cheng et al. 2010). However, the uCreERT transgene has a low level of spontaneous activation even without TM-induction (Bleul et al. 2006; Matsuda & Cepko 2007), causing gradual excision of the loxP-floxed FoxN1 gene with time, resulting in progressive loss of FoxN1 with age in uCreERT-fx/fx mice. The spontaneous deletion of FoxN1 was evaluated in the different aged thymi of the uCreERT-fx/+ and uCreERT-fx/fx mice by PCR assay (Fig. 1B). The FoxN1 ΔE5&6 deletion was increased with age (arrows in Fig. 1B) and was seen in both uCreERT-fx/+ heterozygous and uCreERT-fx/fx homozygous mice.
Figure 1. Spontaneous FoxN1 deletion with age in the thymi of different aged uCreERT-fx mice.
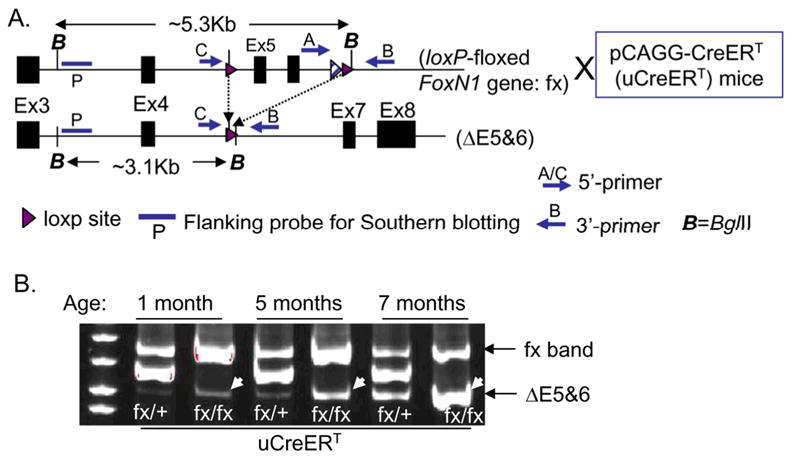
(A) Schematic of loxP-floxed FoxN1 allele crossed with uCreERT mice, before (top) and after (bottom) FoxN1 excision (i.e. deletion of exons 5 and 6 of FoxN1 locus, ΔE5&6). (B) A representative experiment of spontaneous FoxN1 deletion with age in the thymi of 1-, 5- and 7-month-old uCreERT-fx/+ and uCreERT-fx/fx mice, detected by three-primer PCR.
To determine whether spontaneous FoxN1 recombination causes a decrease in FoxN1 expression at the protein level, we carried out immunofluorescence staining with FoxN1 antibody on cryosections. As shown in Figure 2, we demonstrated, for the first time, that the FoxN1 positive (FoxN1+) TECs (at protein level) were reduced with age in the naturally aged thymus (Fig. 2A), though the FoxN1 mRNA was reported to be reduced with age in these mice (Ortman et al. 2002). Importantly, the reduction of FoxN1+ TECs occurred much earlier in the uCreERT-fx/fx mice (Figs. 2B and D), which could be seen as an overt decrease starting at the age of 3 months. By 6–9 months of age, uCreERT-fx/fx mice expressed FoxN1+ TECs at a level equivalent to a 22-month-old WT mouse (Figs. 2B and D). Distribution of FoxN1+ TECs in cortex and medulla in 1-month-old uCreERT-fx/fx thymus were almost the same as that in young WT thymus (Fig. 2C and our previous paper (Cheng et al. 2010)). The results suggest that loss of FoxN1+ TECs is accelerated with age in the uCreERT-fx/fx mice compared with the naturally aged mice, due to spontaneous uCreERT activation.
Figure 2. FoxN1+ TECs were reduced in an age-dependent manner in WT and uCreERT-fx/fx mice.

A representative result shows frozen sections of WT (1-, 12-, 18-, and 22-month-old) (A), and uCreERT-fx/fx (1-, 3-, 6-, 9-month-old) (B) mouse thymi co-stained with keratin-8 (green) and FoxN1 (red) antibodies, and analyzed with NIH software image-J to determine the proportion of positive areas for both markers (bar graph) (D). Means and SEMs are shown. This experiment was done with 2–3 animals (9-month-old uCreERT-fx/fx mouse was only done with 1 animal) in each group, with essentially identical results. (C), A representative result shows the distribution of FoxN1+ TECs in the medulla and cortex of uCreERT-fx/fx thymu from a 1-month-old mouse. Two consecutive tissue sections through a serial sectioning on separate slides were co-stained with K5 (red, to determine medullary region) vs. K8 (green, to determine cortical region) on one slide, and with FoxN1 (red) vs. K8 (green) on the other slide. Through comparative analysis of these two slides, we obtained the distribution of FoxN1+ TECs in the two regions. Dotted lines indicate cortical-medullary borders. The proportion of FoxN1+ TECs in the medulla and cortex is 0.54% vs. 0.33%, respectively, analyzed by Image-J software. C = cortex; M = medulla.
Accelerated decreases in thymus size, and TEC and thymocyte numbers in uCreERT-fx/fx mice correlated with age
To answer whether progressive loss of FoxN1+ TECs with age will accelerate thymic involution, we observed thymic size changes in uCreERT-fx/fx mice and found that the size was gradually reduced at an accelerated rate, showing significant differences from 3 months of age when compared to fx/fx-only (without Cre-recombinase) and uCreERT-fx/+ littermates (Fig. 3B and data not shown). The 3-month-old uCreERT-fx/fx mice showed a thymic size equivalent to that of ~14-month-old WT mice (Fig. 3A and data not shown). By 5–6 months old, the uCreERT-fx/fx mouse thymus was as small as the thymus of a 22-month-old WT mouse (Figs. 3A and B). The absolute number of total CD45− TSCs, total thymocytes, particularly DP and CD4+SP thymocytes (Figs. 3C and D), were found to negatively correlate with ages and showed an accelerated decline in the uCreERT-fx/fx mice. The age-related decline of DP and CD4+SP absolute cell numbers were steeper in uCreERT-fx/fx mice providing proof that loss of FoxN1+ TECs accelerated thymic aging. However, the proportions of thymocytes expressing CD4 and CD8 in all groups of mice were less affected (data not shown). This is consistent with the changes in the naturally aged thymus, where there is a reduction in absolute thymocyte number, but no alterations in the proportions of major thymocyte subpopulations (Thoman 1995; Aspinall 1997).
Figure 3. Accelerated thymic involution, and rapidly reduced cell numbers in uCreERT-fx/fx mice correlated with age.

(A) A gross representation of thymic size in WT mice at 2, 9, 14, and 22 months of age and fx/fx-only (without Cre-recombinase) mice at 9 months of age. (B) A gross representation of thymic size in fx/fx-only (without Cre-recombinase), uCreERT-fx/+, and uCreERT-fx/fx mice at 1, 3, and 5 months of age. (C) The linear regression for DP thymocyte numbers correlated with age. (D) The linear regression for CD4+SP numbers correlated with age. Both C and D panels include 25 fx/fx-only (without Cre-recombinase) mice, each circle = one mouse, p = 0.008 in DP cells (panel C) and p = 0.52 in CD4+SP cells (panel D); 32 uCreERT-fx/+ mice, each triangle = one mouse, p = 0.004 in DP cells (panel C) and p = 0.004 in CD4+SP cells (panel D); and 16 uCreERT-fx/fx mice, each square = one mouse, p = 0.01 in DP cells (panel C) and p = 0.03 in CD4+SP cells (panel D).
Since in the uCreERT-fx/+ mice, one copy of floxed-FoxN1 gene undergoes a spontaneous gradual deletion over time (Fig. 1B), this also produces thymic involution with age. This is consistent with prior findings of mildly reduced thymic size in FoxN1+/nu heterozygous nude mice with a congenital loss of one FoxN1 copy, despite normal thymic structure (Scheiff et al. 1978; Kojima et al. 1984), and reflects a haploinsufficient feature of the FoxN1 gene. But the involution in uCreERT-fx/+ heterozygous mice did not occur as early or to as severe a degree as was seen in uCreERT-fx/fx homozygous mice (samples in Fig. 3B middle column, and Figs. 3C and D), which suggests that the thymic phenotype is sensitive to FoxN1 gene dosage (Chen et al. 2009). Fx/fx-only mice that lacked Cre-recombinase showed normal thymic size, similar to WT mice (Figs. 3A and B), indicating that the phenotype was due to FoxN1 gene recombination and not due to the loxP-floxed (fx) allele.
Accelerated changes in thymic micro-architecture in uCreERT-fx/fx mice resembled that of the naturally aged thymus
In our previous report (Gui et al. 2007), we analyzed the architecture of the naturally aged thymus with antibodies to Keratin-5 (K5) and K8, and found that the aged thymus (>18 months old) showed a disorganized cortex and medulla, with poorly defined boundaries between cortical and medullary regions (corticomedullary junction, CMJ) and a mosaic pattern of intermingled medullary TECs (mTECs, presenting K5+) and cortical TECs (cTECs, presenting K8+). The uCreERT-fx/fx mice also showed these phenotypes with age, but it significantly occurred as early as at age of 3–6 months of age in these mice (Fig. 4), including the changes in gross and micro-architectures observed by H&E staining (Fig. 4A), and sparse K5+ mTECs and mingled K5 and K8 staining with an indistinct CMJ (Fig. 4B). The results enhanced the fact that the uCreERT-fx/fx mice undergo accelerated thymic aging due to loss of FoxN1+ TECs with age.
Figure 4. Microanatomic changes in thymic architecture with age.

(A) A representative result shows H&E staining of fx/fx-only (without Cre-recombinase), uCreERT-fx/+, and uCreERT-fx/fx thymi at 1, 3, and 5 months of age. (B) A representative result of immunofluorescence staining with antibodies to Keratin-8 (green) and keratin-5 (red) from fx/fx-only (without Cre-recombinase), uCreERT-fx/+, and uCreERT-fx/fx thymi at 1, 3, and 5 months of age. M = medulla; C = cortex. This experiment was done in at least two biological replicates (i.e. 2 mice) in each genotype and each age, and at least 2 tissue sections per thymus have been observed, and a grand total of ~36 animals have been observed.
Reduced MHC-IIhiUEA-1hi mTECs in uCreERT-fx/fx mice resembled those found in the naturally aged thymus
The thymus in C57BL/6 WT mice normally undergoes initial involution from ~3 months of age, but the observable shrinkage is pronounced from ~12 months of age and is characterized by reduced thymic size and decreased cellularity of thymocytes and TECs. Detailed information on specific thymic phenotypes during aging is limited, because in the aged thymus, except for decreased cellularity, all T-cell subsets are present and the proportions of T-cell subsets do not change (Thoman 1995; Aspinall 1997; Chidgey et al. 2007). Furthermore, although the percentage of double negative 1 thymocytes has been reported to be relatively increased by some investigators (Thoman 1995; Aspinall 1997), it is not a specific thymic aging phenotype. Thymic aging has also been associated with development of a poorly defined CMJ (Takeoka et al. 1996), but evaluation of this feature is somewhat various.
Since we found that the percentage of MHC-IIhiUEA-1hi mature mTECs was reduced in the conditional FoxN1 knockout mice in our parallel paper (Cheng et al. 2010), and this subpopulation was reported to have a high FoxN1 expression in WT mice (Chen et al. 2009), as well as aged mice are known to have gradually reduced FoxN1 expression (Ortman et al. 2002), we hypothesized that reduction of MHC-IIhiUEA-1hi subpopulation may be related to natural thymic aging and should show in the naturally aged thymus. Indeed, flow cytometry analysis showed that the percentage of MHC-IIhiUEA-1hi mature mTECs did decline with age in WT mice (Figs. 5A and B). Coinciding with the flow cytometry results, our immunohistology results showed a similar reduction in UEA-1+ TECs during the thymic aging process (Fig. 5C). These changes paralleled the steady reduction in thymic size with age in WT mice (Fig. 3A). Mice of the fx/fx-only, without Cre-recombinase, had thymic size and MHC-IIhiUEA-1hi mTEC numbers that were similar to those of WT mice (Figs. 5C and D), indicating again that the loxP-floxed allele will not induce phenotype changes, and the fx/fx-only mice can be used as controls in place of WT mice.
Figure 5. Decrease in MHC-IIhiUEA-1hi mTECs with age in WT and fx/fx-only thymi.

(A) A representative flow cytometry plot of staining for CD45−MHC-IIhiUEA-1hi mTECs (gate A) in thymi from WT mice of different ages. (B) Summarized results of staining for CD45−MHC-IIhiUEA-1hi mature mTECs in WT mice of different ages. * p < 0.05 and ** p < 0.01, compared with 1–2-month-old mice. (C) Immunofluorescence staining for MHC-II (green) versus UEA-1 (red), and K5 (green) versus UEA-1 (red), from young (2-month-old), middle-aged (9-month-old), and aged (14- and 22-month-old) WT mice, as well as from fx/fx-only middle-aged (9-month-old) mice. Pictures taken at 20x magnification of objective lens, M = medulla; C = cortex. (D) Comparison of CD45−MHC-IIhiUEA-1hi mature mTEC subpopulation (gate A) in fx/fx-only and in WT mice at 9 months of age. For panel C, the experiment included 2–3 biological replicates (animals numbers) in each group with essentially identical results.
To determine whether the decline of MHC-IIhiUEA-1hi mTECs in naturally aged mice is more closely related to the gradual reduction in FoxN1 expression, we observed the changes of MHC-IIhiUEA-1hi mTECs in uCreERT-fx/fx mice, which have a spontaneous FoxN1 deletion. We found that the loss of MHC-IIhiUEA-1hi subset in uCreERT-fx/fx mice was accelerated, which was significantly reduced at 3–6 months of age (Figs. 6A and C). The slope of decline was steeper than those observed in fx/fx-only and uCreERT-fx/+ mice, (Fig. 6B). We noticed that onset was various in individual mice and complete penetrance of this phenotype needs a span of several months, but this is a valuable phenotype related to thymic aging and FoxN1+ TEC decline.
Figure 6. Accelerated loss of MHC-IIhiUEA-1hi mTECs with gradual FoxN1-deletion in uCreERT-fx/fx mice during aging.

(A) A representative flow cytometry plot of staining for CD45−MHC-IIhiUEA-1hi mTECs (gate A) in thymi from uCreERT-fx/fx mice at ~1, ~3, and ~6 months of age. (B) Summarized results of MHC-IIhiUEA-1hi mTEC absolute numbers in fx/fx-only (without Cre-recombinase), uCreERT-fx/+, and uCreERT-fx/fx mice with linear regression analysis. r2 = correlation of cell numbers to age; p < 0.05 = significant correlation. (C) t-test of MHC-IIhiUEA-1hi mTEC absolute numbers at 3–6 months of age from the three groups of mice as in panel B.
Exogenous FoxN1 could partially, but significantly, rescue natural aging-related thymic involution, increase thymocyte number and elevate peripheral CD4+ T-cell function
If gradual decline of FoxN1 expression is a potential cause of age-related thymic involution, then, enhancement of FoxN1 expression in the middle-aged and/or aged thymus should retard further thymic involution and/or rejuvenate thymic function. For a rapid test of this hypothesis, we performed intrathymic injection of either FoxN1-cDNA bearing vector or the empty vector into middle-aged (9–12 months old) and aged (18 months old) age-matched paired WT mice. The results showed that: 1), non-viral polyethylenimine(PEI)-mediated FoxN1-cDNA plasmid delivery (Boussif et al. 1995) can transform ~20–30% of thymic cells based on GFP expression (data not shown). Although GFP+ cells included both thymocytes and TECs, FoxN1 only provides functionality in TECs and not in thymocytes. 2), injection of FoxN1-cDNA plasmid into thymic anlage and/or peri-thymus of nude mice, which have a germline mutation of FoxN1 with athymia phenotype, could also partially rescue the thymic phenotype. Number of thymocytes was significantly increased and mature CD4+SP and CD8+SP T cells could be found in the FoxN1-cDNA infused nude mice (Fig. 7A). The results suggest that a FoxN1-cDNA plasmid, when injected using a PEI in vivo delivery approach, can work well as an intrathymic injection system. 3), comparing the age-matched paired FoxN1-cDNA plasmid-and empty vector-infused groups, the thymic size and number of thyomocytes in the FoxN1-cDNA plasmid-infused group were always larger or more than that in the empty vector-infused age-matched mice for both ages tested (two examples shown in Fig. 7B). 4), total thymocyte number either in middle-aged or aged mice was significantly increased after infusion of FoxN1-cDNA plasmid (Figs. 7C and D). 5), the percentage of intra-cellular IL-2+ peripheral CD4+ T cell population cannot be elevated in response to co-stimulation of CD3 and CD28 antibodies in the aged mice (Zhu et al. 2007) (Fig. 8A, the second row), whereas it was significantly elevated in aged mice following two infusions of the FoxN1-cDNA plasmid, given every three weeks, for a total incubation of six weeks, compared with the group infused with empty vector.
Figure 7. Effect of FoxN1-cDNA plasmid on FoxN1-null (nude) mice and partial rescue of age-related thymic involution in WT mice via intrathymic injection of exogenous FoxN1 cDNA.

(A) Left panel: Absolute thymocyte numbers from young FoxN1-null (nude) mice six weeks after two injections (3-week intervals) of FoxN1-cDNA plasmid, empty vector, or PBS; Right panel: Representative flow cytometric plots of CD4 versus CD8 staining of nude mouse thymocytes six weeks after two injections (3-week intervals) of FoxN1-cDNA plasmid, empty vector, or PBS. (B) Gross appearance of thymic size from FoxN1-cDNA-infused WT mice with enlarged thymus (arrows) and empty vector-infused WT mice with original-sized thymus (arrow heads) at 12 and 18 months of age. (C) Absolute thymocyte numbers from middle-aged (9–12 months old) WT mice, one month after FoxN1-cDNA plasmid or empty vector input. (D) Absolute thymocyte numbers from aged (18 months old) WT mice, 6 weeks after two injections (3-week intervals) of FoxN1–cDNA plasmid or empty vector.
Figure 8. Increased IL-2 production in peripheral CD4+ T cells of aged mice via intrathymic infusion of exogenous FoxN1-cDNA.

(A) A representative FACS result, from top to bottom, shows IL-2+CD4+T-cell subset in young mouse spleen (2 months old), aged mouse spleen (18 months old), empty vector-infused aged mouse spleen (18 months old), and FoxN1-cDNA-infused aged mouse spleen (18 months old) without stimulation (left panels) and in response to CD3 and CD28 antibodies (right panels). (B) A summarized % of splenic IL-2+CD4+T cells of the above four groups in response to CD3 and CD28 antibodies.
The partial rescue of natural aging-related thymic involution and improved thymic function, thereby partially rejuvenating peripheral CD4+ T cell function in the aged mice resulting from supplying aged WT mice with exogenous FoxN1 confirms that shortage of FoxN1 in the thymus is a key issue related to T-cell immune system aging.
DISCUSSION
Under the physiological condition, thymic aging is regulated by changes of gene expression. These changes should be accounted for as epigenetic changes including chromatin remodeling and DNA modifications, rather than genetic changes, since there is no DNA sequence change. However, by studying which genes are expressed and how changes in expression affects rates of thymic aging by using a genetic model should provide insights on single gene function in aging. A conventional gene knockout model cannot be used in this type of aging study since suddenly shutting down a gene does not mimic a natural aging scenario, whereas the conditional spontaneous gene deletion model can be used for this study because, like natural aging, this deletion model generates a process with progressive/gradual declining gene expression. We, herein, established such a genetic model by using a loxP-uCreERT approach to study changes in expression of the FoxN1 gene with thymic aging, and found that loss of epithelial cell-autonomous gene FoxN1 is a key to induction and acceleration of age-related thymic involution. This was confirmed not only by spontaneously deleting FoxN1 with age to accelerate thymic aging phenotypes via the loss-of-function approach, but also by supplying exogenous FoxN1 cDNA to partially, but significantly, rescue naturally aging-related thymic involution and improve naturally aged thymic function, through a rapid test of the gain-of-function approach.
Although FoxN1 mRNA has been reported to decrease with age (Ortman et al. 2002), it has long been speculated whether decline of FoxN1 expression is a cause of age-related thymic involution or an effect due to thymic epithelial cell deterioration during the thymic aging process. The results in this paper demonstrate that gradual loss of FoxN1 through uCreERT spontaneous activation caused accelerated thymic aging, including atrophic thymic size, disrupted thymic micro-architecture—especially an indistinct CMJ, accelerated reduction in number of TECs—prominent decline in MHC-IIhiUEA-1hi mTEC subset in percentage and numbers, and sharply reduced number of thymocytes—particular in DP and CD4+SP subsets. All these phenotypes resemble those of the naturally aged thymus. On the other hand, input of exogenous FoxN1 cDNA locally rescued the naturally aged thymic phenotype, including reversed thymic involution, increased number of total thymocytes, and rescued peripheral CD4+ T-cell function in response to co-stimulation by CD3 and CD28 antibodies. All these results provided evidence that loss of FoxN1 is a potential cause of age-related thymic involution in mice.
Age-related thymic involution may be through several potential mechanisms, in which there is defect in interactions between TECs and LPCs. Thymic atrophy/involution can be triggered by defects in hematopoietic cell function, such as knockout of T-cell receptor rearrangement gene and depletion of T-cell progenitors (Gruver et al. 2007), or defects in non-hematopoietic cell function, such as alteration of cytokines produced by TECs of the stroma (Lynch et al. 2009) and knockout of FgfR2-IIIb gene (Revest et al. 2001). There are two opposing views of the mechanisms that mediate the initial changes in age-related thymic involution. One holds that LPCs, including hematopoietic stem cells (HSCs), and their downstream multipotent progenitors (MPPs) and early thymic progenitors (ETPs), in aged animals develop cumulative intrinsic defects with age to trigger thymic involution (Min et al. 2004). The other maintains that aging causes dysfunction of basic thymic microenvironmental cells, composed primarily of TECs, causing secondary changes in T-cell precursors—thymocytes, thereby triggering thymic involution (Min et al. 2007; Zhu et al. 2007). Based on our studies in this report, we favor the hypothesis that the dominant defect in age-related thymic involution is primarily derived from TEC dysfunction in the thymus, which in turn cause age-related thymic lymphopoietic insufficiency, but we do not rule out that LPCs will eventually develop an irreversible intrinsic defect during aging. Indeed, we found that aged (>22 months old) murine bone marrow cannot efficiently compete with its young counterpart to develop equivalent mature T cells under the same microenvironment in an in vivo competitive model (data unpublished), which is consistent with other group’s experiment (Zediak et al. 2007), although it has the capacity to do so in a young microenvironment with a non-competitive style (Zhu et al. 2007). However, the intrinsic defect in bone marrow occurs chronologically later than the thymic involution. We believe that aging causes TEC dysfunction at first, and then additionally causes the LPC intrinsic defect. Both TEC and LPC defects amplify the age-related thymic involution.
Generally, there is not one specific phenotype which can be used to define thymic aging except for thymic involution. In our studies, we found that decreases in one of the mTEC subsets—MHC-IIhiUEA-1hi population and FoxN1+ TECs can be used to define thymic aging, since both TEC subsets were found to decrease in inverse proportional to an increase in the age of the thymus. The MHC-IIhiUEA-1hi TECs are a mature mTEC subset. The loss of this subset may be due to these cells being more sensitive to loss of FoxN1 and/or they fail to be continuously replaced by TEC progenitors in situ. This is related to FoxN1 function in the postnatal thymus, which we reported in our parallel paper (Cheng et al. 2010).
Recently, there has been an increased interest in rejuvenation strategies for the immune system (Chidgey & Boyd 2006; Lynch et al. 2009). Restoration of dysfunctional thymic epithelial cells is an effective strategy for rejuvenation of the T-cell immune system. Administration of keratinocyte growth factor can promote mitosis in differentiated TECs; administration of a non-redundant cytokine Interlukin-7, mainly produced by TSCs in the thymus, can augment thymocyte proliferation, whereas administration of FoxN1 cDNA may have potential benefits for promoting TEC progenitor differentiation and maintaining mature mTEC survival. Incorporation of multiple strategies may succeed in creating rejuvenating conditions that more closely resemble conditions found in the natural physiological state, resulting in a more effective strategy.
In summary, age-related thymic involution may occur and be regulated through several potential mechanisms including defects in hematopoietic and non-hematopoietic cells. However, alteration of epithelial cell-autonomous gene expression, such as expression of FoxN1, in TECs may be considered as a key factor of age-related thymic involution for several reasons: 1), decrease of FoxN1 expression with age causes, while providing exogenous FoxN1 prevents progressive TEC impairment and thymic involution 2), TEC defect occurs earlier than hematopoietic stem cell intrinsic deterioration 3), and infusion of young LPCs into the aged thymus cannot reverse aged TEC defects, while replacement of aged TEC meshwork with young one can promote aged LPCs to produce normal levels of T cells (Zhu et al. 2007). However, it is unlikely that changes in only one or two genes can fully explain the complex process of age-related thymic involution, in which there may be multiple defects in the interactions between TECs and LPCs. Therefore, we will continue to decipher these mechanisms from understanding how aging impairs epithelial cell-autonomous gene expression and determining what are the downstream genes targeted by FoxN1.
EXPERIMENTAL PROCEDURES
Mice and genotyping
The FoxN1fx (fx) mice were generated as described in our parallel paper (Cheng et al. 2010), in which exons 5 and 6 (DNA binding domain) of the FoxN1 locus are flanked by two loxP sites. We crossbred fx/fx homozygous mice with pCAGG-CreER™ mice (Jackson Lab #004682) (Hayashi & McMahon 2002), carrying a tamoxifen(TM)-inducible ubiquitous Cre (which we refer to here as uCreERT), then backcrossed them with fx/fx mice to obtain fx/fx and fx/+ carrying uCreERT mice. All young mice were genotyped by PCR at 3–4 weeks of age, using DNA obtained from the tails. Multiple-primer PCR (PCR was performed with 3 primers of A, B, and C, in one reaction) was used for genotyping and to check fx locus deletion (Figure 1). Primer locations are shown in Figure 1A (arrows): 5′-primer-A: 5′-cca acc tcc tgg gga cat ga-3′ and 3′-primer-B: 5′-tag gag gag ggg agc gcc ta-3′, which produce 566-bp and 648-bp amplicons for the WT and floxed-FoxN1 (fx) loci, respectively, as well as 5′-primer-C: 5′-gtg ggc ttt tca cca tcc ta-3′, which produces a ~444-bp amplicon in the mutant locus with deleted exons 5 and 6 of FoxN1 (denoted as “ΔE5&6”), with Primer-B. Aged (≥18 months old) WT mice were purchased from National Institutes for Aging (Bethesda, MD). All animal experiments were done according to the protocols approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at Tyler, in accordance with guidelines of the National Institution of Health, USA.
FoxN1+ TECs and micro-architecture determined by histological and immunofluorescent analysis
For immunofluorescence staining, cryosections (6 μm) were fixed in cold acetone, and blocked with donkey serum in TBS containing 0.5% BSA or with an endogenous biotin-blocking kit (Invitrogen) if a biotinylated antibody was used. Primary antibodies were rabbit anti-mouse FoxN1 (Itoi et al. 2007) or rabbit anti-mouse K5 (Covance), and rat anti-mouse K8 (Troma-1 supernatant) or biotinylated-anti-UEA-1 (Vector Labs). Secondary reagents were Cy3-donkey anti-rabbit IgG, FITC-donkey anti-rat IgG, Cy3-streptavidin (all from Jackson Immunoresearch), or AlexaFluor-488-anti-rabbit IgG (Invitrogen), respectively. For hematoxylin and eosin (H&E) staining, thymi were fixed, cut into 5 μm-thick paraffin sections, and stained with H&E.
Number and proportions of subpopulations of thymocytes and TECs analyzed by flow cytometry
Single-cell suspension of thymocytes, prepared with cell strainers (Zhu et al. 2007), were stained with combinations of either fluorochrome-conjugated anti-mouse-CD3, -CD4, and -CD8, or fluorochrome-conjugated anti-mouse-CD25, –CD44, -lineage cocktail markers (Zhu et al. 2007). Enzymatically (Collagenase-V/DNase-I) dissociated thymic cells (Gui et al. 2007) were stained with combinations of fluorochrome-conjugated anti-mouse-CD45, -MHC-II (M5/114), -Ly51 (6C3), and either FITC-anti-mouse-EpCam (G8.8) (BioLegend) or FITC-UEA-1 (Vector Labs). Data was acquired with a dual-laser FACS Calibur system and analyzed using CellQuest and FlowJo software.
Intrathymic transformation of FoxN1 cDNA
FoxN1-cDNA, preceded by a Kozak sequence to enhance expression and followed by the internal ribosome entry site (IRES) and the green fluorescent protein (GFP) reporter gene, is driven by the CMV promoter in pADTrack vector (kindly provided by Dr. Brissette (Prowse et al. 1999; Weiner et al. 2007)). The control vector is an empty pADTrack plasmid. FoxN1-cDNA plasmid was delivered in vivo by a non-viral polyethylenimine (PEI)-mediated method (Boussif et al. 1995; Neu et al. 2005). A mixture of 40 μg plasmid and 6.4 μl of PEI (Genesee Scientific Cop.) at ionic balance N/P ratio = 8, in ~25μl volume was intrathymically injected into each middle-age or aged mouse at three sites of the thymic lobes under anesthesia and suprasternal notch surgery (Zhu et al. 2007). The middle-aged mice were injected once, and one month after injection the thymi were analyzed by flow cytometry, whereas the aged mice were injected twice at 3-week intervals, 3 weeks after the follow-up injection the thymi were analyzed by flow cytometry.
Analysis of intra-cellular IL-2 in peripheral CD4+ T cells in response to co-stimulation of CD3 and CD28 antibodies
Spleen cells were freshly isolated from either FoxN1-cDNA plasmid or empty vector infused 18-month-old aged mice, which were injected intrathymically every 3 weeks over a 6 week period for a total of 2 injections. The erythrocytes were depleted with ACK (Ammonium-Chloride-Potassum) lysis buffer (pH7.2, 0.15 mM NH4Cl/1.0 mM KHCO3/0.1 mM Na2EDTA). The spleen cells (2 × 106/well) were cultured with anti-mouse CD3ε and CD28 antibodies (2μg/ml each) supplemented with GolgiStop (4μg/ml) for 5 hours in a 48-well plate. The harvested cells were stained for surface CD4, then fixed with 1% PFA (Paraformaldehyde)/PBS overnight, permeabilized with 0.1% Triton-X100 in 0.1% sodium citrate, pH 7.2, then stained with IL-2 antibody intracellularly. The results were analyzed by a FACS Calibur. All Antibodies and GolgiStop were purchased from BD Pharmingen (San Diego, CA).
Statistics
Comparisons were made by the unpaired Student’s t-test. p < 0.05 was considered statistically significant. Correlations were analyzed by linear regression using Prism-4 software (GraphPad).
Acknowledgments
Funding support: This work was supported by the NIAID/NIH and NIA/NIH grants (R01AI081995, R21AG031880, and R21AI079747) to D-M S.
We thank Dr. Janice L. Brissette (Massachusetts General Hospital and Harvard Medical School) for providing FoxN1 cDNA in pADTrack vector; Lili Cheng (UTHSCT) for great technique supports.
Abbreviations
- cTEC/mTEC
cortical/medullary thymic epithelial cells
- DP
double positive
- fx
loxP-floxed-Foxn1
- K
keratin
- LPC
lymphohematopoietic progenitor cell
- SP
single positive
- TSC/TEC
thymic stromal/epithelial cell
- uCerERT
ubiquitously-expressed tamoxifen-inducible Cre-recombinase
- UEA-1
Ulex europaeus agglutinin-1
- WT
wild type
Footnotes
Author Information The authors declare that they have no competing financial interests.
References
- Adriani M, Martinez-Mir A, Fusco F, Busiello R, Frank J, Telese S, Matrecano E, Ursini MV, Christiano AM, Pignata C. Ancestral founder mutation of the nude (FOXN1) gene in congenital severe combined immunodeficiency associated with alopecia in southern Italy population. Ann Hum Genet. 2004;68:265–268. doi: 10.1046/j.1529-8817.2004.00091.x. [DOI] [PubMed] [Google Scholar]
- Amorosi S, D’Armiento M, Calcagno G, Russo I, Adriani M, Christiano AM, Weiner L, Brissette JL, Pignata C. FOXN1 homozygous mutation associated with anencephaly and severe neural tube defect in human athymic Nude/SCID fetus. Clin Genet. 2008;73:380–384. doi: 10.1111/j.1399-0004.2008.00977.x. [DOI] [PubMed] [Google Scholar]
- Aspinall R. Age-associated thymic atrophy in the mouse is due to a deficiency affecting rearrangement of the TCR during intrathymic T cell development. J Immunol. 1997;158:3037–3045. [PubMed] [Google Scholar]
- Aspinall R, Andrew D. Thymic atrophy in the mouse is a soluble problem of the thymic environment. Vaccine. 2000;18:1629–1637. doi: 10.1016/s0264-410x(99)00498-3. [DOI] [PubMed] [Google Scholar]
- Bleul CC, Corbeaux T, Reuter A, Fisch P, Monting JS, Boehm T. Formation of a functional thymus initiated by a postnatal epithelial progenitor cell. Nature. 2006;441:992–996. doi: 10.1038/nature04850. [DOI] [PubMed] [Google Scholar]
- Bodey B, Bodey B, Jr, Siegel SE, Kaiser HE. Involution of the mammalian thymus, one of the leading regulators of aging. In Vivo. 1997;11:421–440. [PubMed] [Google Scholar]
- Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xiao S, Manley NR. Foxn1 is required to maintain the postnatal thymic microenvironment in a dosage-sensitive manner. Blood. 2009;113:567–574. doi: 10.1182/blood-2008-05-156265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Guo J, Sun L, Fu J, Barnes PF, Metzger D, Chambon P, Oshima RG, Amagai T, Su DM. Postnatal tissue-specific disruption of transcription factor FoxN1 triggers acute thymic atrophy. J Biol Chem. 2010:285. doi: 10.1074/jbc.M109.072124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidgey A, Dudakov J, Seach N, Boyd R. Impact of niche aging on thymic regeneration and immune reconstitution. Semin Immunol. 2007;19:331–340. doi: 10.1016/j.smim.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Chidgey AP, Boyd RL. Stemming the tide of thymic aging. Nat Immunol. 2006;7:1013–1016. doi: 10.1038/ni1006-1013. [DOI] [PubMed] [Google Scholar]
- Coffer PJ, Burgering BM. Forkhead-box transcription factors and their role in the immune system. Nat Rev Immunol. 2004;4:889–899. doi: 10.1038/nri1488. [DOI] [PubMed] [Google Scholar]
- Cunningham-Rundles C, Ponda PP. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nat Rev Immunol. 2005;5:880–892. doi: 10.1038/nri1713. [DOI] [PubMed] [Google Scholar]
- Frank J, Pignata C, Panteleyev AA, Prowse DM, Baden H, Weiner L, Gaetaniello L, Ahmad W, Pozzi N, Cserhalmi-Friedman PB, Aita VM, Uyttendaele H, Gordon D, Ott J, Brissette JL, Christiano AM. Exposing the human nude phenotype. Nature. 1999;398:473–474. doi: 10.1038/18997. [DOI] [PubMed] [Google Scholar]
- Gray DH, Seach N, Ueno T, Milton MK, Liston A, Lew AM, Goodnow CC, Boyd RL. Developmental kinetics, turnover, and stimulatory capacity of thymic epithelial cells. Blood. 2006;108:3777–3785. doi: 10.1182/blood-2006-02-004531. [DOI] [PubMed] [Google Scholar]
- Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J Pathol. 2007;211:144–156. doi: 10.1002/path.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruver AL, Sempowski GD. Cytokines, leptin, and stress-induced thymic atrophy. J Leukoc Biol. 2008 doi: 10.1189/jlb.0108025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui J, Zhu X, Dohkan J, Cheng L, Barnes PF, Su DM. The aged thymus shows normal recruitment of lymphohematopoietic progenitors but has defects in thymic epithelial cells. Int Immunol. 2007 doi: 10.1093/intimm/dxm095. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Henson SM, Snelgrove R, Hussell T, Wells DJ, Aspinall R. An IL-7 fusion protein that shows increased thymopoietic ability. J Immunol. 2005;175:4112–4118. doi: 10.4049/jimmunol.175.6.4112. [DOI] [PubMed] [Google Scholar]
- Itoi M, Tsukamoto N, Amagai T. Expression of Dll4 and CCL25 in Foxn1-negative epithelial cells in the post-natal thymus. Int Immunol. 2007;19:127–132. doi: 10.1093/intimm/dxl129. [DOI] [PubMed] [Google Scholar]
- Kojima A, Saito M, Hioki K, Shimanura K, Habu S. NFS/N-nu/+ mice can macroscopically be distinguished from NFS/N- +/+ littermates by their thymic size and shape. Exp Cell Biol. 1984;52:107–110. [PubMed] [Google Scholar]
- Ladi E, Yin X, Chtanova T, Robey EA. Thymic microenvironments for T cell differentiation and selection. Nat Immunol. 2006;7:338–343. doi: 10.1038/ni1323. [DOI] [PubMed] [Google Scholar]
- Lynch HE, Goldberg GL, Chidgey A, Van den Brink MR, Boyd R, Sempowski GD. Thymic involution and immune reconstitution. Trends Immunol. 2009;30:366–373. doi: 10.1016/j.it.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min D, Panoskaltsis-Mortari A, Kuro OM, Hollander GA, Blazar BR, Weinberg KI. Sustained thymopoiesis and improvement in functional immunity induced by exogenous KGF administration in murine models of aging. Blood. 2007;109:2529–2537. doi: 10.1182/blood-2006-08-043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min H, Montecino-Rodriguez E, Dorshkind K. Reduction in the developmental potential of intrathymic T cell progenitors with age. J Immunol. 2004;173:245–250. doi: 10.4049/jimmunol.173.1.245. [DOI] [PubMed] [Google Scholar]
- Nehls M, Kyewski B, Messerle M, Waldschutz R, Schuddekopf K, Smith AJ, Boehm T. Two genetically separable steps in the differentiation of thymic epithelium. Science. 1996;272:886–889. doi: 10.1126/science.272.5263.886. [DOI] [PubMed] [Google Scholar]
- Nehls M, Pfeifer D, Schorpp M, Hedrich H, Boehm T. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature. 1994;372:103–107. doi: 10.1038/372103a0. [DOI] [PubMed] [Google Scholar]
- Neu M, Fischer D, Kissel T. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J Gene Med. 2005;7:992–1009. doi: 10.1002/jgm.773. [DOI] [PubMed] [Google Scholar]
- Ortman CL, Dittmar KA, Witte PL, Le PT. Molecular characterization of the mouse involuted thymus: aberrations in expression of transcription regulators in thymocyte and epithelial compartments. Int Immunol. 2002;14:813–822. doi: 10.1093/intimm/dxf042. [DOI] [PubMed] [Google Scholar]
- Petrie HT. Role of thymic organ structure and stromal composition in steady-state postnatal T-cell production. Immunol Rev. 2002;189:8–19. doi: 10.1034/j.1600-065x.2002.18902.x. [DOI] [PubMed] [Google Scholar]
- Prowse DM, Lee D, Weiner L, Jiang N, Magro CM, Baden HP, Brissette JL. Ectopic expression of the nude gene induces hyperproliferation and defects in differentiation: implications for the self-renewal of cutaneous epithelia. Dev Biol. 1999;212:54–67. doi: 10.1006/dbio.1999.9328. [DOI] [PubMed] [Google Scholar]
- Revest JM, Suniara RK, Kerr K, Owen JJ, Dickson C. Development of the thymus requires signaling through the fibroblast growth factor receptor R2-IIIb. J Immunol. 2001;167:1954–1961. doi: 10.4049/jimmunol.167.4.1954. [DOI] [PubMed] [Google Scholar]
- Rossi SW, Jeker LT, Ueno T, Kuse S, Keller MP, Zuklys S, Gudkov AV, Takahama Y, Krenger W, Blazar BR, Hollander GA. Keratinocyte growth factor (KGF) enhances postnatal T-cell development via enhancements in proliferation and function of thymic epithelial cells. Blood. 2007;109:3803–3811. doi: 10.1182/blood-2006-10-049767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiff JM, Cordier AC, Haumont S. The thymus of Nu/+ mice. Anat Embryol (Berl) 1978;153:115–122. doi: 10.1007/BF00343368. [DOI] [PubMed] [Google Scholar]
- Su DM, Navarre S, Oh WJ, Condie BG, Manley NR. A domain of Foxn1 required for crosstalk-dependent thymic epithelial cell differentiation. Nat Immunol. 2003;4:1128–1135. doi: 10.1038/ni983. [DOI] [PubMed] [Google Scholar]
- Takeoka Y, Chen SY, Yago H, Boyd R, Suehiro S, Shultz LD, Ansari AA, Gershwin ME. The murine thymic microenvironment: changes with age. Int Arch Allergy Immunol. 1996;111:5–12. doi: 10.1159/000237337. [DOI] [PubMed] [Google Scholar]
- Taub DD, Longo DL. Insights into thymic aging and regeneration. Immunol Rev. 2005;205:72–93. doi: 10.1111/j.0105-2896.2005.00275.x. [DOI] [PubMed] [Google Scholar]
- Thoman ML. The pattern of T lymphocyte differentiation is altered during thymic involution. Mech Ageing Dev. 1995;82:155–170. doi: 10.1016/0047-6374(95)01597-s. [DOI] [PubMed] [Google Scholar]
- Weiner L, Han R, Scicchitano BM, Li J, Hasegawa K, Grossi M, Lee D, Brissette JL. Dedicated epithelial recipient cells determine pigmentation patterns. Cell. 2007;130:932–942. doi: 10.1016/j.cell.2007.07.024. [DOI] [PubMed] [Google Scholar]
- Youm YH, Yang H, Sun Y, Smith RG, Manley NR, Vandanmagsar B, Dixit VD. Deficient ghrelin receptor-mediated signaling compromises thymic stromal cell microenvironment by accelerating thymic adiposity. J Biol Chem. 2009;284:7068–7077. doi: 10.1074/jbc.M808302200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zediak VP, Maillard I, Bhandoola A. Multiple prethymic defects underlie age-related loss of T progenitor competence. Blood. 2007;110:1161–1167. doi: 10.1182/blood-2007-01-071605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Gui J, Dohkan J, Cheng L, Barnes PF, Su DM. Lymphohematopoietic progenitors do not have a synchronized defect with age-related thymic involution. Aging Cell. 2007 doi: 10.1111/j.1474-9726.2007.00325.x. [DOI] [PubMed] [Google Scholar]