

Abstract
Study Objectives:
REM sleep is suppressed during infection, an effect mimicked by the administration of cytokines such as interleukin-1 (IL-1). In spite of this observation, brain sites and neurochemical systems mediating IL-1-induced suppression of REM sleep have not been identified. Cholinergic neurons in the brainstem laterodorsal tegmental nucleus (LDT) are part of the neuronal circuitry responsible for REM sleep generation. Since IL-1 inhibits acetylcholine synthesis and release, the aim of this study was to test the two different, but related hypotheses. We hypothesized that IL-1 inhibits LDT cholinergic neurons, and that, as a result of this inhibition, IL-1 suppresses REM sleep.
Design, Measurement, and Results:
To test these hypotheses, the electrophysiological activity of putative cholinergic LDT neurons was recorded in a rat brainstem slice preparation. Interleukin-1 significantly inhibited the firing rate of 76% of recorded putative cholinergic LDT neurons and reduced the amplitude of glutamatergic evoked potentials in 60% of recorded neurons. When IL-1 (1 ng) was microinjected into the LDT of freely behaving rats, REM sleep was reduced by about 50% (from 12.7% ± 1.5% of recording time [after vehicle] to 6.1% ± 1.4% following IL-1 administration) during post-injection hours 3-4.
Conclusions:
Results of this study support the hypothesis that IL-1 can suppress REM sleep by acting at the level of the LDT nucleus. Furthermore this effect may result from the inhibition of evoked glutamatergic responses and of spontaneous firing of putative cholinergic LDT neurons.
Citation:
Brambilla D; Barajon I; Bianchi S; Opp MR; Imeri L. Interleukin-1 inhibits putative cholinergic neurons in vitro and REM sleep when microinjected into the rat laterodorsal tegmental nucleus. SLEEP 2010;33(7):919-929.
Keywords: Cytokines, patch clamp, acetylcholine, glutamate, GABA, adenosine
REM SLEEP OF MAMMALS IS SUPPRESSED DURING INFECTION.1 ONE POTENTIAL FUNCTION OF REM SLEEP SUPPRESSION DURING INFECTION IS TO FACILITATE the generation of fever.1 The adaptive role of fever in surviving infection has been amply demonstrated.2 REM sleep suppression observed during infection is mimicked in different animal models by the administration of cytokines such as interleukin-1 (IL-1) or tumor necrosis factor (TNF), levels of which are increased during infection.3 Brain neuronal circuits and neurochemical systems mediating cytokine-induced suppression of REM sleep have not yet been investigated.
Although interleukin-1 (IL-1) was originally described as a product of the peripheral immune system, there is now ample evidence that IL-1, IL-1 receptors, and the IL-1 receptor antagonist are constitutively expressed in normal brain.4 IL-1 modulates behaviors such as feeding, reproduction, social exploration, locomotor activity, and sleep.3 IL-1 effects on NREM sleep are dose dependent: IL-1 enhances NREM sleep at low doses, whereas it inhibits this sleep phase at higher doses.5,6 On the other hand, IL-1 suppresses REM in different animal species, across a wide range of doses.5–7 Whereas mechanisms mediating IL-1 effects on NREM sleep have been investigated at length,1,3,8 to date no studies have been conducted to determine brain area(s) and neurotransmitter system(s) involved in IL-1-induced suppression of REM sleep.
It has long been known that REM sleep is generated within the brainstem, where cholinergic REM sleep-promoting (REM-on) neurons are located.9 REM-on neurons, which fire at higher rates during REM sleep than during wakefulness or NREM sleep,9 have been described within the laterodorsal and pedunculopontine tegmental nuclei (LDT/PPT). The electrical stimulation of LDT enhances REM sleep,10 whereas neurotoxic lesion of the LDT/PPT nuclei reduces REM sleep in amounts proportional to the loss of cholinergic neurons.11 Although LDT/PPT REM-on neurons have only been putatively identified as cholinergic,12,13 LDT/PPT cholinergic neurons do project to the pontine reticular formation (PRF),14,15 where acetylcholine (ACh) release is greater during REM sleep than other vigilance states.16 Furthermore, administration of cholinomimetics into the PRF induces a state of cortical activation and muscle atonia considered an equivalent of REM sleep,17 whereas microinjection of muscarinic antagonists into the PRF inhibits REM sleep.18 Finally, LDT/PPT cholinergic neurons are activated during periods of enhanced REM sleep.19
Interleukin-1 inhibits ACh release in vivo in the hippocampus,20 as well as ACh synthesis in vitro in cultured pituitary cells.21 Moreover, IL-1 increases acetylcholinesterase activity and mRNA expression in both neuron-glia co-cultures and in vivo in the rat cortex.22
The aim of the present study was to test two distinct, yet related hypotheses. We hypothesized that IL-1 inhibits the electrophysiological activity of cholinergic LDT neurons, and that, because of this inhibition, the LDT represents one brain region where IL-1 acts to suppress REM sleep. To this purpose, (1) effects of IL-1 on firing rate and evoked synaptic responses of putative cholinergic LDT neurons were investigated in a rat brainstem slice preparation, and (2) alterations in sleep induced by IL-1 microinjection into the LDT were determined in freely behaving rats.
METHODS
All procedures involving animals and their care were conducted in conformity with institutional guidelines that are in compliance with European Union (EEC Council Directive 86/609, OJ L 358,1; 12 December 1987) and Italian (D.L. n.116, G.U. suppl. 40, 18 February 1992) laws and policies, as well as with the United States Department of Agriculture Animal Welfare Act and the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Substances
IL-1 (rat recombinant IL-1β expressed in Escherichia coli) was purchased from Euroclone (West York, UK). Lyophilized IL-1 was dissolved in pyrogen-free saline containing 0.1% bovine serum albumin, aliquoted, frozen, and stored at −20°C until use. Drugs used in the in vitro experiments were DL-noradrenaline hydrochloride (NA), DL-2-Amino-5-phosphonopentanoic acid (APV), (-)-bicuculline methiodide (BMI) and 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX), all obtained from Sigma (St. Louis, MO, USA). Drugs were aliquoted, stored at −20°C, and dissolved just prior to use in artificial cerebrospinal fluid (aCSF) of the following composition: 124 mM NaCl, 2 mM KCl, 3 mM KH2PO4, 26 mM NaHCO3, 1.3 mM MgCl2, 2.5 mM CaCl2, 10 mM glucose (final pH 7.4). Drugs were then applied via bath perfusion. The stock of DNQX was dissolved in dimethyl sulfoxide (DMSO) before being added to aCSF (final concentration of DMSO < 0.1%).
In Vitro Experiments
Preparation of brainstem slices
Male Sprague-Dawley rats (25–50 g of body weight; 20–25 days old at time of experiment; Charles River, Italy) were anesthetized with isoflurane, and decapitated. The brains were then rapidly removed and placed in ice-cold (4°C) aCSF, continuously bubbled with an O2-CO2 mixture (95%-5%, respectively). Coronal sections were then cut (400 μm thick) with a vibratome (752 Vibroslice, Campden Instruments Ltd, Loughborough, UK) from a block of tissue containing the LDT and kept in ice-cold carbogenated aCSF. Three slices were taken from each animal for subsequent recording and transferred to a holding chamber. The slices were then incubated at room temperature in carbogenated aCSF and were left to recover for at least 1 h. After recovery, slices were individually transferred to a warmed (32°C) submersion-type slice recording chamber, through which carbogenated aCSF was continuously superfused at a rate of 2.5 mL/min.
Recording methods
Neurons were visualized with a differential interference contrast (DIC) and infrared (IR)-filtered light Axioscope 2-FS plus microscope (Carl Zeiss, Oberkochen, Germany) equipped for in vitro electrophysiological applications. The microscope was equipped with a high-resolution digital camera (AxioCam MRm, Carl Zeiss) interfaced to a computer and controlled by AxioVison AC imaging software (Carl Zeiss). Cells were selected for whole-cell recording from a region identified at low magnification as LDT (Figure 1A). Neurons were recorded in current-clamp mode using a high-impedance amplifier (Axoclamp-2B, Axon Instrument, Union City, CA, USA) connected to a digitizer (Digidata 1322A, Axon Instrument), interfaced to a computer. Axon Instrument software (pClamp 8.2) was used for the on- and off-line data acquisition and analysis. The patch-clamp recording micropipettes were obtained from borosilicate capillaries (o.d. = 1.5 mm; i.d. = 0.86 mm; Clark Electromedical Instruments, Pangbourne, UK) pulled by a micropipette puller (Model P97, Sutter Instrument, Novato, CA, USA). Recording micropipettes filled with electrolytic solution (120 mM K-gluconate, 10 mM KCl, 3 mM MgCl2, 10 mM HEPES, 2 mM MgATP, 0.2 mM NaGTP; pH 7.2, adjusted with KOH) had resistances between 5 and 10 MOhm.
Figure 1A-C.

Morphological, electrophysiological, and pharmacological identification of putative cholinergic neurons within the laterodorsal tegmental nucleus (LDT). (A) Low (right) and high (left) magnification images obtained by differential interference contrast microscopy (DIC) and infrared filtered light (IR) of an impaled neuron within the laterodorsal tegmental nucleus (LDT). DTg: dorsal tegmental nucleus, 4 V: fourth ventricle. Calibration bar, 50 μm. (B) Representative current clamp recording of a putative cholinergic LDT neuron, showing a low-threshold burst response following hyperpolarization (top trace, voltage; bottom trace, current). The burst is shown on an expanded time bases in the dashed line box. Please note the hyperpolarization-activated nonspecific cation current Ih indicated by arrow. (C) Continuous recording of a representative LDT cholinergic neuron. Reversible hyperpolarization of membrane potential and block of spontaneous firing activity were induced by application of noradrenaline (NA). The effect was reversible upon washout D-H—Immunofluorescence identification of cholinergic neurons within the LDT. (D) High magnification image of an impaled neuron within the LDT of a rat brain slice preparation. Image was obtained by differential interference contrast microscopy (DIC) and infrared filtered light (IR). Calibration bar, 20 μm. (E) Post-recording fluorescence image of the same neuron shown in panel A and containing lucifer yellow (visualized in green). (F) Positive immunolabeling (visualized in red) for choline acetyltransferase (ChAT) of the same neuron shown in panels D and E. (G) Superimposition of images from panels E and F showing that the LDT recorded neuron was cholinergic. (H) Whole-cell patch-clamp recording in current clamp mode from the same neuron (shown in panels D-G) in response to the application of noradrenaline (NA). NA reversibly hyperpolarized membrane potential and blocked spontaneous firing activity.
The impact of IL-1 on spontaneous firing rates and the amplitude of evoked postsynaptic potentials (evPSPs) was determined. Interleukin-1-induced changes in LDT neuron discharge rate were evaluated using a continuous (gap-free) recording protocol. Evoked PSPs were elicited using a bipolar stimulating electrode (FHC, Bowdoinham, ME, USA) connected to an isolation unit (SC-100; Winston Electronics, Millbrae, CA) delivering single square wave current pulses (0.8-5 mA; 200 μs duration; 0.2 Hz). The bipolar electrode was placed on the surface of the slice close to the dorsolateral border of the ipsilateral LDT (between 0.28 and −0.30 mm relative to interaural line.)23 The stimulated region lies between the cuneiform nucleus (rostrally) and the parabrachial nucleus (caudally). All evoked responses, were recorded at membrane potential of −70 mV.
Putative cholinergic LDT neurons were identified on the basis of their morphology,24 as well as electrophysiological16,25 and pharmacological properties26 (Figures 1A-C). To validate the use, in our experimental conditions, of these morphological, electrophysiological, and pharmacological criteria for the identification of putative cholinergic LDT neurons, the cholinergic nature of a subset of already identified neurons was verified by use of immunofluorescence (Figures 1D-H). To this purpose, lucifer yellow CH dipotassium salt (0.1%; Sigma) was added to the micropipette solution for the labeling of recorded neurons, and recordings were performed as previously described. After recording, brain slices were fixed in buffered 4% paraformaldehyde overnight, and free-floating sections were assayed for choline acetyltransferase (ChAT) immunofluorescence, which marks cholinergic neurons. Briefly, samples were permeabilized in 0.3% Triton X-100 in Tris buffered saline (TBS-T) and following blockage of nonspecific sites with swine serum, incubated with a goat anti-ChAT primary antibody (Chemicon International, Temecula, CA, USA), diluted 1:100 in TBS-T for 24 h at 4°C. After several rinses in TBS-T, slices were incubated with a TRITC-conjugated donkey anti-goat secondary antibody (Jackson Immunoresearch, West Grove, PA, USA) diluted 1:250 in TBS-T for 2 h at room temperature, collected on silane-coated slides, and mounted with Mowiol 4-88 (Calbiochem, La Jolla, CA, USA). Controls were treated as described above but omitting the primary antibody. Brain slices were observed with a ViCo (Video Confocal) microscope (Nikon Eclipse 80i) equipped with selective barrier filter sets (TRITC: EX 540-525/DM 565/BA 605-655; Lucifer Yellow: EX 425-440/DM 460/BA 540-50). Images were acquired by a digital camera (Nikon Digital Sight 5MC) and image acquisition software provided with the instrument.
Application of test substances
Test substances, dissolved in carbogenated aCSF just prior to use and applied via bath perfusion, arrived in the recording chamber 3 min after application. NA (30 μM) and IL-1β (25 pg/mL, 1.4 pM) were applied for 1 and 3 minutes, respectively. A GABAergic antagonist (BMI) and glutamatergic antagonists (APV and DNQX) were applied for 15 min. Complete washout of previous effect was obtained between application of different substances. Firing rates were sampled off line, in 1-min intervals, 2 min prior to administration of test substances (control condition), when change in firing rate was maximal in comparison to control condition (between 1 and 6 min after IL-1 arrival in the recording chamber), and during washout when firing rate returned to the value closest to control condition. Amplitudes of evPSPs were sampled in control conditions (before substance application), 10 min after arrival of test substances in the recording chamber, and during washout when amplitude reached the closest value to control condition.
Data Acquisition and Analysis
All data are expressed as means ± SEM. Data were statistically analyzed by the nonparametric Kolmogorov-Smirnov test, one-way ANOVA, and Tukey or t tests, as appropriate. An α level of P < 0.05 was taken as indicating a statistically significant difference between experimental conditions.
In Vivo Experiments
Animals and surgery
Adult male Sprague-Dawley rats (225–275 g of body weight; 50–56 days old at time of surgery; Charles River, Calco CO, Italy) were housed individually in environmentally controlled chambers, maintained at 22 ± 1°C with a 12:12 h light: dark cycle. Food and water were available ad libitum. Rats were anesthetized (isoflurane 5.0% in 50/50% O2/N2O during induction, then adjusted to 1.5% during surgery), positioned in a stereotaxic apparatus, and surgically prepared for chronic polygraphic recordings. Stainless steel screws placed over frontal, parietal, and occipital cortices served as electroencephalographic (EEG) recording and ground electrodes. A calibrated 30-kW thermistor (Omega Engineering, Stamford, CT) was implanted between the dura mater and the skull over the parietal cortex to measure cortical brain temperature (Tcort). A stainless steel guide cannula (length = 1.5 cm; outer diameter = 0.5 mm) was stereotaxically placed 3 mm above the LDT. To avoid damage to the midline structures, cannulae were implanted at a 30° angle to the sagittal plane. The following stereotaxic coordinates were used (adapted from27): anteroposterior = −0.3 mm from interaural line, lateral = 3.15 mm, dorsoventral = 6.4 mm below the dura mater (tooth bar = −3.3 mm). Insulated leads were routed from the screws and the thermistor to a plastic pedestal (Plastics One Inc., Roanoke, VA) that was cemented in place with dental acrylic (Isocryl; Lang Dental Supply, Wheeling, IL). The incision was treated topically with Polysporin (polymyxin B sulfate-bacitracin zinc) and animals were placed under heat lamps and monitored until recovery from anesthesia. On the surgical day, the animals were injected with a broad spectrum antibiotic (benzyl penicillin, 5000 IU/rat) and an analgesic (flunixin 2.5 mg/kg). The analgesic treatment was continued on the first post-surgical day. On the third post-surgical day, the rats were connected to the recording apparatus (see later) via a flexible tether connected to the Teflon pedestal, which allowed relatively unrestricted movement within the cage. For the next 5 days the animals were adapted to the experimental procedures.
Apparatus and recording
Gross body activity was detected using an infrared sensor housed in an observation unit that also contained a camera (BioBserve GmbH, Bonn, Germany). Movements detected by the infrared sensor were converted to a voltage output, the magnitude of which was directly related to the magnitude of the movements detected. Signals from the EEG recording electrodes and from the thermistor were fed into a Grass (Quincy, MA) polygraph in the adjacent room. The EEG was amplified (factor of 3,000) and analog bandpass filtered between 0.1 and 40 Hz (frequency response: ± 3 dB; filter frequency roll off: 12 dB/octave). These conditioned signals, as well as the voltage output from the infrared sensor, were digitized with 12-bit precision at a sampling rate of 128 Hz (PCI-6023E; National Instruments, Austin, TX) and collected using custom software written in LabView (National Instruments). The digitized signals were stored as binary computer files until subsequent analyses. Movement values were integrated into 1-s bins. Post-acquisition determination of vigilance state was done by visual scoring of 12-s epochs using custom software, as previously described.28 Briefly, the rat's behavior was classified (by a scorer blind to the experimental condition) as either wakefulness (W), NREM sleep, or REM sleep. Wakefulness was characterized by low amplitude EEG and body movements. During NREM sleep, EEG amplitude was increased, the contribution of delta (1-4 Hz) frequency bands to the total EEG power increased, and body movements were absent or very brief. Complete absence of movements, and low amplitude EEG dominated by a regular theta rhythm (6-9 Hz) characterized REM sleep.
Experimental Protocol
Each animal was injected unilaterally with both vehicle (pyrogen-free saline containing 0.1% bovine serum albumin) and IL-1, thus serving as its own control. In a pilot study aimed to determine which IL-1 doses were effective in modifying sleep-wake patterns when microinjected into the LDT, 8 animals were divided in 2 groups of 4. Animals in the first group were microinjected with 0.25 and 0.5 ng IL-1, whereas animals in the second group received 1 and 4 ng IL-1. These IL-1 doses where chosen on the basis of a previous study investigating the effects of IL-1 microinjection into the dorsal raphe nucleus (DRN).29 Since no changes were observed in either sleep-wake patterns or Tcort values after microinjections of IL-1 0.25 and 4 ng, the effects of IL-1 0.5 and 1 ng were further investigated in the study and the number of animals used was increased accordingly. A total of 8 animals were microinjected with 0.5 ng IL-1, whereas 10 animals received 1 ng IL-1.
Experiments were scheduled randomly with an interval of at least 3 days between microinjections. IL-1 and vehicle were administered at light onset in a volume of 100 nL, using a stainless steel needle (length = 1.8 cm; outer diameter = 0.3 mm), connected via polyethylene tubing to a Hamilton micro syringe and inserted through the guide cannula. The needle extended 3 mm past the tip of the guide cannula, reaching the LDT. Microinjections were performed over a 1-min period. After the microinjection the needle was left in place for 3 min. Polygraphic recordings began immediately after the microinjections and continued for 24 hours.
At the end of the experiments rats were euthanized with an isoflurane overdose and dye (trypan blue) was microinjected (100 nL) into the microinjection site; the brains were then removed and fixed. The position of the dye spot was determined from 50 μm thick, frozen, neutral red stained sections. Location of the microinjection sites was determined before scoring of polygraphic recordings for determination of vigilance states. The microinjection sites were grouped in 2 locations, either within the central portion of the LDT or more rostrally. In the 18 animals used in the study, 12 microinjection sites were located within the central portion of the LDT (see Results), whereas 6 microinjection sites more rostral, within or adjacent to the dorsal raphe nucleus (DRN) or the ventrolateral periaqueductal grey (vlPAG; see Results). At this level, the LDT is of smaller size.
Statistical Analysis
Results are presented as means ± SEM. Tests for statistical significance were performed using SPSS for Windows (SPSS, Inc., Chicago, IL, USA). Values were compared by mixed model analysis for repeated measures using a first order autoregressive covariance structure. For these analyses, the individual rat was the subject variable, time (hours) was the repeated measure, and manipulation (vehicle vs. IL-1) was the fixed effect. Experimental variables (amount of time spent in NREM sleep, REM sleep, wakefulness, Tcort values) were the dependent variables. An a level of P < 0.05 was used to indicate a statistically significant difference.
RESULTS
In Vitro Results
Eighty-two neurons were recorded. Sixty of these neurons were identified as putatively cholinergic on the basis of morphological, electrophysiological, and pharmacological criteria (Figures 1A-C). The cholinergic nature of a subset (n = 5) of these 60 neurons was confirmed by use of immunofluorescence (Figures 1D-H), although IL-1 effects were not determined in these neurons. Thirty-one of the 55 morphologically, electrophysiologically, and pharmacologically identified putative cholinergic neurons were used in the study: in 17 neurons IL-1 effects on firing rate were tested, while in the other 14 neurons changes induced in evPSP amplitude by application of IL-1 (n = 5), BMI (n = 3), and APV + DNQX (n = 6) were evaluated.
Identification of LDT cholinergic neurons
Recorded neurons were all within the LDT, as visualized at low magnification with a DIC/IR microscope (Figure 1A). Cholinergic LDT neurons, characterized by a medium-large diameter, are usually larger than other LDT neurons.24 Mean long and short diameters of neurons identified in this study as putatively cholinergic (n = 60) on the basis of their electrophysiological and pharmacological properties (see later) were 25.6 ± 2.9 μm and 14.2 ± 1.8 μm, respectively (Figure 1A).
Although cholinergic LDT neurons are not electrophysiologically homogeneous30–33 and their electrophysiological properties may depend on such factors as the animal species and the age of the donor animal,30–32 at least a subgroup of LDT cholinergic neurons display rebound bursts of action potentials, and Ih depolarizing cation current,16,25 which we observed in our putatively cholinergic neurons (Figure 1B). As far as the pharmacological properties of cholinergic LDT neurons are concerned, 90% of these neurons are hyperpolarized by NA, and their firing is inhibited by the same treatment, via activation of the adrenergic α2 receptor.26 NA (30 μM) administration (Figure 1C) reduced mean firing rate of our putative cholinergic neurons from 4.2 ± 0.3 Hz (before NA administration) to 0.4 ± 0.1 Hz and induced a significant hyperpolarization of membrane potential from −43.8 ± 0.8 mV (before NA administration) to −52.6 ± 1.1 mV (n = 55).
Since non-cholinergic LDT neurons also display rebound bursts of action potentials30–32 and about one-third of non-cholinergic neurons are inhibited by NA application,26 we cannot rule out the possibility that some of the neurons we recorded were not cholinergic. For this reason, the neurons investigated in this study are indicated as putatively cholinergic. On the other hand, in our experimental conditions, a subset of recorded neurons (n = 6) were identified because containing lucifer yellow (Figures 1D and E). Five of these 6 neurons were also ChAT positive (Figures 1F and G). Morphological and electrophysiological properties, as well as the pharmacological response (Figure 1H) of the 5 ChAT positive neurons did not statistically differ from those reported in the 55 neurons, which were identified as cholinergic on the basis of morphological, electrophysiological, and pharmacological criteria (see previous paragraph). The only ChAT negative neuron did not exhibit a rebound burst of action potentials, nor a lh, was not inhibited by NA, and its long and short diameters were 17.6 mm and 10.4 mm, respectively.
IL-1 administration decreased spontaneous firing rates of LDT cholinergic neurons and amplitude of evPSPs
Bath application of 25 pg/mL (1.4 pM) IL-1 reduced firing rate of putative cholinergic LDT neurons (Figure 2A). Responding neurons were identified on the basis of a statistical variation in firing rate as determined by the Kolmogorov-Smirnov test.34,35 As a group (Figure 2B), the firing rates of the 13 responding neurons (76%) of the 17 identified putative cholinergic LDT neurons were significantly reduced from 4.3 ± 0.7 Hz prior to IL-1 administration to 3.2 ± 0.6 Hz following IL-1. Mean firing rate recovered to 4.0 ± 0.7 Hz after the washout. Although IL-1-induced inhibition of firing started within seconds of arrival in the recording chamber (Figure 2A, exploded view), the maximal decrease in firing was recorded in responding neurons between 1 and 3 min after cessation of IL-1 application, a time course slower than responses to NA.
Figure 2.

Interleukin-1 (IL-1) inhibited the firing rate of laterodorsal tegmental nucleus (LDT) putative cholinergic neurons whole-cell recorded in vitro. (A) mean spike frequency histogram (calculated in 5-s bins) showing the effects of bath perfusion of 25 pg/mL IL-1 on instantaneous firing rates of a putative cholinergic neuron, whole-cell patch-clamp recorded in current clamp mode. The black bar denotes the time during which IL-1 is present in the recording chamber. The exploded view (above) depicts actual spiking in the designated portion of the record with an expanded time scale. Firing rates during selected 10-s periods are shown above spiking. Responding neurons were identified on the basis of a statistical variation in firing rate as determined by the Kolmogorov-Smirnov test.34,35 (B) summary of IL-1 effects on the firing rates of 13 responding putative cholinergic LDT neurons. The thick line represents the mean ± SE for all neurons; thin lines display firing rates of the individual neurons. **P < 0.05 vs control (CTR); *P < 0.05 vs washout (WASH) condition (one-way ANOVA and Tukey test). (C) mean spike frequency histogram (calculated in 5 s bins) showing the effects of bath perfusion of heat-inactivated 25 pg/mL IL-1 on instantaneous firing rates of a putative cholinergic neuron, recorded as in A.
We adopted the conservative approach of including neurons in statistical analysis if discharge rates differed between IL-1 and washout conditions, even if firing rates did not differ between pre-administration control and IL-1 conditions. To establish the specificity of IL-1 effects on firing of putative cholinergic LDT neurons, heat-inactivated IL-1 (90°C, 30 min) was administered to 8 of the 17 neurons in which IL-1 was also tested (Figure 2C). Firing rate was not modified by heat-inactivated IL-1 (4.0 ± 0.5 Hz before and 3.9 ± 0.5 Hz after denatured IL-1 administration) irrespective of whether neurons responded (n = 5) or not (n = 3) to subsequent IL-1 administration.
Bath application of 25 pg/mL (1.4 pM) IL-1 significantly reduced the amplitude of evPSPs in 3 responding neurons out of 5 recorded putative cholinergic LDT neurons (Figure 3A). These evPSPs were glutamatergic, as they were abolished (Figure 3B) by perfusion with the NMDA (N-methyl-D-aspartic acid) and non-NMDA antagonists APV (50 μM) and DNQX (20 μM). Bath application of the GABAA antagonist BMI did not modify evPSP amplitude (Figure 3C). Heat inactivated IL-1 was also used to determine the specificity of IL-1 effects in the stimulation protocol adopted above. The amplitude of evPSPs was not affected by heat-inactivated IL-1 in seven recorded putative cholinergic LDT neurons (Figure 3D).
Figure 3.
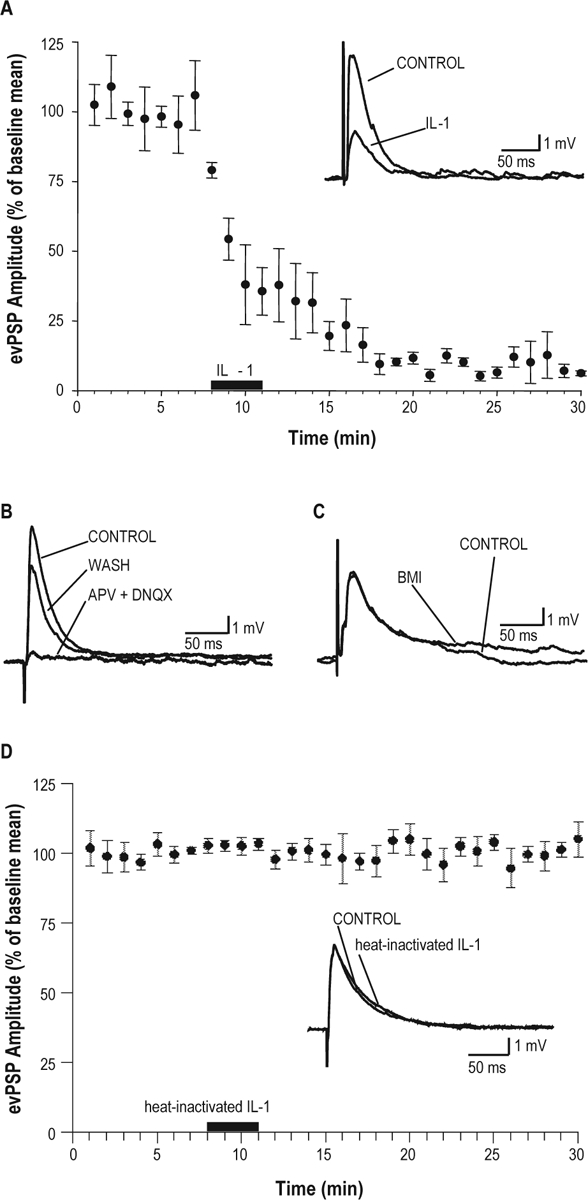
Interleukin-1(IL-1) inhibited the amplitude of glutamatergic evoked post-synaptic potentials in putative cholinergic neurons in a laterodorsal tegmental nucleus (LDT) slice preparation. (A) Time course of inhibition of the depolarizing post-synaptic potentials (evPSPs) amplitude induced by IL-1 administration (25 pg/mL, as depicted by the black bar on the x-axis) in 3 responding of 5 recorded neurons (Student t-test). Inset: IL-1-induced inhibition of the depolarizing evPSP amplitude in a representative putative cholinergic LDT neuron, 10 min after beginning of IL-1 application. (B) Depolarizing evPSPs are glutamatergic. Administration of NMDA and non-NMDA glutamate receptor antagonists [dl-2-amino-5-phosphonopentanoic acid (APV, 50 μM) and 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX, 20 μM)] completely blocked the depolarizing evPSP. (C) Amplitude of evPSPs is not affected by GABAA antagonism. Administration of the GABAA receptor antagonist [(–)-bicuculline methiodide (BMI 10 μM)] did not modify the amplitude of depolarizing evPSP. (D) IL-1 effect on evPSPs is specific. Heat-inactivated IL-1 (25 pg/mL) did not affect the amplitude of evPSP in putative cholinergic LDT neurons (n = 7). Inset: evPSP amplitude recorded prior and 10 min after beginning of heat-inactivated IL-1 application in a representative LDT cholinergic neuron. All evPSPs shown in A (inset), B, C, and D (inset) were recorded at Vm = −70 mV and represent the average of 6 consecutive evPSP for each condition.
In Vivo Results
IL-1 inhibited REM sleep amount and increased tcort when microinjected into the central portion of the LDT
During post-injection hours 3-4, microinjection of 1 ng IL-1 into the central portion of the LDT (n = 6; Figure 4) decreased amount of time spent in REM sleep from 12.7% ± 1.5% of recording time (after vehicle) to 6.1% ± 1.4% (Figure 5A). The amount of time spent in REM sleep was also reduced during post-injection hours 8-9 from 18.6% ± 1.8% of recording time (after vehicle) to 9.3% ± 2.0% (P < 0.01; Figure 5A). During post-injection hours 3-4 and post-injection hours 8-9, the decrease in REM sleep amount was due to a significant decrease in the number of REM sleep bouts/hour from 5.6 ± 0.5 (after vehicle) to 2.6 ± 0.4.
Figure 4.

Location of microinjection sites. Upper panel: schematic representation of location of microinjection sites. Coronal sections (left) and corresponding and enlarged details of the areas of interest (right) [from27, with modifications]. Distances are from the interaural line. Microinjection sites within the central portion of the laterodorsal tegmental nucleus (LDT) (n = 12) are represented by open circles. More rostral microinjection sites (n = 6) are represented by open triangles. Lower panel: microphotograph of a histological section showing site of microinjection (indicated by an arrow) into the LDT. Section shown corresponds approximately to the section at –0.12 mm from interaural line shown in the upper panel. DRN: dorsal raphe nucleus; DTg: dorsal tegmental nucleus; LDT: laterodorsal tegmental nucleus; Me5: mesencephalic trigeminal nucleus; scp: superior cerebellar peduncle; vlPAG: ventrolateral periaqueductal gray. Calibration bar, 500 mm.
Figure 5.

Interleukin-1 (IL-1) microinjected into the laterodorsal tegmental nucleus (LDT) inhibited REM sleep. Changes induced in amount of time spent in wakefulness, NREM sleep, and REM sleep by 1 ng interleukin-1 (IL-1) microinjection into the central portion of the laterodorsal tegmental nucleus (A; n = 6) of freely behaving rats or more rostrally (B; n = 4). Each animal received vehicle (pyrogen-free saline containing 0.1% bovine serum albumin; open circles) and 1 ng IL-1 (filled circles). Symbols depict means ± SEM. The dark bars on the x-axis indicate the dark portion of the light-dark cycle. **P ≤ 0.01 vs. vehicle.
Amounts of time spent in wakefulness and NREM sleep were not significantly affected by microinjection of 1 ng IL-1 into the central portion of the LDT (Figure 5A). Microinjection of 0.5 ng IL-1 within the central portion of the LDT (n = 6) did not significantly alter sleep-wake activity (data not shown).
Microinjection of 0.5 ng IL-1 into the central portion of the LDT induced a significant 0.7 ± 0.2°C Tcort increase (treatment – control) that peaked in post-injection hour 3. Tcort was not modified by microinjection of 1 ng IL-1 into the central portion of the LDT.
Effects of IL-1 rostral microinjection
When microinjections missed the central portion of the LDT, they were located more rostrally (Figure 4). In this region, microinjection of 1 ng IL-1 (n = 4) induced a significant increase in amount of time spent in REM sleep during post-injection hours 7-12 from 10.4% ± 1.2% of recording time (after vehicle) to 17.0% ± 1.0% (Figure 5B). Amount of time spent in other vigilance states (Figure 5B), as well as Tcort were not modified by rostral microinjections of 1 ng IL-1 (Figure 4). In animals receiving 0.5 ng IL-1, microinjections were placed rostrally only in 2 animals of 8 tested, so the effect of this dose was not statistically evaluated under this condition.
DISCUSSION
Results of the present study show that IL-1 (1) inhibits firing rate of putative cholinergic LDT neurons in vitro; (2) reduces the amplitude of glutamatergic evPSPs in the same neurons; and (3) when microinjected into the LDT of freely behaving rats, reduces REM sleep. These results for the first time identify one specific brain area where IL-1 can act to inhibit REM sleep. Moreover, the present results suggest that putative cholinergic LDT neurons may represent one neuronal cell type mediating IL-1-induced suppression of REM sleep, because IL-1 inhibits firing of these neurons. The inhibition of firing rate of putative cholinergic LDT neurons by IL-1 may be due to inhibition of excitatory glutamatergic responses.
Since cholinergic LDT neurons are part of the neuronal circuitry responsible for REM sleep generation,9 results of this study provide a neurophysiological basis for REM sleep suppression induced by IL-1. Moreover, results of the present study complement and extend extant literature demonstrating IL-1 inhibitory effects on cholinergic neurons in other brain areas.20–22 Cholinergic LDT neurons receive dense glutamatergic innervation, originating in part from neurons in the pontine reticular formation and its oral part (pontine reticular nucleus-oral part, PnO).16 Our results demonstrate that IL-1 inhibits evoked glutamatergic responses in putative cholinergic LDT neurons. Therefore, IL-1 may reduce firing of cholinergic LDT neurons by inhibiting an excitatory drive. These results agree with previously published data showing that IL-1 induces a profound decrease of evoked glutamatergic excitatory responses in other brain regions, such as in hippocampal CA1 pyramidal neurons.36
Interleukin-1 stimulates adenosine production,37,38 which exerts tonic inhibitory control on cholinergic LDT neurons.23,39 In the LDT, increased extracellular adenosine levels inhibit glutamate release via actions on presynaptic adenosine A1 receptors.34 Involvement of local glial cells in the control of adenosine-mediated inhibition of glutamate release34 may further amplify and extend the duration of IL-1 effects. These previous observations may explain why in this study IL-1-induced inhibition of firing of putative cholinergic LDT neurons peaks few minutes after cessation of IL-1 application to the bath perfusate, and why IL-1-induced inhibition of evoked glutamatergic responses is not washable within 30 min. The observation that IL-1-induced inhibition of glutamatergic responses in the hippocampus is mediated by adenosine36 also supports such a hypothesis. We determined IL-1 effects on firing rate during basal, unstimulated conditions, whereas post-synaptic potentials were electrically evoked. These different aspects of the protocol may explain why IL-1 effects on evEPSs amplitude do not wash out within 30 min, whereas IL-1 effects on firing rates do.
In rats, suppression of REM sleep induced by intracerebroventricular (ICV) administration of IL-1 is apparent by the first or second post-injection hour and may last up to 8 hours.5,6,40–42 Furthermore, REM sleep of rats is completely abolished during some periods after ICV administration of IL-1.42 In contrast to effects on REM sleep of ICV administration, results of this study indicate that microinjection of IL-1 directly into the LDT of rats does not completely abolish REM sleep, and REM sleep suppression is somewhat delayed. Although this present study demonstrates that REM sleep is reduced by about 50% during effective periods after microinjection of IL-1 into the LDT, these effects are not apparent until the third post-injection hour. Availability of exogenously applied IL-1 to other structures involved in the regulation/modulation of REM sleep may explain differences between ICV administration and microinjections into LDT in the time course and magnitude of responses. Substances administered ICV are rapidly available to the entire brain,43 including the hypothalamus and brainstem, and structures such as the choroid plexus. These brain regions are characterized by high concentrations of IL-1 receptors.44 On the other hand, when IL-1 is microinjected into the LDT, it has only local access to cells, and is not available to other brain areas involved in REM sleep regulation/modulation, or in regions where IL-1 could induce its own synthesis or the synthesis of other immunomodulators.45,46 The lack of availability to disparate brain regions is one likely explanation for the delay in REM sleep suppression induced by IL-1 microinjection into the LDT, relative to rapid onset of effects after ICV administration.
Availability of IL-1 may also explain differences in responsiveness of putative cholinergic LDT neurons in vitro and behavioral effects of IL-1 in freely-behaving animals. Neurons from which in vitro recordings are obtained are located in the superficial layers of the slice preparation, within 150 μm of the slice surface. Since IL-1 is provided in the bath perfusate, recorded neurons are continuously exposed to IL-1, at a constant concentration and in the absence of proteases. During in vitro recordings, effects of IL-1 on a single neuron are determined, whereas actions on a vast population of neurons in a given brain area are necessary to alter behavior of a freely moving animal. Furthermore, proteases in brain degrade IL-1, which may reduce concentrations of biologically active IL-1 and as a consequence diminish the number of neurons upon which IL-1 may act. As such, the inhibition of a relatively few neurons in vivo may be ineffective in altering behavior. In the experimental conditions of the present study, we speculate the delay in REM sleep suppression after microinjection of IL-1 reflects a requirement for IL-1 to induce its own synthesis,45,46 which would result in concentrations high enough so that IL-1 could spread and inhibit a greater number of neurons that may be necessary to produce a change in behavior. The balance between IL-1 degradation by proteases and the synthesis of new IL-1 may also explain the second phase of IL-1-induced REM sleep suppression, i.e., the inhibition that occurs during post-injection hours 8-9. Biphasic NREM sleep responses to IL-1 administration have been consistently reported,5,6,40,42 an effect hypothesized to reflect the synthesis of new IL-1 protein. Additional experiments are necessary to determine if such hypotheses are valid.
This present study demonstrates that when microinjections miss the central portion of the LDT and are placed more rostrally, in a region that includes the rostral margins of the LDT, the dorsal raphe nucleus (DRN), and the ventrolateral periaqueductal gray (vlPAG), REM sleep increases. These results indicate that effects of microinjection of IL-1 into the brainstem are site-specific. Serotonergic DRN neurons are REM-off and their inhibition is permissive for REM sleep manifestation.9 IL-1 inhibits serotonergic DRN neurons in vitro,29,35 and as such, increases in REM sleep after microinjection of IL-1 into or close to the DRN may result from inhibition of REM-off serotonergic neurons. In addition, GABAergic neurons within the vlPAG have been previously considered to be important for the inhibition of brainstem REM-off aminergic neurons and, as such, for REM sleep generation.9 On the other hand, GABAergic neurons in the vlPAG inhibit neurons of the sublaterodorsal nucleus,47 which have been proposed to represent executive REM-on neurons. Interleukin-1 potentiates GABA effects, acting at both pre- and post-synaptic levels.35,36,48–52 As such, data in the present study would be consistent with the hypothesis that at least a subset of GABAergic neurons in the vlPAG facilitate REM sleep suppression and may be considered part of the REM-off circuitry.
Microinjection of IL-1 into the LDT induces a transient and modest increase in Tcort. Observations that, under the conditions of this study, changes in Tcort and changes in REM sleep are induced by different doses of IL-1 (0.5 and 1 ng, respectively) suggest that these two biological responses are dissociable. Observations that hypothalamic warming (1°C) does not alter REM sleep,53 whereas increasing ambient temperature from 23°C to 29°C doubles the amount of REM sleep,54 suggest that REM sleep suppression induced by microinjection of IL-1 into the LDT is not a byproduct of an increase in brain temperature. Observations that Tcort is not altered when microinjections miss the LDT demonstrate that the increase in Tcort induced by IL-1 administration into the LDT is due to specific actions on the LDT.
Stimulation of the brain cholinergic system promotes heat dissipation.55,56 Cholinergic stimulation of the anterior hypothalamus and preoptic area reduces body or brain temperature,57–61 whereas blockade of muscarinic M2 receptors in the same area increases brain temperature.62 Since hypothalamic thermoregulatory areas receive a dense cholinergic innervation originating from the LDT/PPT nuclei,63 results of this study showing that IL-1 inhibits firing of LDT cholinergic neurons may account for the increase in Tcort induced by IL-1 microinjection into the LDT. Such a hypothesis is further supported by data showing that IL-1 inhibits the central cholinergic system at multiple levels.20–22
REM sleep is suppressed during infection.1 Although little, if any, effort has focused on determining the functional significance, it has been hypothesized1 that suppression of REM sleep during infection may contribute to survival of the host, and that the relationship between altered sleep and fever during infection is a critical determinant of outcome. The adaptive role of fever in surviving infection has been amply demonstrated.2 The major thermoregulatory effector mechanisms (i.e., shivering, panting) are impaired during REM sleep.64 Generation of a febrile response is an active process requiring an elevated thermoregulatory set point, reduction of heat loss, and increased heat production (i.e., shivering). As such, one potential function of suppression of REM sleep during infection may be to facilitate the generation of fever,1 since the suppression of REM sleep allows shivering and, as a consequence, the increase in body temperature. IL-1-induced fever is generated at the hypothalamic level,65 whereas results of the present study suggest that IL-1-induced suppression of REM sleep is mediated, in part, at the brainstem level. Thus, IL-1 acts at different brain regions to regulate/modulate REM sleep and body temperature. By acting independently on different brain regions, IL-1 is likely to play an important role in orchestrating the collective of host defense mechanisms.
DISCLOSURE STATEMENT
This was not an industry supported study. Dr. Opp has received research support from Sepracor.
ACKNOWLEDGMENTS
This work was supported, in part, by the National Institutes of Health grant MH 64843 (MRO, LI). The authors thank Dr. Francesca Arnaboldi for technical assistance in immunofluorescence identification of recorded neurons.
REFERENCES
- 1.Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci. 2009;10:199–210. doi: 10.1038/nrn2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kluger MJ, Kozak W, Conn CA, Leon LR, Soszynski D. The adaptive value of fever. Infect Dis Clin North Am. 1996;10:1–20. doi: 10.1016/s0891-5520(05)70282-8. [DOI] [PubMed] [Google Scholar]
- 3.Opp MR. Cytokines and sleep. Sleep Med Rev. 2005;9:355–64. doi: 10.1016/j.smrv.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines: neuromodulators in normal brain? J Neurochem. 2000;74:457–71. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- 5.Gemma C, Imeri L, De Simoni MG, Mancia M. Interleukin-1 induces changes in sleep, brain temperature, and serotonergic metabolism. Am J Physiol. 1997;272:R601–6. doi: 10.1152/ajpregu.1997.272.2.R601. [DOI] [PubMed] [Google Scholar]
- 6.Opp MR, Obal F, Jr, Krueger JM. Interleukin 1 alters rat sleep: temporal and dose-related effects. Am J Physiol. 1991;260:R52–8. doi: 10.1152/ajpregu.1991.260.1.R52. [DOI] [PubMed] [Google Scholar]
- 7.Olivadoti MD, Opp MR. Effects of i.c.v. administration of interleukin-1 on sleep and body temperature of interleukin-6-deficient mice. Neuroscience. 2008;153:338–48. doi: 10.1016/j.neuroscience.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obal F, Jr, Krueger JM. Biochemical regulation of non-rapid-eye-movement sleep. Front Biosci. 2003;8:d520–50. doi: 10.2741/1033. [DOI] [PubMed] [Google Scholar]
- 9.McCarley RW. Neurobiology of REM and NREM sleep. Sleep Med. 2007;8:302–30. doi: 10.1016/j.sleep.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Thakkar M, Portas C, McCarley RW. Chronic low-amplitude electrical stimulation of the laterodorsal tegmental nucleus of freely moving cats increases REM sleep. Brain Res. 1996;723:223–7. doi: 10.1016/0006-8993(96)00256-9. [DOI] [PubMed] [Google Scholar]
- 11.Webster HH, Jones BE. Neurotoxic lesions of the dorsolateral pontomesencephalic tegmentum-cholinergic cell area in the cat. II. Effects upon sleep-waking states. Brain Res. 1988;458:285–302. doi: 10.1016/0006-8993(88)90471-4. [DOI] [PubMed] [Google Scholar]
- 12.El Mansari M, Sakai K, Jouvet M. Unitary characteristics of presumptive cholinergic tegmental neurons during the sleep-waking cycle in freely moving cats. Exp Brain Res. 1989;76:519–29. doi: 10.1007/BF00248908. [DOI] [PubMed] [Google Scholar]
- 13.Steriade M, Pare D, Datta S, Oakson G, Curro DR. Different cellular types in mesopontine cholinergic nuclei related to ponto-geniculo-occipital waves. J Neurosci. 1990;10:2560–79. doi: 10.1523/JNEUROSCI.10-08-02560.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitani A, Ito K, Mitani Y, McCarley RW. Morphological and electrophysiological identification of gigantocellular tegmental field neurons with descending projections in the cat: I. Pons. J Comp Neurol. 1988;268:527–45. doi: 10.1002/cne.902680405. [DOI] [PubMed] [Google Scholar]
- 15.Semba K, Reiner PB, Fibiger HC. Single cholinergic mesopontine tegmental neurons project to both the pontine reticular formation and the thalamus in the rat. Neuroscience. 1990;38:643–54. doi: 10.1016/0306-4522(90)90058-c. [DOI] [PubMed] [Google Scholar]
- 16.Semba K. The mesopontine cholinergic system: a dual role in REM sleep and wakefulness. In: Lydic R, Baghdoyan HA, editors. Handbook of behavioral state control cellular and molecular mechanisms. Boca Raton: CRC Press; 1999. pp. 161–80. [Google Scholar]
- 17.Baghdoyan HA. Schwarz WJ. Sleep science: integrating basic research and clinical practice. Basel: Karger; 2002. Cholinergic mechanisms regulating REM sleep; pp. 88–116. [Google Scholar]
- 18.Imeri L, Bianchi S, Angeli P, Mancia M. Selective blockade of different brain stem muscarinic receptor subtypes: effects on the sleep-wake cycle. Brain Res. 1994;636:68–72. doi: 10.1016/0006-8993(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 19.Maloney KJ, Mainville L, Jones BE. Differential c-Fos expression in cholinergic, monoaminergic, and GABAergic cell groups of the pontomesencephalic tegmentum after paradoxical sleep deprivation and recovery. J Neurosci. 1999;19:3057–72. doi: 10.1523/JNEUROSCI.19-08-03057.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rada P, Mark GP, Vitek MP, et al. Interleukin-1 beta decreases acetylcholine measured by microdialysis in the hippocampus of freely moving rats. Brain Res. 1991;550:287–90. doi: 10.1016/0006-8993(91)91330-4. [DOI] [PubMed] [Google Scholar]
- 21.Carmeliet P, Van DJ, Denef C. Interleukin-1 beta inhibits acetylcholine synthesis in the pituitary corticotropic cell line AtT20. Brain Res. 1989;491:199–203. doi: 10.1016/0006-8993(89)90106-6. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Liu L, Kang J, et al. Neuronal-glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J Neurosci. 2000;20:149–55. doi: 10.1523/JNEUROSCI.20-01-00149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arrigoni E, Rainnie DG, McCarley RW, Greene RW. Adenosine-mediated presynaptic modulation of glutamatergic transmission in the laterodorsal tegmentum. J Neurosci. 2001;21:1076–85. doi: 10.1523/JNEUROSCI.21-03-01076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ford B, Holmes CJ, Mainville L, Jones BE. GABAergic neurons in the rat pontomesencephalic tegmentum: codistribution with cholinergic and other tegmental neurons projecting to the posterior lateral hypothalamus. J Comp Neurol. 1995;363:177–96. doi: 10.1002/cne.903630203. [DOI] [PubMed] [Google Scholar]
- 25.Greene RW, Rainnie DG. Mechanisms affecting neuronal excitability in brainstem cholinergic centers and their impact on behavioral state. In: Lydic R, Baghdoyan HA, editors. Handbook of behavioral state control: cellular and molecular mechanisms. Boca Raton: CRC Press; 1999. pp. 277–96. [Google Scholar]
- 26.Williams JA, Reiner PB. Noradrenaline hyperpolarizes identified rat mesopontine cholinergic neurons in vitro. J Neurosci. 1993;13:3878–83. doi: 10.1523/JNEUROSCI.13-09-03878.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. fifth edition. Burlington, MA: Elsevier Academic Press, Inc.; 2005. [Google Scholar]
- 28.Opp MR. Rat strain differences suggest a role for corticotropin-releasing hormone in modulating sleep. Physiol Behav. 1997;63:67–74. doi: 10.1016/s0031-9384(97)00390-9. [DOI] [PubMed] [Google Scholar]
- 29.Manfridi A, Brambilla D, Bianchi S, Mariotti M, Opp MR, Imeri L. Interleukin-1beta enhances non-rapid eye movement sleep when microinjected into the dorsal raphe nucleus and inhibits serotonergic neurons in vitro. Eur J Neurosci. 2003;18:1041–9. doi: 10.1046/j.1460-9568.2003.02836.x. [DOI] [PubMed] [Google Scholar]
- 30.Kamondi A, Williams JA, Hutcheon B, Reiner PB. Membrane properties of mesopontine cholinergic neurons studied with the whole-cell patch-clamp technique: implications for behavioral state control. J Neurophysiol. 1992;68:1359–72. doi: 10.1152/jn.1992.68.4.1359. [DOI] [PubMed] [Google Scholar]
- 31.Kang Y, Kitai ST. Electrophysiological properties of pedunculopontine neurons and their postsynaptic responses following stimulation of substantia nigra reticulata. Brain Res. 1990;535:79–95. doi: 10.1016/0006-8993(90)91826-3. [DOI] [PubMed] [Google Scholar]
- 32.Leonard CS, Llinas R. Electrophysiology of mammalian peduncolopontine and laterodorsal tegmental neurons in vitro: implications for the control of REM sleep. In: Steriade M, Biesold D, editors. Brain cholinergic system. New York: Oxford University Press; 1990. pp. 205–23. [Google Scholar]
- 33.Luebke JI, Greene RW, Semba K, Kamondi A, McCarley RW, Reiner PB. Serotonin hyperpolarizes cholinergic low-threshold burst neurons in the rat laterodorsal tegmental nucleus in vitro. Proc Natl Acad Sci U S A. 1992;89:743–7. doi: 10.1073/pnas.89.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brambilla D, Chapman D, Greene R. Adenosine mediation of presynaptic feedback inhibition of glutamate release. Neuron. 2005;46:275–83. doi: 10.1016/j.neuron.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 35.Brambilla D, Franciosi S, Opp MR, Imeri L. Interleukin-1 inhibits firing of serotonergic neurons in the dorsal raphe nucleus and enhances GABAergic inhibitory post-synaptic potentials. Eur J Neurosci. 2007;26:1862–9. doi: 10.1111/j.1460-9568.2007.05796.x. [DOI] [PubMed] [Google Scholar]
- 36.Luk WP, Zhang Y, White TD, et al. Adenosine: a mediator of interleukin-1beta-induced hippocampal synaptic inhibition. J Neurosci. 1999;19:4238–44. doi: 10.1523/JNEUROSCI.19-11-04238.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sperlagh B, Baranyi M, Hasko G, Vizi ES. Potent effect of interleukin-1 beta to evoke ATP and adenosine release from rat hippocampal slices. J Neuroimmunol. 2004;151:33–39. doi: 10.1016/j.jneuroim.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Zhu G, Okada M, Yoshida S, et al. Involvement of Ca(2+)-induced Ca2+ releasing system in interleukin-1beta-associated adenosine release. Eur J Pharmacol. 2006;532:246–52. doi: 10.1016/j.ejphar.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 39.Rainnie DG, Grunze HC, McCarley RW, Greene RW. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science. 1994;263:689–92. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imeri L, Bianchi S, Mancia M. Muramyl dipeptide and IL-1 effects on sleep and brain temperature after inhibition of serotonin synthesis. Am J Physiol. 1997;273:R1663–8. doi: 10.1152/ajpregu.1997.273.5.R1663. [DOI] [PubMed] [Google Scholar]
- 41.Imeri L, Ceccarelli P, Mariotti M, Manfridi A, Opp MR, Mancia M. Sleep, but not febrile responses of Fisher 344 rats to immune challenge are affected by aging. Brain Behav Immun. 2004;18:399–404. doi: 10.1016/j.bbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 42.Opp MR, Imeri L. Rat strains that differ in corticotropin-releasing hormone production exhibit different sleep-wake responses to interleukin 1. Neuroendocrinology. 2001;73:272–84. doi: 10.1159/000054644. [DOI] [PubMed] [Google Scholar]
- 43.Proescholdt MG, Hutto B, Brady LS, Herkenham M. Studies of cerebrospinal fluid flow and penetration into brain following lateral ventricle and cisterna magna injections of the tracer [14C]inulin in rat. Neuroscience. 2000;95:577–92. doi: 10.1016/s0306-4522(99)00417-0. [DOI] [PubMed] [Google Scholar]
- 44.Schobitz B, De Kloet ER, Holsboer F. Gene expression and function of interleukin 1, interleukin 6 and tumor necrosis factor in the brain. Prog Neurobiol. 1994;44:397–432. doi: 10.1016/0301-0082(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 45.Dinarello CA, Ikejima T, Warner SJ, et al. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. 1987;139:1902–10. [PubMed] [Google Scholar]
- 46.Taishi P, Churchill L, De A, Obal F, Jr, Krueger JM. Cytokine mRNA induction by interleukin-1beta or tumor necrosis factor alfa in vitro and in vivo. Brain Res. 2008;1226:89–98. doi: 10.1016/j.brainres.2008.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–94. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- 48.De A, Churchill L, Obal F, Jr, Simasko SM, Krueger JM. GHRH and IL1beta increase cytoplasmic Ca(2+) levels in cultured hypothalamic GABAergic neurons. Brain Res. 2002;949:209–12. doi: 10.1016/s0006-8993(02)03157-8. [DOI] [PubMed] [Google Scholar]
- 49.Feleder C, Arias P, Refojo D, Nacht S, Moguilevsky J. Interleukin-1 inhibits NMDA-stimulated GnRH secretion: associated effects on the release of hypothalamic inhibitory amino acid neurotransmitters. Neuroimmunomodulation. 2000;7:46–50. doi: 10.1159/000026419. [DOI] [PubMed] [Google Scholar]
- 50.Miller LG, Galpern WR, Dunlap K, Dinarello CA, Turner TJ. Interleukin-1 augments gamma-aminobutyric acidA receptor function in brain. Mol Pharmacol. 1991;39:105–8. [PubMed] [Google Scholar]
- 51.Tabarean IV, Korn H, Bartfai T. Interleukin-1beta induces hyperpolarization and modulates synaptic inhibition in preoptic and anterior hypothalamic neurons. Neuroscience. 2006;141:1685–95. doi: 10.1016/j.neuroscience.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 52.Zeise ML, Madamba S, Siggins GR. Interleukin-1 beta increases synaptic inhibition in rat hippocampal pyramidal neurons in vitro. Regul Pept. 1992;39:1–7. doi: 10.1016/0167-0115(92)90002-c. [DOI] [PubMed] [Google Scholar]
- 53.McGinty D, Szymusiak R, Thomson D. Preoptic/anterior hypothalamic warming increases EEG delta frequency activity within non-rapid eye movement sleep. Brain Res. 1994;667:273–7. doi: 10.1016/0006-8993(94)91506-7. [DOI] [PubMed] [Google Scholar]
- 54.Szymusiak R, Satinoff E. Maximal REM sleep time defines a narrower thermoneutral zone than does minimal metabolic rate. Physiol Behav. 1981;26:687–90. doi: 10.1016/0031-9384(81)90145-1. [DOI] [PubMed] [Google Scholar]
- 55.Lin MT, Wang HC, Chandra A. The effects on thermoregulation of intracerebroventricular injections of acetylcholine, pilocarpine, physostigmine, atropine and hemicholinium in the rat. Neuropharmacology. 1980;19:561–5. doi: 10.1016/0028-3908(80)90027-1. [DOI] [PubMed] [Google Scholar]
- 56.Rodrigues AG, Lima NR, Coimbra CC, Marubayashi U. Intracerebroventricular physostigmine facilitates heat loss mechanisms in running rats. J Appl Physiol. 2004;97:333–8. doi: 10.1152/japplphysiol.00742.2003. [DOI] [PubMed] [Google Scholar]
- 57.Imeri L, Bianchi S, Angeli P, Mancia M. Stimulation of cholinergic receptors in the medial preoptic area affects sleep and cortical temperature. Am J Physiol. 1995;269:R294–9. doi: 10.1152/ajpregu.1995.269.2.R294. [DOI] [PubMed] [Google Scholar]
- 58.Kirkpatrick WE, Lomax P. Temperature changes following iontophoretic injection of acetylcholine into the rostral hypothalamus of the rat. Neuropharmacology. 1970;9:195–202. doi: 10.1016/0028-3908(70)90067-5. [DOI] [PubMed] [Google Scholar]
- 59.Mallick BN, Joseph MM. Role of cholinergic inputs to the medial preoptic area in regulation of sleep-wakefulness and body temperature in freely moving rats. Brain Res. 1997;750:311–7. doi: 10.1016/s0006-8993(96)01400-x. [DOI] [PubMed] [Google Scholar]
- 60.Poole S, Stephenson JD. Effects of noradrenaline and carbachol on temperature regulation of rats. Br J Pharmacol. 1979;65:43–51. doi: 10.1111/j.1476-5381.1979.tb17332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takahashi A, Kishi E, Ishimaru H, Ikarashi Y, Maruyama Y. Role of preoptic and anterior hypothalamic cholinergic input on water intake and body temperature. Brain Res. 2001;889:191–9. doi: 10.1016/s0006-8993(00)03132-2. [DOI] [PubMed] [Google Scholar]
- 62.Imeri L, Bianchi S, Angeli P, Mancia M. Muscarinic receptor subtypes in the medial preoptic area and sleep-wake cycles. Neuroreport. 1996;7:417–20. doi: 10.1097/00001756-199601310-00010. [DOI] [PubMed] [Google Scholar]
- 63.Chiba T, Murata Y. Afferent and efferent connections of the medial preoptic area in the rat: a WGA-HRP study. Brain Res Bull. 1985;14:261–72. doi: 10.1016/0361-9230(85)90091-7. [DOI] [PubMed] [Google Scholar]
- 64.Parmeggiani PL. Thermoregulation and sleep. Front Biosci. 2003;8:s557–67. doi: 10.2741/1054. [DOI] [PubMed] [Google Scholar]
- 65.Dinarello CA. Blocking IL-1 in systemic inflammation. J Exp Med. 2005;201:1355–9. doi: 10.1084/jem.20050640. [DOI] [PMC free article] [PubMed] [Google Scholar]