

Abstract
TiO2nanostructures with fascinating morphologies like cubes, spheres, and rods were synthesized by a simple microwave irradiation technique. Tuning of different morphologies was achieved by changing the pH and the nature of the medium or the precipitating agent. As-synthesized titania nanostructures were characterized by X-ray diffraction (XRD), UV–visible spectroscopy, infrared spectroscopy (IR), BET surface area, photoluminescence (PL), scanning electron microscopy (SEM) and transmission electron microscopy (TEM), and atomic force microscopy (AFM) techniques. Photocatalytic dye degradation studies were conducted using methylene blue under ultraviolet light irradiation. Dye degradation ability for nanocubes was found to be superior to the spheres and the rods and can be attributed to the observed high surface area of nanocubes. As-synthesized titania nanostructures have shown higher photocatalytic activity than the commercial photocatalyst Degussa P25 TiO2.
Keywords: Nanocubes, Nanorods, Nanospheres, Photocatalytic activity, Microwave irradiation, Dye degradation
Introduction
Nanomaterials of transition metal oxides have attracted a great deal of attention from researchers in various fields due to their numerous technological applications [1-4]. Among them, nanocrystalline titania has been attracting increasing attention due to its fascinating properties and potential applications. Titanium dioxide is a versatile material which is being investigated extensively due to its unique optoelectronic and photochemical properties such as high refractive index, high dielectric constant, excellent optical transmittance in the visible and near IR regions as well as its high performance as a photocatalyst for water splitting and degradation of organics [5]. With a band gap of 3.0–3.3 eV, titanium dioxide has been photocatalytically active only under ultraviolet light (wavelength λ < 400 nm) [6]. Titanium dioxide mainly exists in three crystalline phases: anatase, rutile, and brookite [7]. Among the three crystalline forms, anatase titanium dioxide is attracting more attention for its vital use as pigments [8], gas sensors [9], catalysts [10,11], photocatalysts [12-14] in response to its application in environmentally related problems of pollution control and photovoltaics [15]. The properties and catalytic activities of titania strongly depend upon the crystallinity, surface morphology, particle size, and preparation methods. The increased surface area of nanosized TiO2 particles may prove beneficial for the decomposition of dyes in aqueous media. Ohtani et al.[16] proposed that high photocatalytic activity of titania can be achieved by imparting large surface area to adsorb substrates and by making high crystallinity to minimize the photoexcited electron-hole recombination rate. In general anatase titania is observed to be more active compared to its rutile phase. This difference in activity can be due to the high electron-hole recombination rate observed in rutile titania. Many synthetic methods have been reported for the preparation of nanotitania, including sol–gel reactions [17-19], hydrothermal reactions [20,21], non-hydrolytic sol–gel reactions [22,23], template methods [24-26], reactions in reverse micelles [27], and microwave irradiation. Nanotitania with various morphologies and shapes such as nanorods [28], nanotubes [29,30], nanowires [31,32], and nanospheres [33,34] can be produced depending upon the synthetic method used. These different morphologies have different photocatalytic activities. In the present work, we report a simple microwave method to synthesize phase pure anatase and rutile nanotitania with different morphologies viz., cubes, spheres, and rods. Photocatalytic activity studies of the synthesized samples were carried out using the dye, methylene blue in aqueous solution under ultraviolet light irradiation. The photoluminescence (PL) features of the synthesized titania nanostructures were also compared in the present study.
Experimental
Materials
All reagents were purchased from Merck, Germany. Titanium trichloride (15 wt% TiCl3, 10 wt% HC1) was used as the titanium precursor. NH4OH (1.5 M), NaCl (5.0 M), and NH4Cl (5.0 M) were employed for the synthesis. A typical microwave oven (Whirlpool, 1200W) operating at a frequency of 2,450 MHz was used for the synthesis.
Synthesis of TiO2Nanostructures
A general synthetic strategy adopted for the synthesis of titania nanostructures was using TiCl3as Ti precursor by varying the precipitating agents under different pH conditions. The precipitated sol was irradiated in a microwave oven in on and off mode for different durations depending upon the precipitation rate in each case. The completion of the reaction is checked by noting the color change (blue to colorless) of the reaction mixture. The white precipitate formed in each case was aged for 24 h and washed thoroughly with distilled water. The precipitated titania was then dried in an air oven at 100 °C and further calcined in a muffle furnace at 400 °C for 4 h.
In the case of sample 1 (S1) TiCl3 (20.0 mL) was added drop by drop to 200 mL of 1.5 M NH3 (pH = 11) solution [35] and the irradiation was done for 20 min for complete precipitation. In sample 2 (S2), TiCl3 (5.0 mL) was added dropwise with continuous stirring to 200 mL of 5.0 M NaCl solution (pH = 7) [36] and the reaction mixture was irradiated for 60 min for complete precipitation. In sample 3 (S3), TiCl3 (5.0 mL) was added dropwise to 200 mL of 5.0 M NH4Cl solution (pH = 5.9) and irradiated in a similar manner as in the previous case for 60 min.
Characterization of Titania Nanostructures
The X-ray diffraction (XRD) patterns of the titania were recorded on a Brucker D8 advance diffractometer with CuKαradiation. The crystallite size of TiO2was calculated using Debye Scherrer equation,L = kλ/(βcosθ), whereLis the average crystallite size, λ is the wavelength of the radiation, θ is the Bragg’s angle of diffraction, β is the full width at half maximum intensity of the peak andkis a constant usually applied as ~0.89. Scanning electron microscopic images were taken on a JEOL JSM-5600 SEM equipped with energy dispersive X-ray analysis (EDX). High resolution transmission electron micrographs and electron diffraction patterns were recorded using a JEOL JEM-3010 HRTEM microscope at an accelerating voltage of 300 kV. The TEM specimens were prepared by drop casting the sample on the surface of the carbon coated copper grid. The tapping mode AFM images of the samples deposited on a mica sheet were taken using Nanoscope-IV scanning probe microscope. The BET surface area, pore size distribution, and pore volume of the samples were measured on a Micromeritics ASAP 2010 analyzer based on N2adsorption at 77 K in the pressure range from 0.1 to 760 mmHg. The pore size distribution was calculated by the Barrett Joyner Halenda (BJH) method. IR spectra was recorded using Shimadzu 8400S FTIR spectrophotometer in the range of 400–4,000 cm−1. The ultraviolet–visible absorption (UV–vis) spectra were recorded using a UV-2450 Shimadzu UV–visible spectrophotometer. The photoluminescence (PL) spectral measurements were made using Perkin Elmer LS-55 luminescence spectrometer at an excitation wavelength of 325 nm.
Photocatalytic Activity Measurements
Photocatalytic activity of TiO2 was evaluated by the degradation of the dye, methylene blue (MB) in aqueous solution under ultraviolet light irradiation in the presence of as-synthesized TiO2 and the commercial Degussa P25 TiO2. The changes in the concentrations of methylene blue in the aqueous solution were examined by absorption spectra measured on a UV-2450 Shimadzu UV–visible spectrophotometer. Before examining the photocatalytic activity for degradation of aqueous methylene blue, TiO2 sol was prepared. About 100 mg of the synthesized TiO2 was dispersed ultrasonically in 50 mL of deionized water. For photodegradation experiments, 50 mL of 4 × 10−5 M methylene blue solution was added to the as-synthesized titania sol in a quartz reactor. To maximize the adsorption of the dye onto the TiO2 surface, the resulting mixture was kept in the dark for 30 min under stirring conditions [37]. The solution was then irradiated for 180 min using a mercury lamp (100 W, Toshiba SHLS-1002 A). The degradation of the dye was monitored by measuring the absorption maximum of methylene blue at 661 nm at 30 min intervals of reaction.
Results and Discussion
X-ray Diffraction Studies
The X-ray diffraction (XRD) patterns (Fig. 1) of the TiO2particles show that anatase phase is formed when NH4OH (S1) is used whereas the formation of rutile phase is observed when the medium is NaCl (S2) and NH4Cl (S3). The average crystallite size of the S1, S2, and S3 are 12, 10, and 21 nm, respectively. XRD powder pattern of S1 corresponds to anatase phase with lattice constants, a = 3.777 Å and c = 9.501 Å as reported in JCPDS file no. 89-4921. All the peaks in S2 and S3 can be readily indexed to rutile phase with lattice constants a = 4.608 Å, c = 2.973 Å and a = 4.548 Å, c = 2.946 Å, respectively, as reported in JCPDS files, no. 76-0319 and 88-1173. The d-spacing from HRTEM is consistent with the d-spacing from XRD results. The absence of any other peak indicates the phase purity of the synthesized titania.
Figure 1.
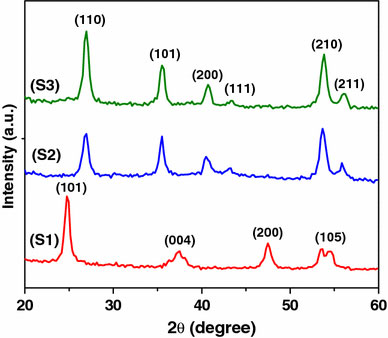
XRD powder patterns of titania synthesized in different medium (S1) NH4OH, (S2) NaCl, and (S3) NH4Cl
BET Surface Area Analysis
Figure 2 shows the N2 adsorption and desorption isotherms of the three titania samples with their corresponding pore size distribution (BJH method) (inset). Type IV isotherm observed with a clear hysteresis at relatively low pressure indicates the mesoporous nature of the sample S1 [38]. Pore size distribution also confirms the mesoporous nature indicating an average pore size around 4 nm. For samples S2 and S3 the hysteresis moves to relatively high pressure indicating a still narrower pore size and is around 2.5 and 2 nm, respectively, as observed from pore size distribution. The crystallite size, BET surface area, pore size, and pore volume values are summarized in Table 1. The surface area of S1, S2, and S3 are 372, 77, and 34 m2 g−1, respectively.
Figure 2.
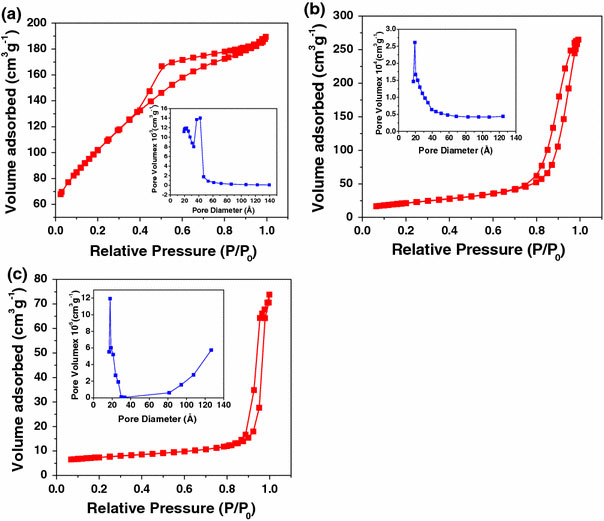
N2adsorption–desorption isotherms and pore size distribution of the synthesized nanotitania (inset)aS1,bS2, andcS3
Table 1.
Textural analysis of mesoporous TiO2Nanostructures
| Sample code | Crystallite size from XRD (nm) | BET surface area (m2g−1) | Pore size (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|---|
| S1 |
12 |
372 |
4 |
0.37 |
| S2 |
10 |
77 |
2.5 |
0.18 |
| S3 | 21 | 34 | 2 | 0.10 |
Electron Microscopic Analysis
SEM images of titania samples are given in Fig. 3. S1 (a), S2 (b), and S3 (c) show a cube-like morphology, spherical morphology, and rod-like morphology, respectively. Agglomerated particles are observed in the SEM images [39]. The high resolution TEM images of the TiO2 nanoparticles synthesized under various reaction conditions are shown in Fig. 4. TEM image of S1 (a) shows the formation of nanocubes with particle size around 25 nm. The HRTEM image (b) shows lattice fringes of the anatase phase. The fringes with d = 0.34 nm match with that of the (101) crystallographic plane of anatase titania. The selected area electron diffraction pattern in the inset of the A confirms that the sample S1 is a single crystalline anatase phase. The high surface area observed for the sample S1 may be due to the highly porous nature of the cubes. Since the sample S1 is not an ordered mesoporous system, mesopores cannot be viewed clearly from HRTEM images. Sample S2 (c) shows the formation of nanospheres of average crystallite size around 8 nm. Corresponding selected area electron diffraction pattern is shown in the inset. The pattern indicates the polycrystalline nature of the sample. Lattice image (d) of these nanospheres shows lattice fringes of the rutile phase with d = 0.32 nm, which matches well with that of (110) plane of rutile titania. Sample S3 (e) shows the formation of nanorods with an average aspect ratio of around 4 nm. Corresponding SAED pattern indicates a polycrystalline nature, which may be due to the diffraction in a bunch of nanorods. The HRTEM image (f) of the rutile nanorods show clear lattice fringes of the rutile phase with d = 0.32 nm, which matches with that of the (110) plane of rutile titania. The TEM results reveal that nano TiO2 with different morphologies like cubes, spheres, and rods can be effectively synthesized by varying the pH in an appropriate media. Figure 5 shows the tapping mode AFM images of the titania cubes (S1), spheres (S2), and rods (S3) which is in good agreement with that of the TEM results [40].
Figure 3.

SEM images of samples S1 (a), S2 (b) and S3 (c)
Figure 4.

HRTEM images of:aS1 (nanocubes) andbcorresponding lattice;dS2 (nanospheres) andecorresponding lattice;gS3 (nanorods) andhcorresponding lattice image. The inset of the figurea,dandgrepresents the selected area electron diffraction pattern of the titania nanostructures
Figure 5.

Tapping AFM micrographs of S1 (a), S2 (b), and S3 (c)
Spectroscopic Analysis
The FTIR spectra of S1, S2, and S3 are shown in Fig. 6. The FTIR spectra shows a broadband around 3,400 cm−1, which is attributed to the O–H stretching mode of the surface adsorbed water molecule. Another band of around 1,600 cm−1 is attributed to the O–H bending mode. The bands around 400–900 cm−1 are due to the Ti–O bond stretching mode of the titania [41-45].
Figure 6.
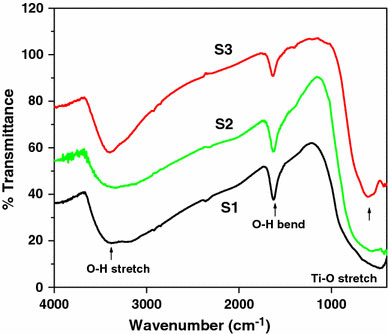
FTIR spectra of TiO2samples S1, S2, and S3
Optical Properties
UV–Visible Absorption Studies
Figure 7 shows the UV-vis absorption spectra of titania nanostructures S1, S2, and S3. The onset of absorption for the three samples is 382, 405, and 415 nm for S1, S2, and S3, respectively. To determine the nature of the band gap, either an indirect or a direct transition, the following power expression for the variation of the absorption coefficient (α) with energy was examined [46,47].
Figure 7.
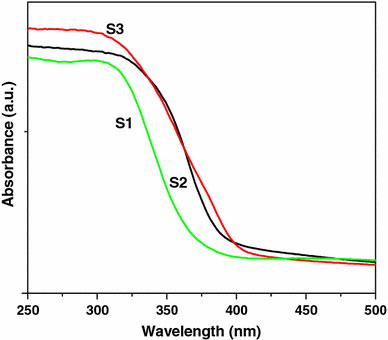
UV–visible absorption spectra of titania samples S1, S2, and S3
where kid is the absorption constant for an indirect (subscript i) or direct (subscript d) transition, n is two for an indirect transition and ½ for a direct transition, hυ is the absorption energy, and Eg is the band gap energy. The absorption coefficient (α) was determined from the equation α = (2.303 × 103)(A)/l by using the measured absorbance (A) and optical path length (l) (1 cm). The band gap (Eg) of a semiconductor can be estimated from the plot of (αhν)2 versus photon energy (hν). The band gap energy is determined by extrapolating the curve to the x-axis, as shown in the Fig. 8[48]. Variation of (αhν)2 with absorption energy (hν) for nanocubes (Fig. 8) gives the extrapolated intercept corresponding to the band gap energy at 3.2 eV, which is in agreement to the onset energy observed in the absorption spectrum, confirming that the band gap is attributed to the indirect transition. The band gap energy of nanocubes (S1) is significantly higher as compared to that of nanospheres (S2, 3.17 eV) and nanorods (S3, 3.15 eV). For pure anatase, the significant increase in the absorption wavelength (λ) (lower than 380 nm) can be assigned to the intrinsic band gap absorption [49]. The band gap (Eg) is estimated to be 3.2 eV, which is in good agreement with the reported value for anatase (3.2–3.3 eV). The absorption spectrum of rutile shows a lower absorption and the calculated band gap is around 3.17 and 3.15 eV, respectively, for the samples S2 and S3. However, rutile nanostructures show a slightly higher band gap than the reported value (3.0–3.1 eV). The higher band gap may be due to the smaller particle size. The band gap (Eg) and absorption onset (λ max) values are summarized in Table 2.
Figure 8.
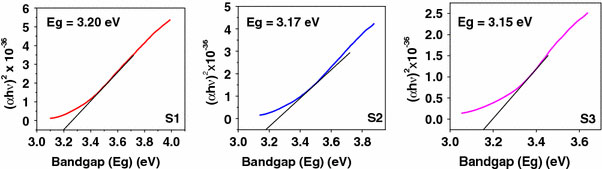
A plot of (αhυ)2versus photon energy (hυ) of the synthesized nanotitania
Table 2.
Summary of band gap and absorption onset of the synthesized nanotitania
| Sample code | Band gap (Eg) eV | Absorption onset (λmax) |
|---|---|---|
| S1 |
3.20 |
382 |
| S2 |
3.17 |
405 |
| S3 | 3.15 | 415 |
Photoluminescence Studies
Figure 9 shows the photoluminescence (PL) emission spectra of titania nanostructures measured at room temperature. The PL emission spectra are observed with an excitation wavelength around 325 nm, exhibiting a strong structural emission band around 360 nm with broad shoulders beyond 380 nm. At a higher wavelength around 500 nm emission due to the trapped or excess surface states is observed. The excited state of TiO2 can be considered as Ti3+…O− and the subsequent emission may be due to the transfer of electron from the excited state (Ti3+) to (O−) leading to the formation of Ti4+O22−. Therefore the strong emission in the region of 360–363 nm is assigned to the exciton emission originating from the recombination of a hole with an electron, whereas the weak and broad emission peaks in the region of 400–500 nm is just a surface state emission originating from the trapped or excess surface states [50-52].
Figure 9.
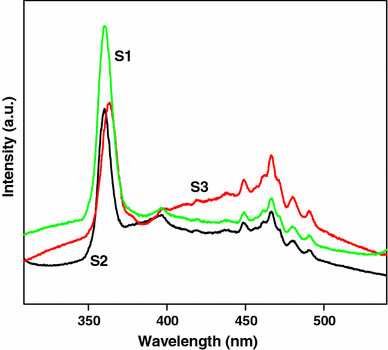
PL emission spectra of titania nanostructures S1, S2, and S3
Mechanistic Aspects
Three different morphologies obtained under Microwave (MW) irradiation can be understood in different ways. It may be due to the fast nucleation of Ti(OH)2 under three different pH (basic, neutral and acidic) and its subsequent condensation during reaction, dehydration and calcination. Morphology difference can be attributed to the ion assisted growth of the crystallites which may be different for OH− assisted growth in the case of S1 and Cl− assisted growth in S2 and Cl− and NH4+ assisted growth in S3. The shape evolution originates from the different adsorption capabilities of theses ions in various planes during the growth of the particle [53]. A schematic of shape tuning achieved during the synthesis under three different pH is shown in Scheme 1.
Scheme 1.

A schematic of shape tuning achieved by ion assisted growth for titania nanostructures in different pH
Photocatalytic Activity Studies
Photocatalytic processes involve irradiation of a semiconductor such as TiO2 with energy greater than or equal to the band gap of the semiconductor. This promotes electrons from the valence band to the conduction band, generating photoexcited electrons (e−) and holes (h+). The photoexcited electrons and holes may diffuse to the surface of the semiconductor, followed by interfacial electron transfer to and from the adsorbed acceptor and donor molecules. The holes are involved in the oxidation reactions, typically the mineralization of organic substances present in the solution [54]. In the present work, photocatalytic activity tests were conducted by the degradation of the dye, methylene blue in aqueous solution under ultraviolet light irradiation. Methylene blue (MB) shows a maximum absorption at 661 nm. The absorption peak gradually diminishes upon the ultraviolet light irradiation, illustrating the methylene blue degradation. The concentrations of methylene blue with irradiation time for the three titania nanostructures, Degussa P25 and methylene blue are shown in Fig. 10. It is clear that the anatase titania nanocubes (S1) shows higher photocatalytic activity than the other two rutile nanostructures (S2 and S3). From the degradation studies, it is observed that the photocatalytic activity varies in the order S1 > S2 > S3 > Degussa P25. The three nanostructures synthesized in different media have different phase structure, particle size, and surface area. It is reported that among the three crystalline phases of TiO2, the anatase phase has higher photocatalytic activity [55]. The difference in activity of the synthesized samples is related to their surface area, particle size, and phase. Small crystallite size and mesoporous texture produces high surface area TiO2 and hence can provide more active sites and adsorb more reactive species. Since S1 is purely anatase phase and has the highest surface area among the three samples, it exhibits the highest photocatalytic activity. The appreciable activity observed for the nanorods (34 m2/g) compared to Degussa P25 (50 m2/g) may be due to the preferentially grown 110 planes in the nanorod morphology.
Figure 10.
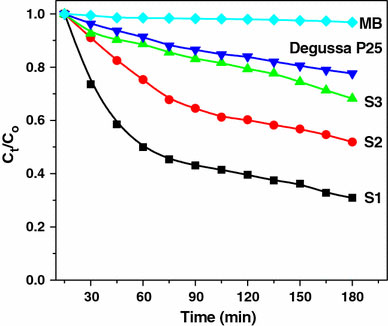
Photocatalytic activity of various TiO2nanostructures for the degradation of methylene blue
Conclusions
Nanotitania with fascinating morphologies, particle size, and surface area can be effectively synthesized by a simple microwave irradiation technique. The morphology of the samples was effectively controlled by changing the pH of the media. The synthesized nano TiO2was structurally and physicochemically characterized. Structural and physicochemical characterization revealed the dependence of photocatalytic activity of nanotitania on different morphologies. The TEM images clearly reveal that the samples have cubical, spherical and rod shaped morphologies. The surface area and porosity of the three titania nanostructures were determined by BET and BJH methods. Anatase nanocubes (S1) exhibit a much higher BET specific surface area than rutile nanospheres (S2) and nanorods (S3). The band gap energy for anatase nanocubes is blue shifted (3.2 eV) compared to that of the rutile nanospheres (S2) and nanorods (S3). The UV–vis absorption and the photoluminescence emission spectral data demonstrated that the indirect transition is the exclusive route for the charge carrier recombination, indicating the strong coupling of wave functions of the trapped exciton pair with lattice phonons. The synthesized mesoporous anatase nanotitania with cubical morphology exhibit higher photocatalytic activity than spherical and rod shaped rutile titania nanostructures. Moreover, the synthesized mesoporous anatase TiO2with BET surface area 372 m2 g−1exhibit much higher photocatalytic activity than the commercial Degussa P25 TiO2photocatalyst in the degradation of the dye, methylene blue in aqueous solution under UV light irradiation. The higher photocatalytic activity of the anatase nanocubes may be due to the higher surface area and the lesser electron-hole recombination rate compared to the rutile nanostructures.
Acknowledgments
We are grateful to Dr. K. George Thomas of Regional Research Laboratory, Trivandrum and Prof. T. Pradeep of Indian Institute of Technology, Chennai for the AFM and HRTEM imaging.
References
- Schmid G. Nanoparticles: From Theory to Application. Wiley-VCH, Weinheim; 2004. [Google Scholar]
- Klabunde KJ. Nanoscale Materials in Chemistry. Wiley Interscience, New York; 2001. [Google Scholar]
- Joo J, Yu T, Kim YW, Park HM, Wu F, Zhang JZ, Hyeon T. J. 2003. p. 6553. COI number [1:CAS:528:DC%2BD3sXjtlWhur4%3D] [DOI] [PubMed]
- Hyeon T, Lee SS, Park J, Chung Y, Na HB. J. 2001. p. 12798. COI number [1:CAS:528:DC%2BD3MXoslSnsrs%3D] [DOI] [PubMed]
- Diebold U. Surf. 2003. p. 53. COI number [1:CAS:528:DC%2BD3sXivVWqtQ%3D%3D]; Bibcode number [2003SurSR..48...53D] [DOI]
- Luo H, Takata T, Lee Y, Zhao J, Domen K, Yan Y. Chem. 2004. p. 846. COI number [1:CAS:528:DC%2BD2cXnsVKhsg%3D%3D] [DOI]
- Sayilkan F, Asilturk M, Erdemoglu S, Akarsu M, Sayilkan H, Erdemoglu M, Arpac E. Mater. 2006. p. 230. COI number [1:CAS:528:DC%2BD2MXhtFOmsLrE] [DOI] [PubMed]
- Balfour JG. Technological Applications of Dispersions. Marcel Dekker, New York; 1994. [Google Scholar]
- Yeh YC, Tseng TY, Chang DA. J. 1989. p. 1472. COI number [1:CAS:528:DyaL1MXmtVOgtLs%3D] [DOI]
- Bond GC, Tahir SF. Appl. 1991. p. 1. COI number [1:CAS:528:DyaK3MXisFClsLc%3D] [DOI]
- Awati PS, Awate SV, Shah PP, Ramaswamy V. Catal. 2003. p. 393. COI number [1:CAS:528:DC%2BD3sXlvVCntrY%3D] [DOI]
- Hagfeldt A, Gratzel M. Chem. 1995. p. 49. COI number [1:CAS:528:DyaK2MXjtF2qtbc%3D] [DOI]
- Hsien YH, Chang CF, Chen YH, Cheng S. Appl. 2001. p. 241. COI number [1:CAS:528:DC%2BD3MXjsleiu7g%3D] [DOI]
- Lizama C, Freer J, Baeza J, Mansilla HD. Catal. Today. 2002. p. 235. COI number [1:CAS:528:DC%2BD38XoslyqtLc%3D] [DOI]
- Serpone N, Texier I, Emeline AV, Pichat P, Hidaka H, Zhao J. J. 2000. p. 145. COI number [1:CAS:528:DC%2BD3cXmsFKrtL0%3D] [DOI]
- Ohtani B, Kakimoto M, Nishimoto S, Kagiya T. J. 1993. p. 265. COI number [1:CAS:528:DyaK3sXksF2ntb0%3D] [DOI]
- Nam HJ, Amemiya T, Murabayashi M, Itoh K. J. Phys. Chem. B. 2004. p. 8254. COI number [1:CAS:528:DC%2BD2cXktlWkur8%3D] [DOI]
- Chu SZ, Inoue S, Wada K, Li D, Haneda H, Awatsu S. J. Phys. Chem. B. 2003. p. 6586. COI number [1:CAS:528:DC%2BD3sXks1ersLw%3D] [DOI] [PubMed]
- Bosc F, Ayral A, Albouy P, Guizard C. Chem. 2003. p. 2463. COI number [1:CAS:528:DC%2BD3sXjsleis7c%3D] [DOI]
- Zhang Q, Gao L. Langmuir. 2003. p. 967. COI number [1:CAS:528:DC%2BD38XpvVantbs%3D] [DOI]
- Wu M, Lin G, Chen D, Wang G, He D, Feng S, Xu R. Chem. 2002. p. 1974. COI number [1:CAS:528:DC%2BD38XhtlOkurg%3D] [DOI]
- Niederberger M, Bartel MH, Stucky GD. Chem. 2002. p. 4364. COI number [1:CAS:528:DC%2BD38XntlagtL8%3D] [DOI]
- Polleux J, Pinna N, Antonietti M, Niederberger M. Adv. 2004. p. 436. COI number [1:CAS:528:DC%2BD2cXis1ejtrg%3D] [DOI] [PubMed]
- Yang P, Zhao D, Margolese DI, Chmelka BF, Stucky GD. Nature. 1998. p. 152. COI number [1:CAS:528:DyaK1cXnslalsLc%3D]; Bibcode number [1998Natur.396..152Y] [DOI]
- Serrano DP, Calleja G, Sanz R, Pizarro P. Chem. 2004. p. 1000. COI number [1:CAS:528:DC%2BD2cXivVGnsb0%3D] [DOI] [PubMed]
- Ostomel TA, Stucky GD. Chem. Commun. (Camb) 2004. p. 1016. COI number [1:CAS:528:DC%2BD2cXivVGnsLk%3D] [DOI] [PubMed]
- Zhang D, Qi L, Ma J, Cheng H. J. 2002. p. 3677. COI number [1:CAS:528:DC%2BD38XptFSlsLw%3D] [DOI]
- Wu JJ, Yu CC. J. Phys. Chem. B. 2004. p. 3377. COI number [1:CAS:528:DC%2BD2cXhtlams7s%3D] [DOI]
- Kasuga T, Hiramatsu M, Hoson A, Sekino T, Niihara K. Adv. 1999. p. 1307. COI number [1:CAS:528:DyaK1MXntVams7s%3D] [DOI]
- Tian ZR, Voigt JA, Liu J, Mckenzie B, Xu H. J. 2003. p. 12384. COI number [1:CAS:528:DC%2BD3sXntlyku74%3D] [DOI] [PubMed]
- Miao Z, Xu D, Ouyang J, Guo G, Zhao X, Tang Y. Nano. 2002. p. 717. COI number [1:CAS:528:DC%2BD38XjsVSiu70%3D]; Bibcode number [2002NanoL...2..717M] [DOI]
- Yi DK, Yoo SJ, Kim DY. Nano. 2002. p. 1101. COI number [1:CAS:528:DC%2BD38XmtVylsL4%3D]; Bibcode number [2002NanoL...2.1101Y] [DOI]
- Zhou Y, Antonietti M. J. 2003. p. 14960. COI number [1:CAS:528:DC%2BD3sXovVOisLk%3D] [DOI] [PubMed]
- Kormann C, Bahnemann DW, Hoffmann MR. J. 1988. p. 5196. COI number [1:CAS:528:DyaL1cXltFKltbg%3D] [DOI] [PubMed]
- Ovenstone J, Yanagisawa K. Chem. 1999. p. 2770. COI number [1:CAS:528:DyaK1MXlslGntL4%3D] [DOI]
- Hosono E, Fujihara S, Kakiuchi K, Imai H. J. 2004. p. 7790. COI number [1:CAS:528:DC%2BD2cXks1arurc%3D] [DOI] [PubMed]
- Zhang D, Yang D, Zhang H, Lu C, Qi L. Chem. 2006. p. 3477. COI number [1:CAS:528:DC%2BD28XmsVSiur8%3D] [DOI]
- Han S, Choi SH, Kim SS, Cho M, Jang B, Kim DY, Yoon J, Hyeon T. Small. 2005. p. 812. COI number [1:CAS:528:DC%2BD2MXntVamu7Y%3D] [DOI] [PubMed]
- Parala H, Devi A, Bhakta R, Fischer RA. J. 2002. p. 1625. COI number [1:CAS:528:DC%2BD38XjvFWjsbc%3D] [DOI]
- Daoud WA, Xin JH. Chem. Commun. (Camb) 2005. p. 2110. COI number [1:CAS:528:DC%2BD2MXjsFCis74%3D] [DOI] [PubMed]
- Wu L, Yu JC, Wang X, Zhang L, Yu J. J. 2005. p. 321. COI number [1:CAS:528:DC%2BD2MXpslSgtA%3D%3D]; Bibcode number [2005JSSCh.178..321W] [DOI]
- Sun J, Gao L, Zhang Q. J. 2003. p. 1677. COI number [1:CAS:528:DC%2BD3sXot1yls7w%3D] [DOI]
- Yan M, Chen F, Zhang J, Anpo M. J. Phys. Chem. B. 2005. p. 8673. COI number [1:CAS:528:DC%2BD2MXivV2mtrw%3D] [DOI] [PubMed]
- Tanaka Y, Suganuma M. Sol–Gel Sci. 2001. p. 83. COI number [1:CAS:528:DC%2BD3MXns1Gnsbk%3D] [DOI]
- Jiang X, Wang Y, Herricks T, Xia Y. J. 2004. p. 695. COI number [1:CAS:528:DC%2BD2cXhtlWmtrk%3D] [DOI]
- Zhao XK, Fendler JH. J. 1991. p. 3716. COI number [1:CAS:528:DyaK3MXitF2jsLo%3D] [DOI]
- Monticone S, Tufeu R, Kanaev AV, Scolan E, Sanchez C. Appl. 2000. p. 565. Bibcode number [2000ApSS..162..565M] [DOI]
- Sreethawong T, Suzuki Y, Yoshikawa S. J. 2005. p. 329. COI number [1:CAS:528:DC%2BD2MXpslSgtQ%3D%3D]; Bibcode number [2005JSSCh.178..329S] [DOI]
- Luo H, Wang C, Yan Y. Chem. 2003. p. 3841. COI number [1:CAS:528:DC%2BD3sXms1arurk%3D] [DOI]
- Yoon M, Seo M, Jeong C, Jang JH, Jeon KS. Chem. 2005. p. 6069. COI number [1:CAS:528:DC%2BD2MXhtFertL7O] [DOI]
- Wang Y, Herron N. J. 1991. p. 525. COI number [1:CAS:528:DyaK3MXntFWqsw%3D%3D] [DOI]
- Daude N, Gout C, Jouanin C. Phys. Rev. B. 1977. p. 3229. COI number [1:CAS:528:DyaE2sXhs1CjtL8%3D]; Bibcode number [1977PhRvB..15.3229D] [DOI]
- Zhu H, Yao K, Wo Y, Wang N, Wang L. Semicond. 2004. p. 1020. COI number [1:CAS:528:DC%2BD2cXosVOisbo%3D]; Bibcode number [2004SeScT..19.1020Z] [DOI]
- Nguyen VNH, Amal R, Beydoun D. J. 2006. p. 57. COI number [1:CAS:528:DC%2BD28Xit1ajtrk%3D] [DOI]
- Kato K, Tsuzuki A, Taoda H, Torii Y, Kato T, Butsugan Y. J. 1994. p. 5911. COI number [1:CAS:528:DyaK2MXitl2gurk%3D]; Bibcode number [1994JMatS..29.5911K] [DOI]