

Abstract
Motivation: The discovery of new and unexpected biomarkers in cardiovascular disease is a highly data-driven process that requires the complementary power of modern metabolite profiling technologies, bioinformatics and biostatistics. Clinical biomarkers of early myocardial injury are lacking. A prospective biomarker cohort study was carried out to identify, categorize and profile kinetic patterns of early metabolic biomarkers of planned myocardial infarction (PMI) and spontaneous (SMI) myocardial infarction. We applied a targeted mass spectrometry (MS)-based metabolite profiling platform to serial blood samples drawn from carefully phenotyped patients undergoing alcohol septal ablation for hypertrophic obstructive cardiomyopathy serving as a human model of PMI. Patients with SMI and patients undergoing catheterization without induction of myocardial infarction served as positive and negative controls to assess generalizability of markers identified in PMI.
Results: To identify metabolites of high predictive value in tandem mass spectrometry data, we introduced a new feature selection method for the categorization of metabolic signatures into three classes of weak, moderate and strong predictors, which can be easily applied to both paired and unpaired samples. Our paradigm outperformed standard null-hypothesis significance testing and other popular methods for feature selection in terms of the area under the receiver operating curve and the product of sensitivity and specificity. Our results emphasize that this new method was able to identify, classify and validate alterations of levels in multiple metabolites participating in pathways associated with myocardial injury as early as 10 min after PMI.
Availability: The algorithm as well as supplementary material is available for download at: www.umit.at/page.cfm?vpath=departments/technik/iebe/tools/bi
Contact: christian.baumgartner@umit.at
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Due to the rapid progress in mass spectrometry (MS) and in the development of related bioinformatics methods in recent years, novel biomarker discovery efforts for cardiovascular diseases are increasingly incorporating MS-based profiling of complex biological mixtures such as blood, urine or tissue (Ackermann et al., 2006; Gerszten and Wang, 2008; Howie-Esquivel and White, 2008; Kell, 2007; Lewis et al., 2008a). Biomarkers have a substantial impact on the care of patients with cardiovascular disease. For example, troponin is an accepted diagnostic marker for myocardial infarction, and B-type naturetic peptide aids in diagnosis and prognostication in myocardial infarction and heart failure (Collinson and Gaze, 2007; Maisel et al., 2008; Sabatine et al., 2005). However, there are no sensitive and specific early biomarkers of myocardial injury, and therefore the complementary power of modern profiling techniques and emerging bioinformatics tools are being utilized for the discovery of new biomarkers.
Modern metabolite profiling to analyze low-molecular weight analytes such as nucleotides, amino acids, organic acids, sugars or peptides is typically performed by nuclear magnetic resonance spectroscopy or tandem mass spectrometry (MS/MS) technologies (Baumgartner and Graber, 2007; Dettmer et al., 2007; Jemal and Xia, 2006). In particular, targeted MS-based platforms, using MS/MS coupled with liquid chromatography (LC), allow analysis of metabolites with high sensitivity and structural specificity, and thus minimize potentially confounding clinical variables. However, such platforms still preclude the analysis of large number of samples. From the clinical perspective, comparisons of metabolite profiles from quantitative targeted assays in disease versus non-disease states may bring forth novel biomarkers that have the potential to substantially improve cardiovascular diagnostics and support risk prediction of future life-threatening events (Lewis et al., 2008a,b; Sabatine et al., 2005).
In general, the process of searching for biomarkers in biological samples is highly data driven and dependent on the application of multivariate data mining and statistical bioinformatics methods (Larrañaga et al., 2006; Osl et al., 2008; Shulaev, 2006). Feature selection is a common method for identifying significant variables in multidimensional biomedical data, typically applied prior to classification and biological interpretation. In addition to traditional null-hypothesis significance testing, widely used data mining methods such as filters, wrappers or more powerful meta-learning approaches are used to substantially reduce the dimensionality of data and allow for the search of those variables that exhibit the best discriminatory ability and prediction. Filters rank variables based on their ability to discriminate predefined cohorts. Different entropy-, correlation- or rule-based evaluation measures such as the information gain, reliefF (RF) or associative voting are available to be applied for feature ranking (Hall and Holmes, 2003; Osl et al., 2008; Saeys et al., 2007). In contrast wrappers use classifiers to evaluate the features' discriminatory ability, exploiting various heuristic paradigms for the search through the feature space to identify reliable predictor subsets, but having the drawback of extensive computational costs. Further improvement in feature selection is achieved by introducing meta-learning models such as embedded or ensemble-based methods (Netzer et al., 2009; Saeys et al., 2008).
In biomarker cohort studies a variety of experimental designs are used, from case–control studies to more complex cross-over or serial sampling designs. In particular serial sampling studies allow patients to serve as their own biological controls and permit investigation of kinetic characteristics of circulating analytes by tracking alterations in levels over time. Standard paired null-hypothesis significance testing or repeated measure analysis are the common methods of making statistical decisions from dependent samples (the null hypothesis H0, which usually states that two groups do not differ is rejected in favor of an alternative hypothesis H1, which typically states that the groups differ) using the P-value as an evaluation criterion for the discriminatory ability of variables. Statistical tests calculate whether confidence in a hypothesis based solely on a sample-based estimate exceeds a significance level, but they do not allow for a general, valid, clinically relevant categorization of selected metabolites because the P-value is a random variable defined over the sample space (size) of the experiment. To help circumvent this problem, alternative feature selection methods appear to be the better choice for selecting and prioritizing variables, but these methods as described above rely on concepts of unpaired testing and thus are not readily applicable to dependent, longitudinal data.
In this article, we introduce a new qualitative and quantitative evaluation model for prioritizing metabolic signatures in independent and dependent populations, and perform receiver operating curve (ROC) analysis to estimate the power of the method to search for highly predictive biomarkers compared with statistical significance testing and other popular feature selection methods. We evaluate the method's paired variant on data obtained from applying a targeted MS-based metabolite profiling platform to serial blood samples drawn from patients undergoing alcohol septal ablation for hypertrophic obstructive cardiomyopathy (HOCM), a human model of planned myocardial infarction (PMI) that faithfully reproduces spontaneous myocardial infarction (Lakkis et al., 1998). Data on patients with spontaneous myocardial infarction (SMI) and patients undergoing catheterization without induction of myocardial infarction (controls) were obtained to evaluate the method from the perspective of an independent test hypothesis.
2 METHODS
2.1 Targeted LC–MS/MS metabolite profiling
We incorporated metabolites in our platform to cover a broad array of metabolic pathways and to include ‘sentinels’ in metabolic pathways known to be involved in human energy homeostasis (i.e. TCA cycle intermediates, purine biosynthesis, etc) and cardiovascular disease. Metabolites were excluded if they were not readily detectable by LC–MS/MS analysis. The detailed protocol of MS analysis was recently published in Lewis et al. (2008b). Briefly, blood samples were drawn during the procedure and collected in K2EDTA-treated tubes. Samples were immediately centrifuged and the supernatant plasma was aliquoted for separation using high-performance LC (HPLC). Three HPLC columns for separating sugars, ribonucleotides, organic acids and amino acids were aligned in sequence with a triple quadrupole mass spectrometer (AB4000Q, Applied Biosystems, MA, USA) using a turbo ion spray LC/MS interface. Targeted MS/MS analysis using selective reaction monitoring conditions was performed allowing monitoring a total of 210 metabolites for each sample. Targeted MS methods using MS/MS coupled with LC permit highly specific identification of analytes. Addition of isotope-labeled internal standards for selected metabolites enabled absolute measurements of analyte concentrations by integrating peak areas for parent–daughter ion pairs in MS/MS spectra.
2.2 A clinical model of PMI
We chose a clinical model of PMI that faithfully represents spontaneous myocardial infarction to study kinetics of small molecules in human plasma over time, as previously reported (Lewis et al., 2008b). We applied targeted metabolite profiling to serial blood samples obtained from patients undergoing alcohol septal ablation for HOCM. Serial sampling before (baseline) and, according to our protocol, at 10, 60, 120 and 240 min after injury allowed patients to serve as their own biological controls and permitted kinetic analyses of circulating metabolites, particularly at early stage after injury.
A total of 31 patients who underwent alcohol septal ablation for the treatment of symptomatic HOCM were enrolled in this study. The number of samples, however, partly differed between timepoints at 10 min (n = 31), 60 min (n = 28), 120 min (n = 25) and 240 min (n = 11). Inclusion criteria as well as detailed patient characteristics can be found in Lewis et al. (2008b). The protocol was approved by the Massachusetts General Hospital Institutional Review Board, Boston, MA and all individuals gave written informed consent.
2.3 Patients with SMI
A cohort of 12 patients undergoing emergency cardiac catheterization for acute spontaneous ST-segment elevation myocardial infarction (SMI) was enrolled within 8 h of symptom onset. Blood samples were drawn during emergency catheterization. A second cohort of nine patients undergoing elective, diagnostic cardiac catheterization for cardiovascular disease without acute symptoms of myocardial ischemia was included in this study as a control cohort. In both groups, blood was drawn prior to the onset of catheterization and at 10 and 60 min after the procedure was completed. A total of 26 samples from multiple timepoints in SMI and 18 samples in controls were included. Note that for some timepoints, not all samples were available for MS analysis.
2.4 Computational approach for biomarker search, prioritization and dynamic analysis
2.4.1 Data preprocessing
An extensive review of generated LC–MS data to specified criteria and laboratory action items was performed to ensure a high level of reliability, completeness, reproducibility and consistency in the data. We carefully examined raw data, determining if all requested metabolites were accounted for, and targeted measurement levels were met, and then correlated data of historical experiments to ensure that analyte levels were stable over time. We applied a three-step preprocessing procedure on technically validated data to provide quality assured datasets for the purpose of biomarker search: (i) of the 210 measured metabolites, we excluded metabolites with more than 20% missing values in the dataset (for each cohort separately) to ensure relatively uniform conditions for statistical analysis; (ii) to avoid distortion of statistical results, outlier detection was performed. We applied a common statistical model using the interquartile ranges (IQRs), defining an outlier as observation outside the range [Q1−k · IQR; Q3+k · IQR], with Q1, Q3 = first and third quartiles, IQR = Q3 − Q1. We set k = 3 to only remove ‘extreme’ outliers in the data. In this study, 4.4% of MS values in PMI and 1.2% in SMI were detected as strong outliers and removed from the datasets using this approach; and (iii) for kinetic analysis, we normalized serial PMI data (four timepoints) to the metabolites' baseline levels, denoted in percent changes.
2.4.2 Prioritization model for paired and unpaired dichotomous samples
We developed a new univariate feature selection model for the search and prioritization of metabolic signatures using serial PMI and SMI data.
-
The model's variant addressing the paired test problem: because main features of a diagnostic test [sensitivity, specificity or the area under the ROC (AUC)] are not defined for paired testing, we first introduced a new, objective measure for expressing the discriminatory ability in dependent samples. Therefore the discriminance measure for a paired test problem DA is defined as the percent change of metabolite levels in a cohort in one direction versus baseline. As an example, if 50% of cohort subjects yield increasing or, respectively, decreasing metabolite levels, DA is calculated to be 0.5 (this value corresponds exactly to AUC = 0.5 in unpaired testing), denoting no discrimination. If 75 or 100% of subjects show increasing or, respectively, decreasing levels, DA = 0.75 or 1.0, indicating good or perfect discrimination.
Based on this definition, we developed the model's variant pBI, termed paired Biomarker Identifier, that combines the introduced discriminance measure DA with a biological effect term calculated as the median percent change in metabolite levels at timepoint tx versus baseline, Δchange, divided by the coefficient of variation (CV) in the normalized data:
λ is a scaling factor (λ = 100 by default), DA* is the initial measure DA (range 0.5–1) rescaled between 0 and 1, weighted by the effect term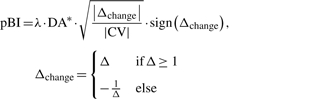
(1) 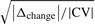 . Note that a CV > 1 is set to 1 by default to consider solely data distributions with smaller variance in normalized data to be interpreted as a positive biological effect. The function sign() determines the direction of change. In summary, pBI is built on four statistical determinants that are DA, magnitude, variance and direction of changes in metabolite levels, permitting a biologically feasible prioritization of metabolic signatures, as we propose, into weak, moderate and strong predictors.
. Note that a CV > 1 is set to 1 by default to consider solely data distributions with smaller variance in normalized data to be interpreted as a positive biological effect. The function sign() determines the direction of change. In summary, pBI is built on four statistical determinants that are DA, magnitude, variance and direction of changes in metabolite levels, permitting a biologically feasible prioritization of metabolic signatures, as we propose, into weak, moderate and strong predictors.To evaluate the power of feature selection, pBI is benchmarked with a paired, one sample, two-tailed significance hypothesis test (Student's t-test or Wilcoxon signed-rank test, the latter if the population is not normally distributed). To our knowledge there are no adequate feature selection methods to be compared with that operate on a paired test hypothesis.
- The model's variant addressing the unpaired test problem: to distinguish between two independent populations we determine the product of the true-positive rates of both classes (TP2) as an objective measure for discrimination. For calculating TP2, classifiers such as support vector machines or logistic regression are used, the latter especially in a biomedical setting (Cristianini and Shawe-Taylor, 2000; Hosmer and Lemeshow, 2000). In a diagnostic test, TP2 is defined as the product of sensitivity and specificity that is an accepted diagnostic parameter tightly associated with the AUC, denoting the probability of correctly classifying a randomly selected true-positive and true-negative subject. The product is now to be interpreted as follows: if sensitivity and specificity is 0.5, TP2 = 0.25, indicating no valuable discrimination (cf. DA = 0.5, paired testing). To guarantee true-positive and true-negative values ≥0.5, we set TP2 = 0 if either sensitivity or specificity is <0.5, thus indicating no discriminatory value, while TP2 = 1.0 depicts perfect discrimination. Analogous to pBI, we developed the variant uBI, termed unpaired Biomarker Identifier that is defined as:
λ is a scaling factor (λ = 100 by default). TP2* is the initial measure TP2 (range 0.25–1) rescaled between 0 and 1, and denotes the discriminatory ability of a metabolite determined from logistic regression analysis in this study (Homser et al., 2000). Analogous to DA, the discriminance measure TP2* is weighted by a biological effect term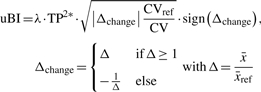
(2) 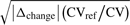 , comprising the parameters Δchange and CV/CVref. The symbol Δchange indicates changes in metabolites calculated as a relative increase or decrease from the levels of a reference group (controls) and is divided by the quotient CV/CVref, denoting changes in the variance of data across the two cohorts where CV < CVref is interpreted as a positive biological effect. CV/CVref values >1 are set to 1 (see also pBI). The function sign() determines the direction of change.
, comprising the parameters Δchange and CV/CVref. The symbol Δchange indicates changes in metabolites calculated as a relative increase or decrease from the levels of a reference group (controls) and is divided by the quotient CV/CVref, denoting changes in the variance of data across the two cohorts where CV < CVref is interpreted as a positive biological effect. CV/CVref values >1 are set to 1 (see also pBI). The function sign() determines the direction of change.  is the mean value of metabolite levels in both classes. It should be noted that through rescaling the initial parameters DA and TP2, the absolute pBI and uBI scores permit a high degree of comparability and generalizability between both feature selection modalities.
is the mean value of metabolite levels in both classes. It should be noted that through rescaling the initial parameters DA and TP2, the absolute pBI and uBI scores permit a high degree of comparability and generalizability between both feature selection modalities.
We benchmarked uBI with two widely used feature selection methods that are the information gain IG [the IG of a feature ai reflects how much information the feature provides on the class attribute cj, and is calculated by IG(ai) = E(cj) − E(cj | ai), where E(cj) is the entropy of the class cj and E(cj | ai) is the conditional entropy of cj given ai] and RF, an instance-based, multivariate feature selection method built on the assumption that useful features have significantly different values for instances of different classes and similar values for instances of the same class as well as standard statistical hypothesis testing using an unpaired, two sample, two-tailed null-hypothesis significance test (Student's t-test or Mann–Whitney U-test if the population is normally versus not normally distributed) (Hall and Holmes, 2003; Osl et al., 2008).
2.4.3 Kinetic mapping
For visualizing dynamic changes in metabolite levels, we employed a 2D pseudocolor representation on serial pBI scores calculated from given metabolites according to the proposed scheme of weak, moderate and strong predictors.
2.4.4 ROC analysis to estimate the discriminatory ability of selected metabolites
To assess the predictive ability of variables to distinguish between two independent samples (e.g. cases versus controls), an established measure of a diagnostic test is the AUC (Fawcett, 2006). It incorporates sensitivity and specificity as the two main features of the test. In the ROC curve, specificity is usually denoted as 1–specificity. An AUC of 1 depicts a test with perfect discrimination, while an AUC of 0.5 denotes an uninformative test (45○ diagonal line in the graph). Alternatively, the product of sensitivity and specificity can be used as a further accepted feature of a diagnostic test. In this study, we also use ROC analysis to compare different feature selection methods and to estimate their power to rank variables according to their discriminatory ability. A χ2-statistics was applied for testing differences between ROC curves being statistically significant.
2.4.5 Cross validation to generalize findings
As PMI and SMI cohorts in this study are of small size, we applied a cross-validation strategy to estimate the degree of reliability of results on a single derivation cohort. Generally, in classification stratified 10-fold cross-validation is an accepted statistical practice of partitioning a sample of data into 10 subsets where each subset is used for testing and the remainder for training, yielding an averaged overall error estimate. For feature selection, the dataset is also subdivided into 10 partitions. The process is repeated 10 times, using nine partitions for generating the feature ranking where rankings of each partitioning are finally aggregated and expressed by a mean ± SD rank (Witten and Frank, 2005). We therefore applied 10-fold cross-validation to the process of feature selection and ranking on PMI data (each timepoint separately) and SMI data to reduce variability in findings. As another layer of validation, we compared the results of both models (PMI versus SMI) to emphasize the methodological and biological plausibility of identified biomarkers.
3 RESULTS
3.1 DA and categorization of metabolites using pBI scores
We applied the new discriminance measure DA to paired testing in order to define three classes of biomarkers of PMI: weak predictors, defined by a DA cutoff of 0.6 (selected metabolites with DA < 0.6 have less or no predictive value), moderate predictors defined by the cutoffs of 0.7 and 0.8 (0.7 ≤DA<0.8), and strong predictors equal to or above the threshold DA = 0.8. Using the transformed measure DA* as implemented in Equation (1), rescaled DA cutoffs (i.e. DA* = 0.2, 0.4 and 0.6) thus allow direct comparison with the corresponding measure TP2* defined for independent samples.
We estimated the strength of the method pBI versus paired Student's t-test/Wilcoxon signed-rank test to select predictors using ROC analysis. DA cutoffs as described above were used to define the dependent variable (dichotomous), and pBI scores and P-values calculated for analytes at all four timepoints served as independent variables. A total of 173 metabolites, comprising sugars, ribonucleotides, organic and amino acids, were included in this analysis after data preprocessing as described in Section 2. Subsequently standard logistic regression analysis was applied to calculate the pBI score thresholds for the proposed categorization scheme. Table 1 and Figure 1 show the detailed results. It can be seen that AUC values ≤0.994 for DA* = 0.2, 0.4 and 0.6 underscore the expected high performance of the method pBI. It was our objective to design a new scoring model that is tightly associated with the discriminance measure DA (DA*), integrating additional metabolic information expressed by the magnitude, variance and direction of metabolite changes, and thus permitting feasible prioritization of candidate biomarkers identified in longitudinal studies. Using paired significance testing, AUC values (0.94, 0.91 and 0.84) were statistically and significantly lower than pBI values (χ 2-statistics, P < 0.001; Table 1), confirming that pBI better ranks variables with regard to their predictive value compared to P-value ratings.
Table 1.
Power of pBI (AUC) and score cuttoffs for analyte categorization on PMI data
| Strong predictors | Moderate predictors | Weak predictors | ||
|---|---|---|---|---|
| DA (DA*) | cutoff | 0.8 (0.6) | 0.7 (0.4) | 0.6 (0.2) |
| |pBI| score | cutoff | 73 | 44 | 21 |
| |pBI| score | AUC | 0.994 | 0.996 | 0.995 |
| P-value | AUC | 0.943 | 0.912 | 0.842 |
| H0: area(pBI) = area(P-value) | χ 2 | 27.5 | 63.1 | 145.5 |
| P > χ2 | 0.0000 | 0.0000 | 0.0000 |
ROC analysis for estimating the power of the method pBI versus paired statistical hypothesis testing (P-value) to select variables according to their predictive value. A χ 2-test was applied to determine statistical significance between ROC curves. Logistic regression analysis was used to estimate pBI score cutoffs for classifying metabolites as weak, moderate and strong predictors. A total of 702 data points, comprising 173 metabolites × four timepoints (excluding 15 NaN values) were used for ROC analysis.
Fig. 1.

ROC curves and AUCs estimated for pBI versus paired statistical hypothesis testing. We used the cutoffs DA* = 0.2 (weak predictors), DA* = 0.4 and 0.6 (moderate and strong predictors, ROC curves not shown) to define the dependent variable for ROC analysis. The inverse P-values and absolute pBI scores were used in this analysis.
As shown in Table 1, the pBI score thresholds were estimated as follows: ≥21 for classifying weak, ≥44 for moderate and ≥73 for strong predictors. It should be noted that pBI may also allow a further prioritization of metabolites beyond the cutoff for ‘strong’ predictors, however relying on the general discriminatory ability of analytes determined by the complexity of the underlying metabolic pathways. For example in less-intricate (monogenic) diseases such as inborn errors of metabolism scores > 100–500 in small sets of metabolites are more likely, and are therefore termed as ‘key’ or ‘primary’ markers, showing extremely elevated levels (up to 10–100-fold higher levels versus reference), and DA or TP2 values close to 1.0 (Baumgartner and Baumgartner, 2006).
3.2 DA and categorization of metabolites using uBI scores
Analogous to pBI, we compared uBI's power to select variables with an unpaired Student's t-test/Mann–Whitney U-test, and two popular entropy- and correlation-based feature selection methods that are the IG and RF using the SMI data (Table 2). Estimating the model's performance for weak predictors (TP2* = 0.2), uBI revealed the largest AUC, achieving statistical significance (P = 0.0269) compared with the other methods. For this predictor class, we estimated an uBI score threshold of 27 that is only 6 score points higher than for pBI. Using TP2* = 0.4, uBI also revealed the largest AUC for the moderate predictor class, however, without achieving statistical significance. The score cutoff for this class was calculated to be 50 (cf. 44 in pBI).
Table 2.
Power of uBI (AUC) and score cutoffs for analyte categorization on SMI data
| Strong predictors | Moderate predictors | Weak predictors | ||
|---|---|---|---|---|
| TP2 (TP2*) | cutoff | 0.7 (0.6) | 0.55 (0.4) | 0.4 (0.2) |
| |uBI| score | cutoff | – | 50 | 27 |
| |uBI| score | AUC | – | 0.990 | 0.994 |
| P-value | AUC | – | 0.980 | 0.971 |
| IG | AUC | – | 0.980 | 0.937 |
| RF | AUC | – | 0.857 | 0.938 |
| H0: area(uBI) = area(P-value) = area(IG) = area(RF) | χ 2 | – | 1.06 | 9.19 |
| P > χ 2 | – | 0.7856 | 0.0269 |
ROC analysis for estimating the power of the method uBI versus unpaired statistical testing (P-value), and IG and RF to rank variables according to their predictive value. A total of 102 metabolites were considered for analysis to estimate the uBI cutoffs for categorization. Too little data were available to perform a representative ROC analysis for TP2* = 0.6.
Not enough data was available beyond TP2* = 0.6 to carry out this analysis. Assuming a positive intercept of roughly 5 score points between PMI and SMI cutoffs, it seems to be likely that the threshold for strong predictors lies in the range of 75–80. However, despite the limited data available for this analysis, uBI yielded the best performance of ranking variables with respect to their discriminatory ability for both the weak and moderate predictor classes. The corresponding ROC curves for the weak predictor class are depicted in Figure 2.
Fig. 2.

ROC curves and AUCs estimated for uBI versus unpaired statistical testing (P-value), IG and RF are depicted. TP2(TP2*) is set to 0.4 (0.2) for defining the weak predictor class. TP2 is the product of sensitivity and specificity. The inverse P-values and absolute uBI scores were used in this analysis.
3.3 Kinetic analysis of metabolites in PMI
We studied peripheral blood samples in a cohort of 31 patients to analyze alterations in analyte levels across multiple timepoints. Figure 3 shows a 2D pseudocolor plot of pBI scores for a selected group of metabolites (amino acids) at 10 min (t10), 60 min (t60) and 240 min (t240) after myocardial injury. Scores are sorted by column t10 in descending order to focus on the investigation of early-appearing biomarker candidates. A 2D plot of additional interesting metabolites is shown in Supplementary Figure A. Within the class of amino acids, e.g. alanine showed decreased concentration levels ≤60 min, while other early metabolites like threonine and serine changes persisted between 10 and 240 min after injury. Interestingly, isoleucine/leucine (Ile/Leu) yielded increased levels as early as 10 min, but decreased levels >60 min after injury and again changes in levels with reverse direction of >240 min. According to the pBI score scheme, all these metabolites revealed moderate predictive value. Tryptophan, phenylalanine and tyrosine classified as strong predictors at the 10- and 60-min timepoint appeared to be promising biomarker candidates. However, these metabolites also changed with cardiac catheterization alone, indicating their lack of specificity for myocardial injury, and therefore were excluded from further analysis.
Fig. 3.

Kinetic map of amino acids on PMI data at 10, 60 and 240 min after myocardial injury using the pBI scores. Red color increments indicate decreasing levels and blue indicates increasing levels.
Interestingly, further candidates of early-appearing metabolites include products of purine and pyrimidine catabolism (ATP, ADP, hypoxanitine, xanitine and malonic acid) being classified as strong predictors, trimethylamine N-oxide (TMNO), which is associated with injury-mediated modulation of dietary compounds, kynurenine and a spectrum of weak predictors including inosine, which has been shown to reduce cardiomyocyte apoptosis (Bäckström et al., 2003; Goldhaber et al., 1982). Figure 4, for example, singles out the dynamic characteristic of hypoxanthine in a bar graph according to the pBI scoring categorization scheme.
Fig. 4.

Kinetic characteristic of hypoxanthine: categorization of pBI scores across the timepoints at 10, 60 and 240 min after PMI are exemplarily shown (height of bars). Values above bars denote median (IQR) of relative changes in levels versus baseline in percent.
3.4 Prioritization of metabolic markers in SMI
We evaluated the uBI model on data provided from patients with SMI presenting for acute coronary angiography versus patients undergoing elective, diagnostic cardiac catheterization without acute coronary syndromes serving as controls. For this analysis, a total of 102 metabolites after data preprocessing were available. Supplementary Figure B shows the prioritized list of metabolites using the uBI score scheme, the values obtained from the methods IG, RF and unpaired statistical testing, the relative increase or decrease of metabolite levels in SMI from those of the reference cohort expressed as median percent change, as well as the product of sensitivity and specificity (TP2) and AUC for each single analyte. Interestingly, malonic acid alone, which is a product of the pyrimidine catabolism, revealed highest predictive value expressed by an AUC = 0.86 and a product of 0.75 (cross-validated) and thus is classified as strong predictor. Not surprisingly, lactic acid, a metabolite related to myocardial anaerobic metabolism, showed a high AUC value of 0.74 as well and was classified as a moderate predictor.
3.5 Validation of metabolic markers in SMI
The exact time of onset of SMIs relative to sample collection was heterogeneous (between 1 and 4 h, 162 ± 102 min), and sample size and number of metabolites were smaller in SMI. However, we found good concordance in direction of changes and absolute pBI and uBI scores in multiple metabolites between SMI and PMI at timepoints 60, 120 and 240 min. Examples of the top-ranked metabolites include: malonic acid [SMI: −97 versus PMI: 84 (60 min), −39 (120 min) and −76 (240 min)] or lactic acid (SMI: 37 versus PMI: −11, 30 and 89). In our recently published study on PMI (Lewis et al., 2008b), we also focused on a group of metabolites that includes aconitic acid, hypoxanthine, TMNO and threonine that changed significantly in a sustained pattern at each of the 60-, 120- and 240-min timepoints after PMI, showing analogous behavior in direction and magnitude of changes in SMI as well.
These results demonstrated that pBI and uBI are feasible models to qualitatively and quantitatively assess altered metabolic patterns in static and dynamic phenotypes, and confirm that metabolic biomarkers derived in the PMI model were similarly changed in the SMI samples, underscoring the effectiveness and the generalizability of this approach.
4 DISCUSSION AND CONCLUSION
A successful metabolic profiling biomarker discovery study in clinical metabolomics relies on a carefully planned experimental design, appropriate clinical study execution, and the use of emerging analytical and bioinformatics tools. We applied a targeted MS-based metabolite platform, which provides high analyte specificity, to blood samples obtained from patients with planned and spontaneous myocardial infarction.
The objective was to search for highly discriminatory metabolic biomarkers participating in pathways associated with myocardial infarction, particularly in the initial hours after myocardial injury when clinically available protein-based biomarkers are not yet elevated. Coupling this technology platform with biostatistics and bioinformatics permits, after careful technical validation and preprocessing of data, the identification, prioritization and verification of biochemicals to characterize the organism's response to an intervention or disease.
To estimate the discriminatory ability of selected single and multiple biomarkers as potential tools for diagnostics, risk prediction or disease screening, we have introduced a new scoring model, termed pBI and uBI, for the univariate search and categorization of altered static and dynamic patterns in multidimensional metabolic data. The proposed data mining method aims at enhancing the predictive power of selected analytes by combining objective measures of discrimination such as DA or TP2 with metabolic determinants expressed by changes in magnitude, variance and direction in metabolite levels versus baseline (pBI) or versus an independent reference group (uBI). It should be emphasized that our model (i) can be applied to both dependent and independent samples, (ii) overcomes the problem of interpreting ‘relative’ P-values that exceed or do not exceed significance levels when applying statistical null-hypothesis testing, (iii) revealed higher performance than statistical testing and other feature selection methods for ranking variables according to their predictive value and (iv) provides a reliable biomarker categorization scheme of high diagnostic and prognostic relevance.
Beyond the utility to study static phenotypes, we chose a serial sampling design in PMI, first to search for correlations and kinetic relations between early- and later-appearing metabolites, thus giving a deeper insight into kinetic mechanisms of metabolites involved in pathways associated with myocardial injury. Secondly, due to the profound degree of interindividual variability observed in multiple cohort studies and limitations of the technology that still suffers from moderate signal-to-noise ratios, serial sampling studies overcome these restrictions because each patient serves as his or her own biological control, which significantly reduces the platform-based variability.
Presently used indicators of myocardial injury such as cardiac troponin or the myocardial isoform of creatine kinase are not readily detectable until at least 4 h after myocardial injury (Zimmerman et al., 1999). In this study, we identified significant metabolite alterations in the PMI cohort that were unambiguously apparent as early as 10 min after injury and validated using a stratified cross-validation strategy. We utilized our scoring model to visualize and prioritize dynamic changes across a spectrum of timepoints using a 2D pseudo color representation that allows for a quick and extensive review for kinetic patterns inherent in the data. It should be noted that because of the small sample size in both PMI and SMI cohorts, statistical reliability of pBI and uBI score thresholds is somewhat limited. Future studies with more data will be necessary to further enhance statistical significance of results. Nonetheless, it could be seen that determined uBI score cutoffs in SMI data show good concordance with pBI cutoffs estimated in PMI, and thus underscore the high degree of comparability and reliability between both scoring model variants.
To assess generalizability, we compared findings in PMI and SMI, serving as another layer of validation that underscores the methodological and biological plausibility demonstrated in this study. A pool of interesting metabolites associated with myocardial injury could be identified and verified, some of which by applying our scoring scheme at the level of weak, moderate or strong predictors, while other metabolites did not fulfill these criteria, but revealed high concordance in direction and magnitude of changes without achieving statistical significance. Nevertheless, differences in predictive value of metabolic biomarker candidates found in PMI versus SMI need to be further discussed from the perspective of heterogeneous sample collection relative to the exact onset of SMI, interindividual variability apparent in regular case–control versus longitudinal studies, and also due to differences in subcellular mechanisms in PMI compared with ‘normal’ patients with myocardial infarction.
In summary, we demonstrated the power of coupling the application of a metabolomics platform to patients with PMI and SMI with efficient statistical bioinformatics methods for aiding in the discovery of new blood biomarkers in disease. Nevertheless, metabolomics remains resource intensive, but with emerging technologies high-throughput metabolite profiling promises to become a reality. We were able to identify a series of metabolites participating in pathways tightly associated with myocardial infarction or human cardiovascular disease in general (i.e. purine and pyrimidine metabolism, the tricarboxylic acid cycle and its upstream contributors, and anaerobic glycolysis), some of which changed as early as 10 min after injury, a time frame in which no currently available clinical biomarkers are present. We confirmed changes in metabolic pathways that are known to be modulated by myocardial injury from animal models of myocardial infarction and highly invasive sampling during cardiopulmonary bypass in humans. For example, we detected sequential purine degradation products (ATP, ADP, hypoxanthine and xanthine), TCA cycle intermediates and intermediates of anaerobic glycolysis (lactic acid) (Bäckström et al., 2003; Goldhaber et al., 1982, Mei et al., 1996, Turgan et al., 1999; Zimmerman et al., 1999). We were also able to find plasma metabolic markers of myocardial injury.
The developed scoring model greatly exceeds traditional feature selection methods including statistical significance testing, and enabled us to identify, categorize and verify candidate biomarkers based on their predictive value in paired and unpaired testing. We propose our new feature selection method as an efficient bioinformatics tool for biomarker discovery in clinical metabolomics to aid in diagnostics and risk prediction of cardiovascular disease.
Funding: Austrian Genome Program GEN-AU, project ‘Bioinformatics Integration Network’ (to C.B. and M.N.); NIH (K23HL091106 to G.D.L. and R01 HL072872, U01HL083141 and R01DK081572 to R.E.G.); the Donald W. Reynolds Foundation (to R.E.G.); Foundation Leducq (to R.E.G.); American Heart Association Fellow-to-Faculty Award (to G.D.L.); Established Investigator Award (to R.E.G.).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Ackermann BL, et al. The role of mass spectrometry in biomarker discovery and measurement. Curr. Drug Metab. 2006;7:525–539. doi: 10.2174/138920006777697918. [DOI] [PubMed] [Google Scholar]
- Baumgartner C, Baumgartner D. Biomarker discovery, disease classification, and similarity query processing on high-throughput MS/MS data of inborn errors of metabolism. J. Biomol. Screen. 2006;11:90–99. doi: 10.1177/1087057105280518. [DOI] [PubMed] [Google Scholar]
- Baumgartner C, Graber A. Data mining and knowledge discovery in metabolomics. In: Masseglia F, Poncelet P, et al., editors. Successes and New Directions in Data Mining. Hershey, PA and London, UK: IGI Global; 2007. pp. 141–166. [Google Scholar]
- Bäckström T, et al. Cardiac outflow of amino acids and purines during myocardial ischemia and reperfusion. J. Appl. Physiol. 2003;94:1122–1128. doi: 10.1152/japplphysiol.00138.2002. [DOI] [PubMed] [Google Scholar]
- Collinson PO, Gaze DC. Biomarkers of cardiovascular damage and dysfunction–an overview. Heart Lung Circ. 2007;16(Suppl. 3):S71–S82. doi: 10.1016/j.hlc.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Cristianini N, Shawe-Taylor J. An Introduction to Support Vector Machines and Other Kernel-based Learning Methods. Cambridge: Cambridge University Press; 2000. [Google Scholar]
- Dettmer K, et al. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006;27:861–874. [Google Scholar]
- Gerszten RE, Wang TJ. The search for new cardiovascular biomarkers. Nature. 2008;451:949–952. doi: 10.1038/nature06802. [DOI] [PubMed] [Google Scholar]
- Goldhaber SZ, et al. Inosine: a protective agent in an organ culture model of myocardial ischemia. Circ. Res. 1982;51:181–188. doi: 10.1161/01.res.51.2.181. [DOI] [PubMed] [Google Scholar]
- Hall MA, Holmes G. Benchmarking attribute selection techniques for discrete class data mining. IEEE Trans. Knowl. Data Eng. 2003;15:1437–1447. [Google Scholar]
- Hosmer DW, Lemeshow S. Applied Logistic Regression. 2. New York, NY: Wiley; 2000. [Google Scholar]
- Howie-Esquivel J, White M. Biomarkers in acute cardiovascular disease. J. Cardiovasc. Nurs. 2008;23:124–131. doi: 10.1097/01.JCN.0000305072.49613.92. [DOI] [PubMed] [Google Scholar]
- Jemal M, Xia YQ. LC-MS development strategies for quantitative bioanalysis. Curr. Drug Metab. 2006;7:491–502. doi: 10.2174/138920006777697927. [DOI] [PubMed] [Google Scholar]
- Kell DB. Metabolomic biomarkers: search, discovery and validation. Expert Rev. Mol. Diagn. 2007;7:329–133. doi: 10.1586/14737159.7.4.329. [DOI] [PubMed] [Google Scholar]
- Lakkis NM, et al. Echocardiography-guided ethanol septal reduction for hypertrophic obstructive cardiomyopathy. Circulation. 1998;98:1750–1755. doi: 10.1161/01.cir.98.17.1750. [DOI] [PubMed] [Google Scholar]
- Larrañaga P, et al. Machine learning in bioinformatics. Brief. Bioinform. 2006;7:86–112. doi: 10.1093/bib/bbk007. [DOI] [PubMed] [Google Scholar]
- Lewis GD, et al. Application of metabolomics to cardiovascular biomarker and pathway discovery. J. Am. Coll. Cardiol. 2008a;52:117–23. doi: 10.1016/j.jacc.2008.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GD, et al. Metabolite profiling of blood from individuals undergoing planned myocardial infarction reveals early markers of myocardial injury. J. Clin. Invest. 2008b;118:3503–3512. doi: 10.1172/JCI35111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisel AS, et al. Timing of immunoreactive B-type natriuretic peptide levels and treatment delay in acute decompensated heart failure: an ADHERE (Acute Decompensated Heart Failure National Registry) analysis. J. Am. Coll. Cardiol. 2008;52:534–540. doi: 10.1016/j.jacc.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Mei DA, et al. Simultaneous determination of adenosine, inosine, hypoxanthine, xanthine, and uric acid in microdialysis samples using microbore column high-performance liquid chromatography with a diode array detector. Anal. Biochem. 1996;238:34–39. doi: 10.1006/abio.1996.0246. [DOI] [PubMed] [Google Scholar]
- Netzer M, et al. A new ensemble-based algorithm for identifying breath gas marker candidates in liver disease using ion molecule reaction mass spectrometry (IMR-MS) Bioinformatics. 2009;25:941–947. doi: 10.1093/bioinformatics/btp093. [DOI] [PubMed] [Google Scholar]
- Osl M, et al. A new rule-based data mining algorithm for identifying metabolic markers in prostate cancer using tandem mass spectrometry. Bioinformatics. 2008;24:2908–2914. doi: 10.1093/bioinformatics/btn506. [DOI] [PubMed] [Google Scholar]
- Sabatine MS, et al. Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation. 2005;112:3868–3875. doi: 10.1161/CIRCULATIONAHA.105.569137. [DOI] [PubMed] [Google Scholar]
- Saeys Y, et al. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23:2507–2517. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- Saeys Y, et al. Proceedings of the European conference on Machine Learning and Knowledge Discovery in Databases. Part II. Vol. 5212. Antwerp, Belgium, Berlin, Heidelberg: Springer; 2008. Robust feature selection using ensemble feature selection techniques; pp. 313–325. of Lecture Notes in Artificial Intelligence. [Google Scholar]
- Shulaev V. Metabolomics technology and bioinformatics. Brief. Bioinform. 2006;7:128–139. doi: 10.1093/bib/bbl012. [DOI] [PubMed] [Google Scholar]
- Turgan N, et al. Urinary hypoxanthine and xanthine levels in acute coronary syndromes. Int. J. Clin. Lab. Res. 1999;29:162–165. doi: 10.1007/s005990050084. [DOI] [PubMed] [Google Scholar]
- Witten IH, Frank E. Data mining: Practical Machine Learning Tools and Techniques. 2. San Francisco: Morgan Kaufmann Publishers; 2005. [Google Scholar]
- Zimmerman J, et al. Diagnostic marker cooperative study for the diagnosis of myocardial infarction. Circulation. 1999;99:1671–1677. doi: 10.1161/01.cir.99.13.1671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.