

Abstract
The surface-initiated ATRP of benzyl methacrylate, methyl methacrylate, and styrene from magnetite nanoparticle is investigated, without the use of sacrificial (free) initiator in solution. It is observed that the grafting density obtained is related to the polymerization kinetics, being higher for faster polymerizing monomer. The grafting density was found to be nearly 2 chains/nm2for the rapidly polymerizing benzyl methacrylate. In contrast, for the less rapidly polymerizing styrene, the grafting density was found to be nearly 0.7 chain/nm2. It is hypothesized that this could be due to the relative rates of surface-initiated polymerization versus conformational mobility of polymer chains anchored by one end to the surface. An amphiphilic diblock polymer based on 2-hydroxylethyl methacrylate is synthesized from the polystyrene monolayer. The homopolymer and block copolymer grafted MNs form stable dispersions in various solvents. In order to evaluate molecular weight of the polymer that was grafted on to the surface of the nanoparticles, it was degrafted suitably and subjected to gel permeation chromatography analysis. Thermogravimetric analysis, transmission electron microscopy, and Fourier transform infrared spectroscopy were used to confirm the grafting reaction.
Keywords: Poly(benzyl methacrylate), Atom transfer radical polymerization, Magnetite nanoparticle
Introduction
The use of material in the nanoparticles form offers many advantages due to the large surface-to-volume ratio [1]. Magnetite nanoparticles (MNs) is one of the most popular nanomaterial known for its biomedical applications because of its low toxicity for living cells and in the view of possibility of selected targeting of tumor area, through external magnetic field. MNs, especially in the size range of 10 nm, is interesting because of its superparamagnetic nature, as it does not retain its residual magnetism after the magnetic field is removed. The superparamagnetic iron oxide nanoparticles are used in a number of biomedical areas such as magnetic resonance imaging [2], targeted drug delivery [3,4], gene delivery systems, and gene therapy [5] as well as targeted hyperthermia of cancers [6]. In all the above applications, it is preferable that MNs are encapsulated with a polymer of interest in order to avoid its agglomeration for various biomedical applications. This is in view of the tendency of nanoparticle to agglomerate, as a result of van der waals attractive forces. The two common modes of preventing the agglomerization and stabilizing the nanoparticles are: (1) electrostatic stabilization and (2) steric stabilization. The electrostatic stabilization results from the coulombic repulsion between the particles caused by the electrical double layer, which inturn is formed by ions adsorbed on the particle surface. The citrate ion is commonly used as the reducing agent as well as an electrostatic stabilizer for gold nanoparticles [7,8]. The stabilization thus brought about is kinetic stabilization and is applicable only to dilute systems [9]. Thus to overcome this disadvantage, steric stabilization is introduced in which the coordination of sterically demanding organic molecules, surfactants, and polymers can act as protecting shields for the steric stabilization of metal colloids. Steric stabilization provides a thermodynamically stable system. Among the stabilizers, polymers are considered to be better steric stabilizing agents [10].
There are two ways of attaching polymer layers to nanoparticulate surfaces namely, “grafting from” and “grafting to”. The shape of the semiflexible polymer chain, in solution, is a sphere. The adsorption or “grafting to” of polymer to a surface produces a monolayer of “spherical” polymer chains. Further adsorption is restricted since the surface concentration is much higher than solution concentration (diffusion barrier) and in addition the “entropic” penalty for stretching away from the surface is high [11]. For example, in a recent publication “click chemistry” was used to anchor an oligomer to silica particle wherein a grafting density of 0.34 chains/nm2[12]. In contrast in the “grafting from” technique, polymer chains are grown from the surface-attached initiator by in situ polymerization via thermal or photochemical means [13] in which the optimum control over the structure of the composite can be achieved with the nanomaterial core and a dense polymer shell. Thus the surface-initiated polymerization i.e., polymerization from a nanoparticle with an active initiator, helps to form a uniform surface coating of the polymer chains on the surface of the particles.
The thickness of the grafted polymer layer increases with increasing polymerization time for a controlled radical polymerization, at fixed monomer concentration. When polymer chains are densely grafted to a surface, steric crowding can force the chains to stretch away from the surface to form a brush. Under this condition, the thickness of the polymer layer should be larger than the radius of gyration of the equivalent free polymer in solution [14,15]. This results in high grafting density as well as the formation of a stable dispersion of the particle in the solvent of interest.
The direct growth of polymer chains can be is accomplished through a monolayer of initiator, which is anchored to the nanoparticles in the first stage using appropriate anchoring chemistry. Several anchoring group chemistry have been reported for the introduction of a monolayer of initiators. The nature of the anchoring group used varies depending on the nature of the nanoparticle. For example, gold nanoparticle [16], magnetite nanoparticle [17], silica nanoparticle [18], and titania nanoparticle [19] require various functional groups as summarized in the Table 1. In the anchoring chemistry commonly used, thiol stabilization of the nanoparticle is restricted to a temperature range below 60 °C as the thiol group is known to cleave from the surface at higher temperatures [20]. The utilization of trichloro and trialkoxy silanes anchoring chemistry is restricted as this functional group can undergo self-condensation reaction [21] to form a polysiloxane film on the surface of the particle. However, a monochlorosilane [22,23] is useful toward the introduction of a monolayer of initiator, followed by the polymerization. However, during the surface modification of the nanoparticle, the reaction of chlorosilanes with free hydroxyl groups on the metal oxide surfaces generates hydrochloric acid as the byproduct, which may affect the modified surfaces [24]. Oleic acid has been used to modify the surface of magnetite. A carboxylic functional group as the anchoring group is reported to be unstable under biological conditions [25]. This is probably due to the weak interaction between the carboxylic acid group with magnetite. The reaction of hydridosilane initiator to the metal oxide particle like titania and zirconia normally resulted in cross-linking of added monolayers instead of grafting on the metal oxides [26]. This cross-linked monolayers (with Si–O–Si bonds) grafted to the metal oxides (via Si–O–MS bonds) were confirmed by FT–IR analysis in which an intense band appeared at ~1,040 cm−1 due to cross-linked siloxane network.
Table 1.
ATRP of methyl methacrylate from the various nanoparticle
| Various anchoring chemistry | Various nanoparticle | Grafting density after polymerization (chain/nm2) |
|---|---|---|
| Thiol |
Gold |
0.3 |
| Choro silane |
Magnetite |
0.1 |
| Triethoxy silane |
Silica |
0.7 |
| Triethoxy silane | Titania | 0.04 |
Initiators with a phosphonic acid anchoring moiety are quite interesting since they are known to form self-assembled monolayer (SAM) by strong covalent binding with the surface hydroxyl (–OH) group on metal oxide (titania and zirconium oxide) surfaces [27]. In comparison to other SAMs, the phosphonic acid-based anchoring is preferable for its three advantages: (1) higher hydrolytic stability [28] under physiological conditions due to chemisorption, (2) easily anchored by sonication [29], and (3) retention of particle property [30], especially in the case of magnetite, after anchoring.
The recent developments in living radical polymerization techniques such as atom transfer radical polymerization (ATRP) [31], nitroxide-mediated polymerization (NMP) [32], and reversible addition-fragmentation transfer polymerization (RAFT) [33] have been considerably applied in the surface modification of the nanoparticles. ATRP is a versatile technique to precisely control the chain length and polydispersity of the polymer, and can be used to synthesize well-defined block copolymers with a range of functionalities since the end-groups remain active [34] at the end of the polymerization. If the ATRP reaction conditions used are mild, a wide range of monomers and macromolecular structures can be used for grafting [35]. Thus, atom transfer radical polymerization (ATRP) [36] that can be performed at ambient temperature is less prone to side reactions and chain transfer, resulting in better control over molecular weight and polydispersion index (PDI) thus enabling the facile synthesis of a wide variety of hybrid materials [34,37].
The synthesis of polystyrene grafted MN nanoparticles, without the addition of sacrificial initiator is reported in the literature [38] but the estimation of grafting density is not reported. The nitroxide-mediated polymerization of styrene [29] was carried out at 125 °C, from a phosphonic acid anchored MN surface. This results in lower grafting density of 0.2 polystyrene chain/nm2, where the density of surface initiator was 0.73 chain/nm2. It can therefore be concluded that 27% of initiator on the magnetite surface participated in the polymerization, with the addition of sacrificial initiator. This low initiator efficiency could be due to the termination between free chains formed in solution (because of the addition of sacrificial initiator) and a surface-bound polymer [39,40]. In comparison, the ambient temperature ATRP of methyl methacrylate from a phosphonic acid anchored magnetite surface results in a grafting density of an initiator 1 chain/nm2 for an initiator grafting density of 2 molecules/nm2. Thus 50% of the surface initiating groups on the magnetite surface participated in the polymerization [41]. This was carried out without sacrificial initiator as well as without the initial addition of Cu(II). The high grafting density obtained in this case could be due to the faster polymerization in comparison to conformational relaxation of the growing chain. In order to explore this hypothesis in detail, we have chosen monomers that polymerize faster as well as slower in comparison with methyl methacrylate and report the results obtained. In this work, the ATRP of benzyl methacrylate at ambient temperature as well as that of styrene at 100 °C is carried out from the magnetite nanoparticle surface, without using the sacrificial initiator [42] to compare its grafting density with that of methyl methacrylate, which was polymerized from the surface of MNs, at ambient temperature [41]. Surface-Initiated polymerization without the use of sacrificial initiator could offer certain advantages such as the elimination of the step associated with the removal of unattached polymer that is formed from the sacrificial initiator. In addition, it could proceed at a faster rate thus facilitating simultaneous growth from the surface sites. A disadvantage of this method is that it is relatively less controlled. To investigate the effect of rate of polymerization on the graft density of polymer chains grown from the MNs surface, the ATRP of benzyl methacrylate, styrene, and MMA were carried out without the addition of sacrificial initiator from a tertiary bromide ATRP initiator anchored to the surface through phosphonic acid anchoring group. In addition, the results from this study are compared with one case where the ATRP initiator is anchored to MNs through carboxylic acid based anchoring group.
Experimental Section
Materials
The inhibitor present in methyl methacrylate (MMA), benzyl methacrylate (BnMA), styrene, and 2-hydroxylethyl methacrylate (Lancaster) were removed by passing through a basic alumina column. The monomer was used immediately after purification. Copper(I) bromide (Aldrich, 99.98%),N,N,N′,N″,N″′-pentamethyldiethyltriamine (Aldrich, 99%), aluminum oxide (activated, basic, for column chromatography, 50–200 μm) were used without purification. Anisole, ethyl methyl ketone, 1-propanol, and DMF (SRL India) were used as received.
Synthesis of ATRP Initiator and its Anchoring to MNs
The synthesis of magnetite nanoparticles as well as the ATRP initiator, 2-bromo-2-methyl-propionic acid 2-phosphonooxy-ethyl ester (1) and its anchoring to the magnetite nanoparticle to get ATRP initiator immobilized magnetite nanoparticle (2) has been reported by us in the literature [41].
Surface-Initiated ATRP of Benzyl Methacrylate from Magnetite Surface
The polymerization was carried out with CuBr (0.01 mmol) and 25 mg of magnetite-ATRP initiator, (2), in a dry Schlenk flask equipped with a magnetic pellet and a rubber septum. Initially, the mixture was subjected to dynamic vacuum for 1 h. This was followed by the addition of the degassed benzyl methacrylate (17.04 mmol) (50% v/v of anisole) such that the mole ratio of [BnMA]:[Initiator]:[CuBr]:[PMDETA] is 1900/1/1/1. The mixture was purged with argon for 15 min. and finally, pentamethyldiethyltriamine ligand (0.01 mmol) was added and the mixture was stirred, at 30 ± 1 °C. After the required time, the polymerization was stopped by diluting the reaction mixture with THF. This was followed by precipitation in excess of hexane. It was then redispersed in ~5 ml of THF and was centrifuged to remove any homopolymer to obtain the hybrid material, (3). This was then analyzed by TGA, TEM, and GPC (after degrafting the polymer from the surface).
Surface-Initiated ATRP of Styrene from Magnetite Surface
CuBr (0.070 mmol) and 50 mg of magnetite-ATRP initiator, (2), were added to a dry Schlenk flask equipped with a magnetic pellet and rubber septum. Initially, the mixture was subjected to dynamic vacuum for 1 h. This was followed by the addition of the degassed styrene (25.6 mmol) (50% v/v of DMF) and PMDETA ligand (0.070 mmol) such that the molar ratio of [Styrene]:[Initiator]:[CuBr]:[PMDETA] is 1400/1/4/4. Then, the flask was purged with argon and the contents were stirred in an oil bath maintained at 100 °C, for the required period, toward the preparation of polymer chains of various molecular weights. At the end of the required period, the polymerization was stopped by dilution with THF and precipitated into excess methanol. The precipitate was redispersed in ~5 ml of THF and centrifuged to remove any homopolymer to obtain the hybrid material, (4). This was characterized by FT–IR, TGA, TEM, and GPC (after degrafting the polymer from the surface) from measurements.
Synthesis of Block Copolymer from Polystyrene Grafted-Magnetite Surface
The polymerization was carried out with the initial addition of CuBr (0.140 mmol), 50 mg of polystyrene-magnetite, (4), ethyl methyl ketone, and 1-propanol (in 70/30% v/v) to a dry Schlenk flask equipped with magnetic pellet and rubber septum. This was followed by the addition of the degassed 2-hydroxyethyl methacrylate (27.86 mmol) and purging with argon for 15 min. Finally, pentamethyldiethyltriamine ligand (0.140 mmol) was added and the mixture was stirred, at 30 ± 1 °C, for 48 h. The polymerization was stopped by opening the septum and diluting the reaction mixture with DMF. This was followed by precipitation in 200 ml of hexane to remove the unpolymerized monomer. It was then vacuum dried, (5), and characterized by FT–IR and TGA analyses.
Surface-Initiated ATRP of MMA from Carboxylic Acid Based Magnetite Surface
The anchoring of 2-bromoisobutyric acid, which is a carboxylic acid based ATRP initiator, to magnetite nanoparticles was performed according to the reported literature [43,44] to get carboxylic immobilized magnetite-ATRP initiator (6). CuBr (0.070 mmol), PMDETA ligand (0.070 mmol), and 50 mg of magnetite-ATRP initiator (6) were added to a dry Schlenk flask equipped with a magnetic pellet and rubber septum. It was degassed using the vacuum line. This was followed by the addition of the degassed methyl methacrylate (27.86 mmol) (50 v/v of anisole) such that the molar ratio of [MMA]:[Initiator]:[CuBr]:[PMDETA] is 335/1/1/1. Then, the flask was purged with argon, and was stirred in an oil bath, maintained at 30 °C. After the desired time, the polymerization was stopped by opening the septum and diluting the reaction mixture with THF. This was followed by precipitation in excess of hexane (200 ml). The precipitate was redispersed in ~5 ml of THF and centrifuged to remove any homopolymer, to obtain the hybrid material, (7). This was characterized by FT–IR, TGA, and GPC analyses.
Method
Thermal analysis was performed using a Mettler Toledo STARe(Switzerland) thermal analysis system under flowing nitrogen atmosphere. The number average molecular weights and polydispersity indices of the degrafted polymer were determined by Waters GPC system. A Waters GPC system with 515 pump (New Jersey, USA; with styragel columns HR3, HR4, HR5) along with Millennium v 2.15 data analyses package was used for the determination of number average molecular weight (Mn) and polydispersity index (PDI). THF was used as an eluent (at a flow rate of 1 ml/min) and narrow molecular weight polystyrene standards were used as the standard. All the measurements were carried out at room temperature. Sample detection was done using a Waters 2414 refractive index detector. Transmission electron microscopy was carried out using a JEOL100CX transmission electron microscope at an acceleration voltage of 100 KeV. Samples were prepared by applying a drop of the nanoparticle solution in THF, to a carbon coated copper grid and imaged after drying. Nicolet 6400 instrument was used for FT–IR analysis. Measurement of magnetization was carried out with a vibrating sample magnetometer (EC&G PARC VSM 155).
Results and Discussion
Surface-Initiated Polymerization of BnMA
Surface-initiated polymerization of benzyl methacrylate was carried out from MNs previously modified with bromide terminated initiating group, which in turn were anchored to the surface through phosphonic acid functional group, as shown in Fig. 1. Polymerization was carried out at ambient temperature, in the presence of CuBr/PMDETA complex, without the addition of sacrificial initiator. In order to find the molecular weight and polydispersity (Mn and Mw/Mn) of the grafted P(BnMA) thus formed, the material isolated after the desired period of polymerization was subjected to degrafting by using concentrated HCl, in the presence of THF (mixture was allowed to stir overnight to obtain free polymer), followed by precipitation and drying. Following this, the molecular weight (Mn) and polydispersity index (Mw/Mn) values of P(BnMA) were measured by GPC. These results are summarized in Table 2. The first observation was that, the Mn (GPC) value did not exceed the expected value of 3 × 105 (g/mol), which is calculated using the Eq. (1). The second observation was that polymerization is uncontrolled.
| (1) |
Figure 1.
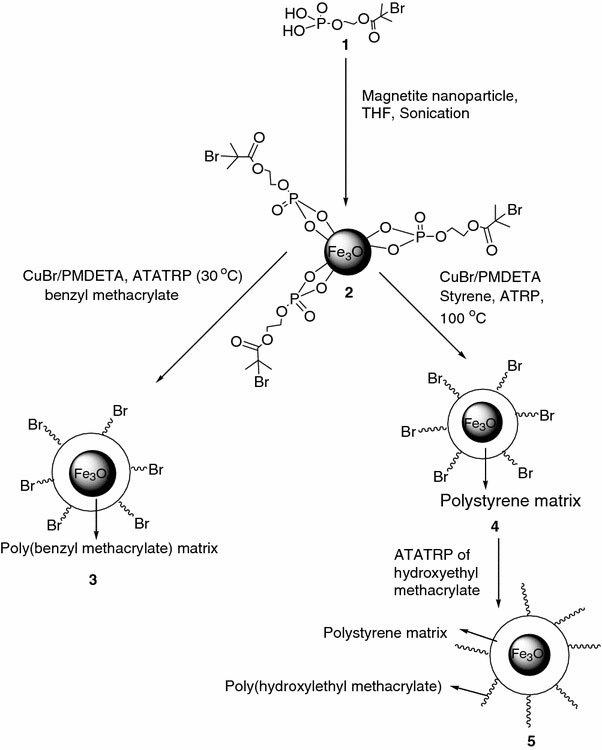
Schematic Illustration depicting the successful grafting of polymer from bromide terminated MNs, through phosphonic acid anchoring group
Table 2.
ATRP of benzyl methacrylate at ambient temperature
| Time (h) | Mn × 103(g/mol) | PDI | % Weight lossa | Grafting densityb | Initiator efficiency |
|---|---|---|---|---|---|
| 1 |
6.3 |
1.47 |
73 |
2.11 |
0.81 |
| 2 |
16.3 |
1.85 |
85 |
1.76 |
0.68 |
| 3 |
22 |
1.80 |
89 |
1.84 |
0.71 |
| 4 |
36.8 |
1.72 |
93 |
1.86 |
0.72 |
| 5 | 46.7 | 1.85 | 95 | 2.06 | 0.79 |
aDetermined by thermogravimetric analysis
bGrafting density calculated using Eq. 2in chains/nm2
This is due to formation of insufficient [Cu(II)], which is expected due to the non-use of sacrificial initiator as well as due to the rapid polymerization of benzyl methacrylate [45]. The initiator efficiency is found to be fairly constant throughout the polymerization but the PDI tends to increase as polymerization proceeds. Control experiments performed from silica nanoparticles demonstrated that the surface-initiated polymerization could be reasonably controlled with higher time of polymerization due to the generation of Cu(II), as established from the kinetics study under similar conditions and this is described in detail in the supporting material. In the control experiments, the PDI decreases with polymerization time suggesting that the generation of Cu(II) helps to bring about control. The variation in PDI with time between the MNs and silica nanoparticles could be due to preferential binding of Cu(II) to the phosphonic acid moiety thus restricting its availability as a persistent radical for the ATRP.
Surface-Initiated Polymerization of Styrene and 2-HEMA Block Copolymer
One of the advantage of ATRP [46,47] is that an alkene monomer like styrene can be polymerized with molecular weight and PDI control, either in bulk or in solution because of reversible Cu(I)/Cu(II) redox process [48]. Polymerizations were carried out in dimethylformide solvent (relative to monomer 50% v/v) as shown in Fig. 1. After the desired time of polymerization, at 100 °C, the PS chains were degrafted from the MNs and analyzed by GPC. These results are summarized in Table 3. In this case, the first observation was that its molecular weight did not exceed the expected molecular weight, which is calculated using Eq. 1 and found to be 1.4 × 105 (g/mol). The second observation was that molecular weight increases regularly with increasing time, which indicated that the molecular weight of the degrafted PS on the surfaces of magnetite can be controlled relatively by the ATRP approach. On the other hand, the PDI of the degrafted PS is considerably broader than that generated by the conventional ATRP of styrene initiated from 2-bromoisobutyrate moiety grafted on magnetite surface, in the presence of sacrificial initiator. The lack of control is due to the low concentration of Cu (II) [47]. This can also be inferred from the lowering of the PDI with increasing polymerization time, as shown in Table 3.
Table 3.
ATRP of styrene at 100 °C
| Time (h) | Mn × 103(g/mol) | PDI | % Weight lossa | Grafting densityb | Initiator efficiency |
|---|---|---|---|---|---|
| 2 |
18 |
2.96 |
64 |
0.48 |
0.18 |
| 4 |
32 |
2.37 |
73 |
0.41 |
0.16 |
| 6 |
41 |
2.54 |
82 |
0.55 |
0.21 |
| 8 |
52 |
2.47 |
91 |
1.01 |
0.39 |
| 10 | 64 | 1.84 | 94 | 1.26 | 0.49 |
aDetermined by thermogravimetric analysis
bGrafting density calculated using Eq. 2in chain/nm2
An amphiphilic diblock polymer based on 2-hydroxylethyl methacrylate was synthesized from the polystyrene monolayer (Fig. 1). This was done to asses the livingness of the PS synthesized via ATRP. The CuBr/PMDETA catalyst of relatively higher concentration was taken, in comparison to the initiating sites, in order to ensure faster initiation in comparison with propagation. This should help in synthesizing the hybrid material with some control over the ratio of hydrophobic to hydrophilic blocks (the block copolymer synthesized will have hydrophobic character due to styrene and hydrophilic character due to 2-hydroxyethyl methacrylate).
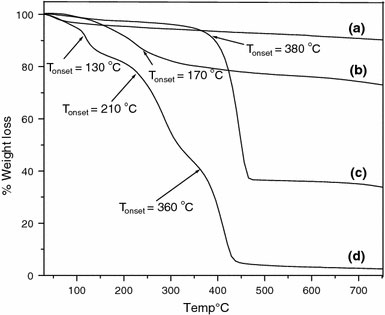
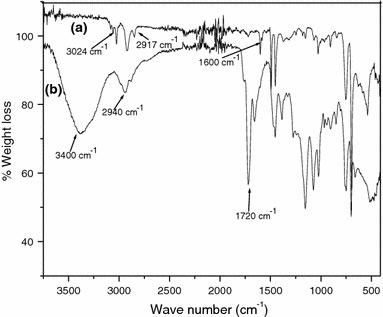
The themogravimetric analysis of MNs as synthesized is shown in Fig. 2a. This shows a 6% weight loss whereas the ATRP initiator anchored magnetite shows a weight loss of around 16%, as shown in Fig. 2b. The polystyrene grafted MNs show 62% weight loss as shown in Fig. 2c, while the poly(styrene-b-2-hydroxyethly methacrylate) shows almost 98% weight loss which is shown in Fig. 2d. The initial weight loss in this case, at ~130 °C can be assigned to the decomposition of initiator moiety on the surface of magnetite and the rapid weight decrease in the second region (the onset at ~210 °C) can be attributed to the decomposition of P(HEMA). The subsequent rapid weight decrease in the third region (the onset at ~360 °C) is attributed to the decomposition of PS, confirming the successful grafting of the block copolymer poly(2-hydroxyethyl methacrylate). The FT–IR of polystyrene grafted MNs display bands at 3,024 cm−1corresponding to the C–H asymmetric stretching of aromatic ring, 2,950 cm−1corresponding to C–H asymmetric aliphatic stretching, and 1,600 cm−1corresponding to C=C stretching of the aromatic ring, as shown in Fig. 3a. The IR spectrum of the block copolymer shows an intense band at 1,720 cm−1, corresponding to the carbonyl group of the poly(2-hydroxyethyl methacrylate) along with the C–H asymmetric stretching at 2,940 cm−1, and the O–H stretching band at 3,400 cm−1, confirming the successful formation of block copolymer as shown in Fig. 3b.
Figure 2.
Thermogravimetric analysis of (a) as synthesized MNs, (b) initiator-immobilized MNs, (c) after grafting PS brush, and (d) after growth of block poly(hydroxyethyl methacrylate)
Figure 3.
FT–IR spectrum of (a) polystyrene grafted MNs and, (b) after polymerization of block poly(hydroxyethyl methacrylate) from the polystyrene grafted MNs
Estimation of Grafting Density of Polymer Grafted MNs using Phosphonic Based Anchoring Group
The graft density (δ) in terms of chains per square nanometer of the surface was calculated from the surface area of magnetite nanoparticles, the molecular weight of the initiator immobilized, and the observed weight loss from the thermogravimetric analysis using the following Eq. 2
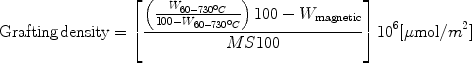 |
(2) |
which is given in the literature [32]. Here, W60–730°C is the weight loss in percentage of immobilized molecules on MNs after grafting, Wmagnetite is the weight loss in percentage for MNs before grafting, M is molar mass of the immobilized molecules on magnetite and S is the surface area of MNs as measured using BET (Brunauer–Emmett–Teller) adsorption isotherms method (found to be 115 m2/g). The ATRP of methyl methacrylate from MNs with the use of sacrificial initiator was reported by Marutani et al. [10]. The use of sacrificial initiator results in the generation of sufficient concentration of the persistent radical, which enables better control of the surface-initiated ATRP. They reported a grafting density of 0.7 chain/nm2, but the big disadvantage associated with this method is the need to remove free polymer, which is formed due to the addition of sacrificial initiator in the polymerization system (by Soxhlet extraction) [19]. The ATRP of poly(ethylene glycol) methyl ether methacrylate from MNs, without the use of sacrificial initiator was reported by Hu et al. [49]. They reported a grafting density of 0.7 chain/nm2. However, they did not report about the control obtained in the polymerizations. The atom transfer radical polymerization of methyl methacrylate from MNs with the initial addition of Cu(II) was reported by Garcia et al. [17]. Cu(II) addition is expected to bring about control in the surface-initiated polymerization by the persistent radical effect. They reported a grafting density of 0.1 chain/nm2. This could be due to the polymerization kinetics being much slower than the conformational rearrangement of the chains at the interface, which may not permit the growth of new chains by restricting the access of the monomer to the initiating sites. One way of testing this hypothesis is to study the grafting density of surface-initiated ATRP, involving monomers with varying rate of propagation. Therefore, we choose to study the grafting density of surface-initiated ATRP involving three different monomers, viz., benzyl methacrylate, styrene, and methyl methacrylate [41]. The results from these study are summarized below.
The thermogravimetric analysis of MNs (in the temperature region room temperature to 750 °C) is shown in Fig. 4a. This shows a 6% weight loss around 100 °C, which is due to the loss of adsorbed water [50]. The ATRP initiator anchored magnetite shows a weight loss of around 16%, as shown in Fig. 4b. From this data, the grafting density was calculated to be ~2.6 molecules/nm2. The P(BnMA) grafted MNs were subjected to thermogravimetric analysis, the results of which is shown in Fig. 5. In this figure, two main weight loss regions can be seen. The first weight loss at around 160 °C can be assigned to the decomposition of the initiator moiety on the surface of magnetite. The significant weight reduction in the second region (the onset at ~240 °C) is attributed to the decomposition of P(BnMA). The grafting density was calculated from the % weight loss along with the corresponding molecular weight data (Table 2) as obtained from GPC measurements. These are summarized in Table 2. The average grafting density following polymerization is found to be 1.92 chains/nm2 and the average initiator efficiency is 0.7 (1.91/2.6). The variation in the initiator efficiency is due to lack of sufficient concentration of Cu(II). However, with time the [Cu(II)] increases and thus fair amount of control is established. The surface-initiated polymerization of MMA, styrene, and n-butyl acrylate, without the use of sacrificial initiator from silica nanoparticles has been reported [51] and this has established that the use of sacrificial initiator is necessary to generate sufficient concentration of Cu(II) for establishing control of the polymerization.
Figure 4.
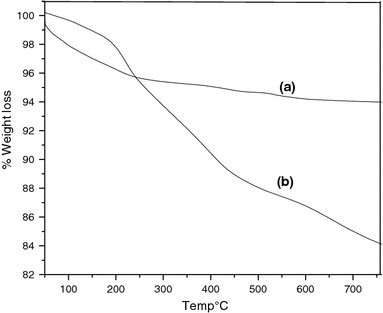
Thermogravimetric analysis of (a) as synthesized MNs, and (b) initiator-immobilized MNs
Figure 5.
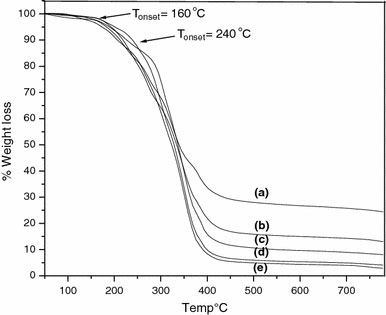
Thermogravimetric analysis of poly(benzyl methacrylate) grafted MNs of molecular weight (a) 6,300 g/mol, (b) 16,300 g/mol, (c) 22,000 g/mol, (d) 36,800 g/mol, and (e) 46,700 g/mol
For the polystyrene grafted MNs, the thermogravimetric analysis data are shown in Fig. 6. The rapid weight decrease in the region (the onset at ~380 °C) is attributed to the decomposition of PS. Thermogravimetric analysis data indicated that the amount of grafted polystyrene increases linearly with increase in molecular weight suggesting that the number of growing chain on the surface of the particle is a constant. The average grafting density as calculated from the TGA data is found to be ~0.74 molecules/nm2, throughout the polymerization time, with an average initiator efficiency of 0.28 as shown in Table 3. The important observation is that not only does the molecular weight but the grafting density and the initiator efficiency also increase with the time of polymerization. The final value of the initiator efficiency is twice of the initial value and the PDI decreases with time of polymerization. Thus this increase in initiator efficiency is due to formation of Cu(II) and indicates that Cu(II) is necessary for better control [40,52]. Thus, it can be concluded that the surface-initiated ATRP of styrene involves slow initiation and is uncontrolled when carried out at 100 °C, resulting in smaller graft density and relatively poor initiator efficiency.
Figure 6.
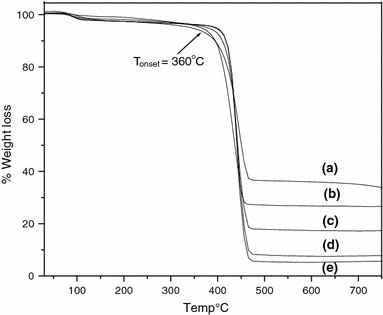
Thermogravimetric analysis of polystyrene grafted MNs of molecular weight (a) 18,000 g/mol, (b) 32,000 g/mol, (c) 41,000 g/mol, (d) 52,000 g/mol, and (e) 64,000 g/mol
Thus, if we compare the grafting density of polymer stabilized MNs using benzyl methacrylate, methyl methacrylate, and styrene, it can found that poly(benzyl methacrylate) resulted in the highest grafting density of about 2 chains/nm2, due to its rapid polymerizing nature. The results are summarized in the Table 4. The polymer graft density of ~2 chains/nm2 is still smaller than the initiator grafting density ~2.6 molecules/nm2. This may be due to the steric blocking of potential initiator sites by the growing chains, which could block the access of the bulky catalyst to the neighboring initiating sites on the magnetite surface [51].
Table 4.
Summary of grafting density results from MNs
| Initiator anchoring chemistry | Monomer | Polymerization | Grafting density in chain(s)/nm2 | Inference |
|---|---|---|---|---|
| Phosphonic acid |
Benzyl methacrylate |
30 °C, ATRP CuBr/PMDETA |
~2.0 |
Fastest polymerization |
| Phosphonic acid |
Methyl methacrylate |
30 °C, ATRP CuBr/PMDETA |
~1.0 |
Faster polymerization |
| Phosphonic acid | Styrene | 100 °C, ATRP CuBr/PMDETA | ~0.7 | Slow polymerization |
Dispersion of Phosphonic Acid Based Polymer Stabilized MNs
The MNs were suspended in chloroform before and after the grafting of the P(BnMA) brush to study the effect on their dispersion, as shown in the photo images of Fig. 7. It can be seen from Fig. 7a, b that MNs and initiator anchored MNs settle down quickly, in chloroform. It can also be seen from the Fig. 7c that the addition of 35 mg of P(BnMA) ofMn = 17,000 to 15 mg of MNs does not result in the formation of stable dispersion even after a waiting period of 1 week. In this case, it was expected that a physisorbed layer of P(BnMA) would provide some stability to the MNs dispersion. The photoimages of MNs (2.5 mg/ml in CHCl3) from which a brush of P(BnMA) was grown is shown in the Fig. 7d. The formation of stable dispersion, in this case, is attributed to the presence of P(BnMA) brush. This particular solution (2.5 mg/ml) was further diluted to 1.25 mg/ml (Fig. 7e), 0.6 mg/ml (Fig. 7f), 0.3 mg/ml (Fig. 7g), and 0.15 mg/ml (Fig. 7h). All these solutions exhibited dispersive stability over a observation period of 1 week. The color gradient observed in Fig. 7d–f is due to the concentration change (progressive dilution). The P(BnMA) grafted MNs were suspended in a variety of solvents namely toluene, acetone, tetrahydrofuran, dichloromethane, and ethyl acetate/water mixture. The photoimages of these are shown in Fig. 8a–d. It is clear from these images that P(BnMA) grafted MNs forms stable dispersion in the above solvents. The blue layer observed in the Fig. 8e is due to the dissolution of Cu(II) present in the polymer layer (formed due to ATRP) in the aqueous layer. The “as synthesized MNs” and initiator-immobilized MNs settled down in H2O–CHCl3mixture, as shown in Fig. 9a, b, respectively, but the polystyrene grafted MNs were partially dispersed in H2O–CHCl3mixture as shown in Fig. 9c. It could be seen in this case as well that PS grafted MNs forms a stable dispersion, especially when diluted sufficiently as shown in Fig. 9d. The poly(2-hydroxylethyl methacrylate-b-styrene) grafted MNs does not form a dispersion in CHCl3in which it is insoluble but it disperses well in DMF in which it is soluble as shown in Fig. 9e, f, respectively.
Figure 7.

Photoimage of polymer grafted MNs in chloroform solvent (a) as synthesized MNs, (b) MNs after grafting of the initiator, (c) poly(benzyl methacrylate) physically mixed with MNs, (d) the poly(benzyl methacrylate) grafted on MNs and, the poly(benzyl methacrylate) with subsequent dilution in chloroform solvent is shown in (e–h)
Figure 8.

Photoimages of poly(benzyl methacrylate) grafted MNs in various organic solvent (a) toluene, (b) acetone, (c) tetrahydrofuran, (d) dichloroform, and (e) ethyl acetate in water
Figure 9.

Photoimages of polystyrene grafted MNs in CHCl3/Water mixture (a) as synthesized MNs, (b) initiator anchored MNs, (c) polystyrene grafted MNs, (d) polystyrene grafted MNs in complete CHCl3solvent, (e) poly(hydroxyethyl methacrylate-block-styrene) grafted MNs in CHCl3solvent, and (f) poly(hydroxyethyl methacrylate-block-styrene) grafted MNs in DMF solvent
The “as synthesized” magnetite nanoparticle shows agglomeration of the particles as shown in the TEM image (Fig. 10a). The formation of stable dispersion when poly(benzyl methacrylate) is grafted to the MNs is also evident from the TEM image as shown in Fig. 10b. The polystyrene grafted MNs forms a stable dispersion in THF, as shown in the TEM images of Fig. 10c, d, respectively. Saturation magnetization of the MNs (after immobilizing phosphonic acid based polymer).
Figure 10.
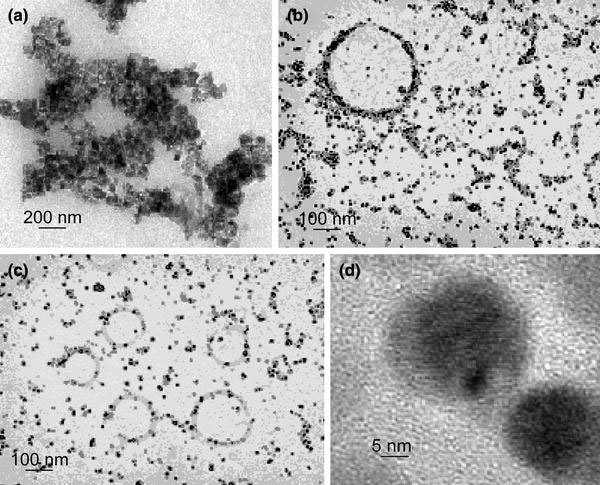
Transmission electron microscopy image of (a) as synthesized MNs, (b) poly(benzyl methacrylate) grafted MNs, (c) polystyrene grafted MNs lower magnification, and (d) higher magnification
The unprotected nanoparticles are well known for their aggregation due to Oswald ripening. This also results in the reduction of the surface energy. When subjected to vibrating sample magnetometer analysis, “as synthesized” MNs show the saturation magnetization value of 1.8 emu/g at ambient temperature, as shown in Fig. 11a. This saturation magnetization value of the nanoparticles is reduced to 0.8 emu/g (of the magnetic material) when initiator is immobilized on the surface of the particle as shown in Fig. 11b. When a polymer is grown from the immobilized surface, the saturation magnetization value is 0.3 and 0.2 emu/g (of the magnetic material) for 1 and 2 h polymerization, respectively as shown in Fig. 11c, d. Upon introduction of the organic layer (initiator or polymer) around MNs, the saturation magnetization per gram of magnetite (as opposed to per gram of composite) is reduced to ~1 emu/g. This may be due to orientation of the magnetic domains, which are restricted in the composite.
Figure 11.
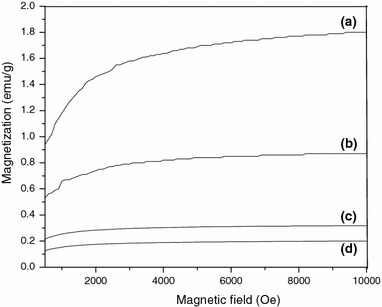
Field dependent magnetization at 25 °C for (a) as synthesized MNs, (b) initiator-immobilized MNs, (c) p(BnMA) grafted MNs after polymerization time of 1 and (d) 2 h
Surface-Initiated Polymerization of MMA from Carboxylic Acid Based Surface Anchored Initiator
To compare the effectiveness of phosphonic acid group as the anchor group, control experiments were performed using carboxylic acid as the anchor group, as shown in Fig. 12. In this case, the ambient temperature ATRP of methyl methacrylate was carried out using CuBr/PMDETA catalytic system without using a sacrificial initiator. It may be noted that the results from the ATATRP of methyl methacrylate from MNs using phosphonic acid anchor group were already reported by us [41]. After the polymerization for the desired period, the poly(methyl methacrylate) was degrafted from the surface of the MNs and the number molecular weight (Mn) and polydispersity index (PDI) were determined as measured by GPC. The results from these experiments are summarized in Table 5. From these results, it is clear that the Mn increases with polymerization time as expected and the PDI decreases, which is due to the generation of higher [Cu(II)] with time. However, the grafting density is poor and is seen to decrease with the polymerization time. Thus –COOH appears to be a poorer anchoring group in comparison with –POOH. The ATATRP of MMA from the tertiary bromide initiating group is much slower in comparison with BnMA as reported earlier by our group [53], and hence the P(MMA) anchored to the MNs could be displacing the initiator molecules anchored to the MNs through a relatively weak, carboxylic acid group.
Figure 12.
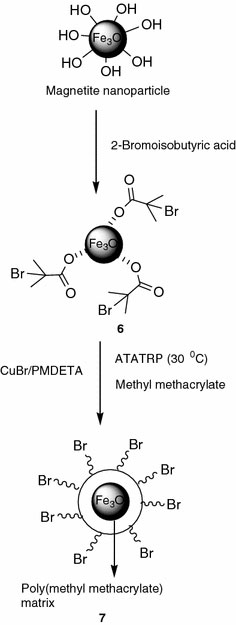
Schematic illustration depicting the grafting of poly(methyl methacrylate) on to the surface of MNs from a carboxylic acid based ATRP initiator
Table 5.
ATRP of methyl methacrylate at ambient temperature
| Time (h) | Mn × 10 (g/mol) | PDI | % Weight lossa | Grafting densityb | Initiator efficiency |
|---|---|---|---|---|---|
| 3 |
23 |
1.73 |
52 |
0.19 |
0.02 |
| 6 |
32 |
1.91 |
54 |
0.15 |
0.02 |
| 9 |
44 |
1.78 |
56 |
0.12 |
0.01 |
| 12 |
56 |
1.45 |
58 |
0.10 |
0.01 |
| 15 | 71 | 1.44 | 64 | 0.11 | 0.01 |
aDetermined by thermogravimetric analysis
bGrafting density calculated using Eq. 2in chain/nm2
The MNs were subjected to thermogravimetric analysis, the results of which are shown in Fig. 13a. The initial weight loss observed, in the vicinity of 100 °C, is due to the continued loss of water. The MNs were also analyzed by thermogravimetric analysis, following the anchoring of the ATRP initiator, the result of which is shown in Fig. 13b. TheTonsetin this case is around 120 °C. The graft density, δ, of the immobilized initiator molecules was calculated using Eq. 2from the thermo-gravimetric analysis data and was found to be 8.6 molecules/nm2. For the polymer grafted MNs, three main weight loss regions are observed in thermogravimetric analysis, as shown in Fig. 13c. The first weight loss at 120 °C, can be assigned to the decomposition of initiator moiety on the surface of magnetite. The subsequent rapid weight decrease in the second region (the onset at ~160 °C) and the significant weight reduction in the third region (the onset at ~240 °C) are attributed to the decomposition of P(MMA). The grafting density as calculated from the TGA data is found to be nearly a constant value of ~0.13 molecules/nm2throughout the polymerization time with an average initiator efficiency of 0.01 as shown in Table 5.
Figure 13.
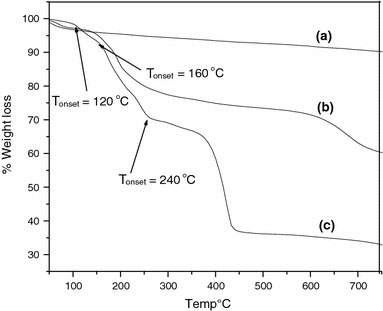
Thermogravimetric analysis of (a) as synthesized MNs, (b) ATRP Initiator anchored MNs, and (c) poly(methyl methacrylate) grafted MNs
Comparison of Initiator Efficiency with Methyl Methacrylate Polymerization from MNs
A comparison of the initiator efficiency for the polymerization of MMA from MNs for various anchoring chemistry is compared in Table 6. It can be seen from this data that the phosphonic acid based anchoring chemistry is superior to chlorosilane, triethoxysilane, and carboxylic acid anchoring chemistry. This is perhaps due to the formation of a stable M–O–P bond in this case [41], which helps in stable dispersion of the nanoparticles as shown in Fig. 14. The other anchoring chemistry, such as chlorosilane anchoring moiety resulted in lower grafting density possibly due to the low stability of M–O–Si bond [54]. In case of triethoxysilane moiety and the ATRP initiator is anchored in two steps [55] that could lead to gelation of the particle and lower grafting density. Even though the carboxylic acid anchoring resulted in very high grafting density [56] for the initiator immobilization but once the polymerization is performed it resulted in the significant lowering of polymer graft density and hence the least initiator efficiency. This could be due to the replacement of the initiator by the ligand as well as by the monomer and the polymer as the interaction between the carboxylic acid group and MNs is weak van der waals.
Table 6.
ATRP of methyl methacrylate from MNs—comparison of grafting density for various anchoring chemistry
| Anchoring chemistry | Grafting density after immobilizing initiator (molecules/nm2) | Grafting density after polymerization of MMA (chain/nm2) | Average initiator efficiency after polymerization |
|---|---|---|---|
| Phosphonic acid |
2.6 |
1.0 |
0.38 |
| Choro silane |
1.5 |
0.1 |
0.06 |
| Triethoxy silane |
5.6 |
0.1 |
0.01 |
| Carboxylic acid | 8.6 | 0.1 | 0.01 |
Figure 14.
The photoimages to show how polymer acts as steric stabilizer for stable dispersion of nanoparticle
Conclusions
Polymer brushes [P(BnMA), PS and P(S-b-2-HEMA)] were grown from the surface of magnetite nanoparticles using ATRP. ATRP from the surface was enabled by initiator with phosphonic acid as well as carboxylic acid anchoring groups. It was inferred that phosphonic acid anchoring system can play a better role in modifying the surface when compared with carboxylic acid anchoring system. To synthesize the polymer brush of the highest grafting density, it is preferable to use the fastest polymerization system i.e., benzyl methacrylate polymerization at ambient temperature. Block copolymerization of 2-hydroxylethyl methacrylate was carried out from the polystyrene monolayer, without using sacrificial initiator, and this confirms the controlled “living” nature of the polymerization. The polymer grafted nanoparticles (stabilized by phosphonic acid anchoring moiety) form stable dispersions in various solvents of interest. Thus the surface-initiated polymerization from the magnetite nanoparticles, without the addition of the sacrificial initiator as well as without the initial addition of Cu(II), results in high grafting density provided the fastest polymerizing system is used. This suggests that under the conditions of the experiment, a polymer brush with higher grafting density can be obtained if polymerization kinetics are faster than conformational rearrangement associated with the grafting chain. This result requires detailed modeling and the same is under study.
(See supplementary material 1)
Supplementary Material
The online version of this article (doi:10.1007/s11671-009-9365-z) contains supplementary material, which is available to authorized users.
Acknowledgments
We thank the Council for Scientific and Industrial Research (CSIR) for sanctioning the project.
References
- Klabunde KJ. Nanoscale Materials in Chemistry. Wiley-Interscience, New York; 2001. [DOI] [Google Scholar]
- Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Nat. 2000. p. 410. COI number [1:CAS:528:DC%2BD3cXis1Gmt7s%3D] [DOI] [PubMed]
- Torchilin VP. Eur. 2000. p. S81. COI number [1:CAS:528:DC%2BD3cXntF2gtbc%3D] [DOI] [PubMed]
- Häfeli UO. Int. 2000. p. 19. [DOI]
- Xiang JJ, Tang JQ, Zhu SG, Nie XM, Lu HB, Shen SR, Li XL, Tang K, Zhou M, Li GY. J. 2003. p. 803. COI number [1:CAS:528:DC%2BD3sXotlSmt70%3D] [DOI] [PubMed]
- Jordan A, Scholz R, Maier-Hauff K, Johannsen M, Wust P, Nadobny J, Schirra H, Schmidt H, Deger S, Loening S, Lanksch W, Felix R. J. 2001. p. 18. [DOI]
- Turkevich J, Kim G. Science. 1970. p. 873. COI number [1:CAS:528:DyaE3cXkvFWrt70%3D]; Bibcode number [1970Sci...169..873T] [DOI] [PubMed]
- Turkevich J, Stevenson PC, Hillier J. Discuss. 1951. p. 55. [DOI]
- Chow MK, Zukoski CF. J. 1994. p. 97. COI number [1:CAS:528:DyaK2cXjtlais7o%3D] [DOI]
- Marutani E, Yamamoto S, Ninjbadkar T, Tsuji Y, Fukuda T, Takano M. Polymer. 2004. p. 2231. COI number [1:CAS:528:DC%2BD2cXhs12jt70%3D] [DOI]
- Caruso F. Adv. 2001. p. 11. COI number [1:CAS:528:DC%2BD3MXhtVyktLg%3D] [DOI]
- Rajesh R, Brittain WJ. Macromolecules. 2007. p. 6217. Bibcode number [2007Mamol..40.6217R] [DOI]
- Prucker O, Rühe J. Macromolecules. 1998. p. 592. COI number [1:CAS:528:DyaK1cXlt1Slug%3D%3D]; Bibcode number [1998Mamol..31..592P] [DOI]
- Auroy P, Auvray L, Leger L. Physica A. 1991. p. 269. COI number [1:CAS:528:DyaK3MXls1Sgsr4%3D]; Bibcode number [1991PhyA..172..269A] [DOI] [PubMed]
- Milner ST. Science. 1991. p. 968. [DOI] [PubMed]
- Ohno K, Koh K, Tsujii Y, Fukuda T. Macromolecules. 2002. p. 8989. COI number [1:CAS:528:DC%2BD38XnvVOisbs%3D]; Bibcode number [2002Mamol..35.8989O] [DOI]
- Garcia I, Zafeiropoulos NE, Janke A, Tercjak A, Eceiza A, Stamm M, Mondragon I. J. 2007. p. 925. COI number [1:CAS:528:DC%2BD2sXitVars7w%3D] [DOI]
- Ohno K, Morinaga T, Koh K, Tsujii Y, Fukuda T. Macromolecules. 2005. p. 2137. COI number [1:CAS:528:DC%2BD2MXhtFSntrw%3D]; Bibcode number [2005Mamol..38.2137O] [DOI]
- Raghuraman GK, Ruhe J, Dhamodharan R. J. 2008. p. 415. COI number [1:CAS:528:DC%2BD1cXpslCjsw%3D%3D] [DOI]
- Mandal TK, Fleming MS, Walt DR. Nano Lett. 2002. p. 3. Bibcode number [2002NanoL...2....3M] [DOI]
- Flesch C, Delaite C, Dumas P, Bourgeat-Lami E, Duguet E. J. 2004. p. 6011. COI number [1:CAS:528:DC%2BD2cXhtVCjtLrM] [DOI]
- Ramakrishnan A, Dhamodharan R, Ruhe J. Macromol. 2002. p. 612. COI number [1:CAS:528:DC%2BD38Xmt1CisLs%3D] [DOI]
- Samuel JDJS, Dhamodharan R, Ruhe J. Macromol. 2002. p. 277. [DOI]
- Raghuraman GK, Dhamodharan R, Prucker O, Rühe J. Macromolecules. 2008. p. 873. COI number [1:CAS:528:DC%2BD1cXntVenuw%3D%3D]; Bibcode number [2008Mamol..41..873R] [DOI]
- Guifeng L, Jinda F, Rong J, Yong G. Chem. 2004. p. 1835. [DOI]
- Fadeev AY, Helmy R, Marcinko S. Langmuir. 2002. p. 7521. COI number [1:CAS:528:DC%2BD38XmtlGksLo%3D] [DOI]
- Stephen M, Roy H, Fadeev AY. Langmuir. 2003. p. 2752. [DOI]
- Matsuno R, Yamamoto K, Otsuka H, Takahara A. Chem. 2003. p. 3. COI number [1:CAS:528:DC%2BD38XptFCht78%3D] [DOI]
- Matsuno R, Yamamoto K, Otsuka H, Takahara A. Macromolecules. 2004. p. 2203. COI number [1:CAS:528:DC%2BD2cXht1Gisbc%3D]; Bibcode number [2004Mamol..37.2203M] [DOI]
- Daou TJ, Begin-Colin S, Greneche JM, Thomas F, Derory A, Bernhardt P, Legare P, Pourroy G. Chem. 2007. p. 4494. COI number [1:CAS:528:DC%2BD2sXovFOgur4%3D] [DOI]
- Zhao H, Kang X, Liu L. Macromolecules. 2005. p. 10619. COI number [1:CAS:528:DC%2BD2MXht1ajsrbN]; Bibcode number [2005Mamol..3810619Z] [DOI]
- Bartholome C, Beyou E, Bourgeat-Lami E, Chaumont P, Zydowicz N. Macromolecules. 2003. p. 7946. COI number [1:CAS:528:DC%2BD3sXnsVehu7o%3D]; Bibcode number [2003Mamol..36.7946B] [DOI]
- Li C, Benicewicz BC. Macromolecules. 2005. p. 5929. COI number [1:CAS:528:DC%2BD2MXltVylsLs%3D]; Bibcode number [2005Mamol..38.5929L] [DOI]
- Ramakrishnan A, Dhamodharan R. Macromolecules. 2003. p. 1039. COI number [1:CAS:528:DC%2BD3sXnt1Slug%3D%3D]; Bibcode number [2003Mamol..36.1039R] [DOI]
- Li D, Sheng X, Zhao B. J. 2005. p. 6248. COI number [1:CAS:528:DC%2BD2MXjtVOjsbs%3D] [DOI] [PubMed]
- Matyjaszewski K, Xia J. Chem. 2001. p. 2291. [DOI] [PubMed]
- Vivek AV, Dhamodharan R. J. 2007. p. 3818. COI number [1:CAS:528:DC%2BD2sXhtV2qt7zM] [DOI]
- Vestal CR, Zhang ZJ. J. 2002. p. 14312. COI number [1:CAS:528:DC%2BD38XosFWhtbs%3D] [DOI] [PubMed]
- Timothy VW, Timothy EP. J. 2001. p. 7497. [DOI] [PubMed]
- von Werne T, Patten TE. J. 1999. p. 7409. [DOI]
- Babu K, Dhamodharan R. Nano. 2008. p. 109. COI number [1:CAS:528:DC%2BD1cXlslejt7c%3D] [DOI]
- Fu GD, Shang Z, Hong L, Kang ET, Neoh KG. Macromolecules. 2005. p. 7867. COI number [1:CAS:528:DC%2BD2MXntVCgsrk%3D]; Bibcode number [2005Mamol..38.7867F] [DOI]
- Wang Y, Teng X, Wang JS, Yang H. Nano Lett. 2003. p. 789. COI number [1:CAS:528:DC%2BD3sXjsVOjtbw%3D]; Bibcode number [2003NanoL...3..789W] [DOI]
- Gelbrich T, Feyen M, Schmidt AM. Macromolecules. 2006. p. 3469. COI number [1:CAS:528:DC%2BD28XjtFWisrg%3D]; Bibcode number [2006Mamol..39.3469G] [DOI]
- Munirasu SM, Dhamodharan R. J. 2004. p. 1053. COI number [1:CAS:528:DC%2BD2cXhsFagt7s%3D] [DOI]
- Wang JS, Matyjaszewski K. J. 1995. p. 5614. COI number [1:CAS:528:DyaK2MXls12js7g%3D] [DOI]
- Coessens V, Pintauer T, Matyjaszewski K. Prog. 2001. p. 337. COI number [1:CAS:528:DC%2BD3MXjvF2itr4%3D] [DOI]
- Wade AB, Matyjaszewski K. Prog. 2007. p. 93. [DOI]
- Hu FX, Neoh KG, Cen L, Kang ET. Biomacromolecules. 2006. p. 809. COI number [1:CAS:528:DC%2BD28XhtlOqsr0%3D] [DOI] [PubMed]
- Blomberg S, Ostberg S, Harth E, Bosman AW, van Horn B, Hawker CJ. J. 2002. p. 1309. COI number [1:CAS:528:DC%2BD38XivVeqsbY%3D] [DOI]
- Pyun J, Jia S, Kowalewski T, Patterson GD, Matyjaszewski K. Macromolecules. 2003. p. 5094. COI number [1:CAS:528:DC%2BD3sXks12mt70%3D]; Bibcode number [2003Mamol..36.5094P] [DOI]
- Ramakrishnan A, Dhamodharan R, Jurgen R. Macromol. 2002. p. 612. COI number [1:CAS:528:DC%2BD38Xmt1CisLs%3D] [DOI]
- Munirasu S, Ruhe J, Dhamodharan R. J. 2006. p. 2848. COI number [1:CAS:528:DC%2BD28XktVOhtb8%3D] [DOI]
- Stephen M, Fadeev AY. Langmuir. 2004. p. 2270. [DOI] [PubMed]
- Raghuraman GK, Dhamodharan R. J. 2006. p. 2018. COI number [1:CAS:528:DC%2BD28XosVegsbk%3D] [DOI] [PubMed]
- White MA, Johnson JA, Koberstein JT, Turro J. J. 2006. p. 11356. COI number [1:CAS:528:DC%2BD28Xot1Kgs7w%3D] [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The online version of this article (doi:10.1007/s11671-009-9365-z) contains supplementary material, which is available to authorized users.