

Abstract
In vitro and epidemiologic studies favor the efficacy of nonsteroidal anti-inflammatory drugs (NSAID) in preventing skin squamous photocarcinogenesis, but there has been relatively little study of their efficacy in preventing the more common skin basal cell carcinoma (BCC) carcinogenesis. We first compared the relative anti-BCC effects of genetic deletion and NSAID pharmacologic inhibition of cyclooxygenase (COX) enzymes in the skin of Ptch1+/− mice. We then assessed the effects of celecoxib on the development of BCCs in a 3-year, double-blinded, randomized clinical trial in 60 (PTCH1+/−) patients with the basal cell nevus syndrome. In Ptch1+/− mice, genetic deletion of COX1 or COX2 robustly decreased (75%; P < 0.05) microscopic BCC tumor burden, but pharmacologic inhibition with celecoxib reduced microscopic BCCs less efficaciously (35%; P < 0.05). In the human trial, we detected a trend for oral celecoxib reducing BCC burden in all subjects (P = 0.069). Considering only the 60% of patients with less severe disease (<15 BCCs at study entry), celecoxib significantly reduced BCC number and burden: subjects receiving placebo had a 50% increase in BCC burden per year, whereas subjects in the celecoxib group had a 20% increase (Pdifference = 0.024). Oral celecoxib treatment inhibited BCC carcinogenesis in PTCH1+/− mice and had a significant anti-BCC effect in humans with less severe disease.
Introduction
The incidence of basal cell carcinomas (BCC) is increasing significantly even in younger individuals (1) despite extensive educational efforts advising reduced exposure to solar UV radiation, the major known cause of skin cancer. Because clinical trials of sunscreens (2), oral retinoids, and β-carotene (3–5) indicate that these interventions fail to reduce BCCs, there is a real need for new preventive approaches. To this end, we are focusing on patients with the basal cell nevus syndrome (BCNS; Gorlin syndrome MIM #109400), who are genetically predisposed to develop many BCCs, often hundreds or even thousands (6). This phenotype makes it possible to obtain statistical power for studying new drugs for BCC chemoprevention with limited numbers of patients over a reasonably short time span. Studying BCC prevention in BCNS patients before studying patients with sporadic BCCs mirrors the testing of colorectal cancer chemopreventive agents in patients with familial adenomatous polyposis coli before conducting longer, more expensive chemoprevention trials in patients with sporadic colorectal adenomas (7, 8). However, it differs from other chemoprevention studies in that it is possible to assess directly the numbers and growth of the actual cancers that are the prevention targets, not simply the numbers of precancerous lesions or surrogate end points (9–12).
BCNS patients are constitutionally heterozygous for one functioning copy of PTCH1, which encodes a primary inhibitor of hedgehog signaling (13, 14). Essentially all sporadic and familial BCC tumors have aberrant hedgehog signaling caused by mutations, frequently of a UV signature type, in genes encoding hedgehog pathway members, most commonly PTCH1. Similarly, Ptch1+/− mice develop many BCCs when exposed to UV or ionizing radiation (IR; refs. 15–18). The development of the same tumors (BCCs) with the same pivotal signaling abnormality (hedgehog activation) in the same tissue (skin) caused by the same environmental insults (UV radiation or IR) as in humans makes these mice a particularly attractive preclinical model for study of prevention and treatment of BCCs.
Prospective studies showing the efficacy of nonsteroidal anti-inflammatory drugs (NSAID) in reducing colorectal polyps and epidemiologic data supporting their anticancer effects have stimulated the testing of their chemopreventive efficacy against many tumors in animal models (19–21) and in humans (22). NSAIDs inhibit the cyclooxygenase (COX) enzymes (COX1 and COX2) responsible for generating prostaglandin-mediated inflammation. Although NSAIDs and COX2-specific inhibitors have shown impressive efficacy in preventing murine squamous cell carcinomas (SCC; refs. 23–27) and some efficacy against human precancerous skin lesions and human SCCs (28–30), there have been minimal data published on the effect of NSAIDs on BCCs, the more common nonmelanoma skin cancer. Some studies suggest that BCCs express high levels of COX enzymes (31), and one case-control study found that polymorphisms in COX2 may modify BCC risk (32).
To test the relative importance of the COX enzymes in BCC carcinogenesis, we first quantitated BCCs in Ptch1+/− mice lacking or overexpressing COX1 or COX2. We next assessed the results of pharmacologic inhibition of COX2 using NSAIDs for the prevention of BCCs in Ptch1+/− mice. Finally, we tested the effects of the COX2-specific inhibitor celecoxib in a randomized, double-blinded, clinical trial in 60 PTCH1+/− humans.
Materials and Methods
Mice
Ptch1+/− mice (15) were used for studies of the prevention of murine BCC carcinogenesis, except as otherwise indicated. The mutant allele was carried on a mixed C57BL/6 DBA/2 background by continuous crossing to B6 D2 F1 breeders (The Jackson Laboratory). Ptch1+/− mice also were bred with mice carrying a K14-COX2 transgene or with C57BL/6 mice carrying mutant COX1 or COX2 genes to produce mice overexpressing or underexpressing COX enzyme. Ptch1+/− K14-COX2 mice were backcrossed to an FVB background given the strain-dependent effects on breeding and tumorigenicity (33). Mice lacking COX1 or COX2 (Ptch1+/− COX1−/− and Ptch1+/− COX2−/− mice; ref. 34) were backcrossed to a mixed C57BL/6 FVBN/J background, as they bred poorly on a C57BL/6 background.
Induction and assessment of BCCs were as described (35). In brief, Ptch1+/− mice at age 6 wk were begun on test diets and at age 8 wk received a single 5 Gy dose of IR (Cs-137) or began thrice weekly treatments with UV radiation, 240 mJ/cm2, which continued until age 12 mo. Standard biopsies of the skin were taken from the lower back (35). BCCs were identified by their histologic appearance and their β-galactosidase staining and were scored manually for number, size, and burden (average cross-sectional area × number of BCCs per mouse). In addition, mice were observed for development of macroscopic tumors in most studies until age 16 mo. As reported previously (36), in Ptch1+/− mice treated with IR, all macroscopic tumors were BCCs and trichoepitheliomas; in Ptch1+/− mice treated with UV, macroscopic tumors included BCCs, SCCs, and spindle cell tumors (“fibrosarcomas”).
Diets
The nonspecific COX inhibitor sulindac and the COX2-specific inhibitor MF-tricyclic (37) were generous gifts from Merck; the COX2-specific inhibitor celecoxib was donated to National Cancer Institute by Searle/Pharmacia/Pfizer. Sulindac was administered to mice in LabAuto Control Chow at 150 ppm. MF-tricyclic was administered in the same chow at 67 ppm. Both were made fresh monthly. Celecoxib was administered in LabAuto Control Chow at 240, 480, or 1,500 ppm and was prepared fresh at 4-mo intervals. Mice were allowed ad libitum access to the solutions and chow.
In vitro studies
Cells from our murine BCC cell lines were incubated with celecoxib at various concentrations for 48 h, and the effects on cell proliferation were assessed using the WST assay as reported previously (38).
Total RNA was extracted from the C5N immortalized keratinocyte and the ASZ001 BCC cell lines using Trizol (Invitrogen), DNase I treated (Promega), and repurified using the RNeasy mini-prep kit (Qiagen). Reverse transcription was carried out using the Taqman Reverse Transcription Reagents kit (Applied Biosystems), and quantitative PCR was carried out by Taqman Real-time PCR (Applied Biosystems). Validated gene-specific primerprobe assays (Applied Biosystems) for 18S rRNA (Hs99999901_s1), COX1 (Mm00477214_m1), and COX2 (Mm01307329_m1; Mm01307334_g1) were used for quantitative PCR.
Human subjects
All aspects of our human studies were reviewed and approved by the Institutional Review Boards of the University of California at San Francisco and Columbia University Medical Center as well as by the Protocol Review Committees of the University of California at San Francisco Comprehensive Cancer Center and the Herbert Irving Comprehensive Cancer Center of Columbia University. We recruited BCNS patients for the clinical trial from all regions of the United States (ClinicalTrials.gov identifier: NCT00023621). Subjects all fulfilled published criteria for the diagnosis of BCNS—any two major criteria or any single major plus two minor criteria (39). In practice, all enrolled subjects had BCCs plus at least one other major criterion—in no instance did the diagnosis rely on minor criteria. In particular, because the study analyzed BCCs, patients enrolling in the study were required to have had at least four histologically verified BCCs during the year before enrollment. Patients were not allowed to use oral NSAIDs (acetaminophen for pain and aspirin for cardioprotective use at 81 mg/d were permitted), corticosteroids, or retinoids or to use topical agents (e.g., 5-fluorouracil, imiquimod, retinoids, and NSAIDs) with possible anti-BCC efficacy except as therapy for individual BCCs.
Study design
The study was designed as a double-blind, placebo-controlled, randomized phase II study of the chemopreventive efficacy of oral celecoxib, 200 mg twice daily for 24 mo, against BCCs in BCNS patients. Based on our preliminary data about the expected numbers of new BCCs per subject and an estimated 20% dropout rate during the study, the trial was constructed to have 80% power to identify a 40% difference in the number of new BCCs in celecoxib-treated subjects as compared with the numbers of new BCCs in placebo-treated subjects. Enrollees, planned 30 per group, were randomized independently at the four study centers with stratification according to their reported numbers of BCCs during the year preceding enrollment (<15 or ≥15 tumors, a natural dichotomization point based on our BCNS registry) to receive capsules containing either placebo or 200 mg celecoxib. Patients returned to a clinical study center in Newport Beach, CA, New York City, NY, San Francisco, CA, or South Euclid, OH, at 3-mo intervals for physical examination, during which their BCCs were measured and counted and their clinical, laboratory, and compliance status were assessed. Enrollment began on May 11, 2001 and was completed on June 12, 2004. In December 2004, when the potential cardiovascular adverse events in persons treated with celecoxib became known (40), all subjects immediately discontinued taking study medicines and continued their quarterly visits to the clinical study centers for the next year or, if sooner, until they had completed the planned 36 mo of study participation. Initially, we planned that all BCCs would be removed at study entry but the large number of tumors in most patients made this impractical. Primary skin care physicians (PSCP) examined the subjects and periodically removed individual tumors, and records of these procedures as well as of the histology of each tumor were collated at the Coordinating Study Center in San Francisco. The number of BCCs removed by PSCPs was only a small fraction of those identified clinically and did not differ between the two treatment groups (P = 0.32).Of the lesions removed, 85% were confirmed histologically as BCCs. Excised BCCs were included in our analysis, and their number and size were carried forward to subsequent study months. At the time of clinical study center visits, ≥4-mm-diameter BCCs (excluding the lower legs) were identified clinically by one of three study dermatologist-investigators (M.A. in Newport Beach, D.R.B. in New York and South Euclid, or E.H.E. in San Francisco), mapped, and photographed. At each study visit, the body maps and/or photographs were compared with those of the previous examination to improve consistency. Adverse events were graded according to the National Cancer Institute Common Toxicity Criteria v 2.0. Fasting blood was taken at each visit while subjects were taking study medications and assayed for complete blood count and comprehensive metabolic panel, and urine was collected for urinalysis. In addition, a negative serum pregnancy test was required for all women of child-bearing potential before their entry into the study.
Statistical methods
In the mouse studies, t tests were used to compare the average number, size, and burden of tumors in biopsies. Nonparametric Wilcoxon rank tests and linear regression methods were used to compare the BCC number, size, and burden among the groups of mice fed celecoxib at 0, 240, 480, and 1,600 ppm. All statistical tests were two-sided.
For the clinical trial, all primary outcomes were analyzed according to the intention-to-treat principle. We counted and measured all ≥4-mm-diameter BCCs at each study visit for a cumulative BCC tumor number or burden (sum of the diameters of all BCCs). A mixed effect model (XTMIXED) was used to accommodate the repeated BCC tumor counts across clinic visits within each subject. The XTMIXED model can account for unbalanced data and missing data points (from dropouts or from missing study visits; some of the subjects did not attend all scheduled study visits). Cumulative BCC number and burden were log transformed to approximate a normal distribution. We calculated a slope (a time-based interaction) or rate of BCC development for the placebo and celecoxib groups and examined whether the slopes differed in the two treatment groups. We fitted a model for drug treatment, age, gender, and baseline BCC (≥15 or <15). Specific interactions were examined between BCC burden and drug treatment. Backward stepwise selection was used to develop a parsimonious model by sequentially removing variables with the highest P values (with the exception of drug treatment group). Each predictor retained in the final model was required to have a P value of ≤0.05. Our XTMIXED model fit the data, as cluster resampled bootstrap analysis (41) and linear regression were used as a check on distributional and model assumptions and showed similar results. Data were entered in Access and analyzed with STATA (StataCorp).
Results
Mouse studies: effects of genetic alterations of COX1 and COX2 on BCCs in Ptch1+/− mice
We first assessed whether genetic manipulation of expression of COXs, the major target of NSAID chemopreventive activity, alters BCC carcinogenesis. We induced BCCs by treating mice with 5 Gy IR at age 8 weeks and assessed BCC number and size in biopsies taken from normal-appearing dorsal skin at age 6 to 7 months. Tumor burden was calculated by multiplying the number of microscopic BCC tumors and their average size. As compared with their COX wild-type sibs, IR-treated Ptch1+/− mice deficient in COX1 or COX2 (Ptch1+/− COX1−/− or Ptch1+/− COX2−/− mice) had marked reductions in their microscopic BCC size and therefore burden (P <0.05; Fig. 1A). Deletion of COX2 decreased BCC burden by 75%, and deletion of COX1 or COX2 also reduced microscopic BCC numbers and size. By contrast, we found that IR-treated transgenic mice overexpressing COX2 in basal keratinocytes of the interfollicular epidermis, follicular outer root sheath, and in BCCs themselves (Ptch1+/− K14-COX2 mice; refs. 34, 36, 42) had a 2-fold increase in microscopic BCC size as compared with that in their COX wild-type sibs (P < 0.05; Fig. 1B). The shortened life span of the COX knockout mice, similar to previous reports (33), precluded analysis of their susceptibility to the growth of macroscopic BCCs. Thus, BCC carcinogenesis in Ptch1+/− mice correlated with COX enzyme expression, with changes in size being especially prominent.
Fig. 1.

A, average BCC number, size, and BCC burden at age 7 mo in standardized skin biopsies of IR-treated Ptch1+/− mice wild-type (n = 24), deleted for COX1 (n = 12) or for COX2 (n = 6). Columns, mean; bars, SE. COX knockout (KO) mice are compared with COX wild-type (WT) mice. *, P < 0.05. B, average BCC number, size, and BCC burden at age 6 mo in standardized skin biopsies of IR-treated Ptch1+/− mice (11) versus Ptch1+/− K14 COX2 transgenic mice (10). Columns, mean; bars, SE. *, P < 0.05.
Mouse studies: effect of pharmacologic inhibition of COX enzymes on murine BCC cell lines and BCC tumors in Ptch1+/− mice
We next assessed whether pharmacologic inhibition of the COX2 enzyme with celecoxib (a COX2-specific inhibitor) could inhibit BCCs in vitro and in vivo. We found that celecoxib at 10 and 20 μmol/L inhibited the proliferation of three murine BCC cell lines (38) by 70% but did not inhibit the proliferation of the nontransformed, non-BCC C5N murine keratinocyte line (P < 0.01; Fig.2; ref.43). By quantitative PCR, COX2 expression was 4-fold higher in ASZ compared with C5N. The correlation in these two cell lines between COX2 expression and celecoxib inhibition of proliferation is consistent with the hypothesis that COX2 participates in driving cell proliferation in the ASZ cells.
Fig. 2.

Cellular proliferation assay. Celecoxib at 10 and 20 μmol/L selectively inhibits BCC cell line proliferation (ASZ, BSZ, and CSZ) compared with non-BCC keratinocytes (C5N). Columns, mean; bars, SE. *, P < 0.05; †, P < 0.01, for difference between BCC versus C5N non-BCC keratinocyte cell line.
Because proliferation of murine BCC cell lines could be inhibited by celecoxib in vitro, we fed Ptch1+/− mice either a standard chow diet (n =19) or a diet with celecoxib at doses of 240 ppm (n = 21),480 ppm (n = 28),or 1,600 ppm (n = 23) starting at age 6 weeks and gave them IR at age 8 weeks. We assessed dorsal skin microscopic BCC number, size, and total burden at age 9 months. Dietary celecoxib at all three doses reduced microscopic BCC burden by ~30%, again with a trend toward greater effect on size than on number (Fig. 3A). Because the effect of celecoxib was not dose dependent over the range tested, we combined data from all three groups of celecoxib-treated mice (n = 72) and found that treatment with celecoxib reduced microscopic BCC burden by 35% (P < 0.05). Thus, pharmacologic inhibition of COX2 with celecoxib was only half as effective in reducing tumor burden as was genetic inhibition of this enzyme. A 480-ppm dose of celecoxib led to a plasma concentration of 1.5 μg/mL, a plasma concentration similar to that achieved in humans taking 200 mg twice daily orally (44, 45). We found that two other oral NSAIDs—the COX1/COX2 inhibitor sulindac (46) and the COX2 inhibitor MF-tricyclic (47)—reduced microscopic BCC burden by 50% to 60% (Fig. 3B).
Fig. 3.

A, IR-induced tumor number, average size, and burden at age 9 mo in standardized biopsies in Ptch1+/− mice fed chow (control, n = 19), 240 ppm celecoxib (n = 21), 480 ppm celecoxib (n = 28), or 1,600 ppm celecoxib (n = 23). Columns, mean; bars, SE. Celecoxib-treated mice are compared with control-treated mice. *, P < 0.05. B, IR-induced tumor number, average size, and burden at age 9 mo in standardized biopsies in Ptch1+/− mice fed chow (control, n = 10), MF-tricyclic (MFTC; n = 10), or sulindac (n = 10). Columns, mean; bars, SE. MF-tricyclic–treated and sulindac-treated mice are compared with control-treated mice. *, P < 0.05; **, P < 0.01.
Given these findings of some antimurine BCC efficacy of celecoxib, we proceeded to test the effects of celecoxib in preventing BCC tumors in patients with BCNS (PTCH1+/−).
Human studies: effect of oral celecoxib on BCCs in BCNS patients
We enrolled 60 BCNS patients in a multicenter, doubleblinded, randomized phase II clinical trial of celecoxib for BCC prevention (Table 1A). Subjects were stratified for number of BCCs at study entry (≥15 or <15) because this was a natural dichotomization point (Fig. 4). Subjects in the celecoxib group were, on average, 5 years older and had eight more BCCs at study entry, but these differences were not statistically significant. The groups did not differ significantly in gender, study site of enrollment, or percentage of subjects with ≥15 BCCs.
Table 1.
Characteristics of 60 basal cell nevus syndrome patients enrolled in clinical trial
| A. Baseline characteristics of study participants (mean ± SD) | ||
|---|---|---|
| Placebo (n = 27) | Celecoxib (n = 33) | |
| Age | 42±12 | 47±12 |
| Number of BCC tumors >4 mm | 24 ± 23 | 32 ± 60 |
| Body mass index (kg/m2) | 31 ± 1.3 | 30 ± 1.1 |
| % Male | 48% | 58% |
| % Caucasian | 96% | 100% |
| % With ≥15 BCCs at baseline | 41% | 39% |
| % Seen at California site | 59% | 55% |
| B. Reason for patient termination | |||
|---|---|---|---|
| Placebo | Celecoxib | Total | |
| Early discontinuation | 4 (14%) | 5 (15%) | 9 (15%) |
| Lost to follow-up | 1 (3.7%) | 0 (0%) | 1 (1.7%) |
| Protocol noncompliance | 6 (22%) | 4 (12%) | 10 (17%) |
| Adverse events | 1 (3.7%) | 3 (9.1%) | 4 (6.7%) |
| Subject withdrew | 1 (3.7%) | 2 (6.1%) | 3 (5.0%) |
| Other | 3 (11%) | 4 (12%) | 7 (12%) |
Fig. 4.
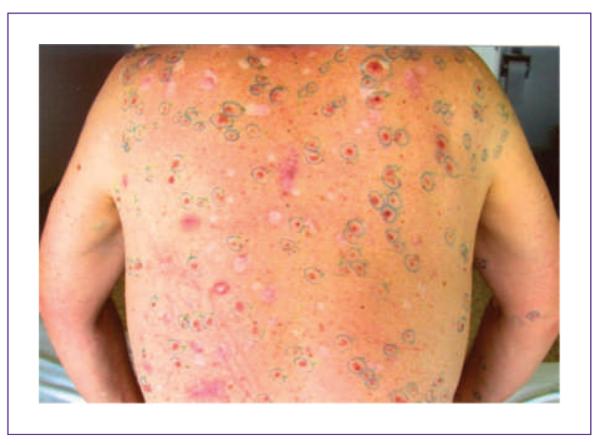
Back of a BCNS subject with ≥15 BCC tumors present at baseline. Active BCC tumors are circled in green.
Subjects were randomized into two study arms, 27 to placebo and 33 to celecoxib, as shown in Fig. 5. Subjects returned every 3 months for total skin examinations and photographic documentation of BCC tumor number and size. At the end of the first year of the trial, 72% of all subjects were still enrolled and 55% completed the first 24 months of the study, during which subjects received either placebo or celecoxib. There was no differential dropout or differences in the stated reasons for dropping out between the placebo and celecoxib arms (Table 1B). In the third year of the study (months 24-36), medication administration was discontinued but we continued to observe patients for BCC numbers. The study was discontinued in December 2004 because of emerging evidence of enhanced cardiovascular risks associated with COX2 inhibitors (48). At that time, 26 patients were still participating in the trial. The majority of these subjects were in the observation arm of study months 24 to 36. We discontinued the study medication before completion of the planned 24 months in nine subjects and continued to observe them for BCC numbers. Statistical analysis was carried out on an intent-to-treat basis because the overall compliance rate was high (85% in placebo group and 91% in the celecoxib group). There was no difference between the placebo and celecoxib groups in the rate or severity of adverse events (Table 1B), and no subject had a serious adverse event or a laboratory-detected abnormality related to study drug. In particular, no patient was known to have had a myocardial infarction or a cerebrovascular accident, and no patient died during the course of the study.
Fig. 5.

CONSORT flowchart for clinical trial.
Initially, we had planned to use changes in the numbers of new BCCs as the primary end point for the study. Once the trial was under way, it became clear that this was impractical because analyses of body maps and of photographs indicated that, despite our best efforts, it was difficult to distinguish new versus preexisting BCCs at each visit. Therefore, we compared changes in the total number of BCCs as the primary outcome. We also assessed the cumulative surface area of these BCCs (BCC burden) for each subject. During the trial, subjects continued to receive care from their PSCPs, who removed visible BCCs using whatever modality they deemed clinically appropriate (surgical excision, electrodesiccation-curettage, or by chemical methods). A total of 13,362 lesions were counted on the entire body excluding the lower legs. A fraction (8.1%) of those lesions identified clinically as BCCs were excised by PSCPs, and the number of BCCs removed did not differ significantly between the two treatment groups (n = 529 treated in placebo versus n = 561 treated in celecoxib group; P = 0.32). Excised BCCs were included in our analysis.
Our primary statistical end point assessed changes in cumulative BCC number or burden during the entire 36 month trial. We determined the percent change in BCC numbers or BCC burden per year for the placebo and celecoxib groups using a linear mixed model for longitudinal data. BCC tumors increased in both groups during the 36 months of the trial (Table 2). Subjects receiving placebo had a 37% increase in BCCs per year (P < 0.001for increase) when lesions were assessed by number or burden (cumulative surface area of BCCs). Subjects in the celecoxib group had a 26% increase in BCCs per year (P < 0.001 for increase) when lesions were assessed by number or burden. Comparing the two groups, the difference in the percent change of BCCs per year (37% versus 26%) was nonsignificant (Pdifference = 0.18) when BCCs were assessed by number. There was a trend toward celecoxib reducing BCC development when BCC tumors were assessed by burden (Pdifference = 0.069).
Table 2.
Percent change in BCCs per year in subjects treated with placebo or celecoxib
| No. subjects | Placebo (95% CI) | Celecoxib (95% CI) | Pdifference | |
|---|---|---|---|---|
| All subjects (BCC number) | 60 | 37% (25-50) | 26% (17-36) | 0.18 |
| ≥15 BCCs at baseline | 24 | 28% (16-43) | 32% (18-48) | 0.74 |
| <15 BCCs at baseline | 36 | 48% (27-71) | 22% (10-36) | 0.043 |
| All subjects (BCC burden) | 60 | 37% (24-50) | 25% (15-36) | 0.069 |
| ≥15 BCCs at baseline | 24 | 33% (19-48) | 33% (18-50) | 0.97 |
| <15 BCCs at baseline | 36 | 50% (28-76) | 20% (6.8-35) | 0.024 |
Abbreviation: 95% CI, 95% confidence interval.
Per our preplanned subgroup analyses, we stratified subjects based on the number of BCC tumors at baseline (≥15 or <15 BCCs) because we anticipated that celecoxib might have differential effects depending on disease severity. Celecoxib reduced the change in BCC numbers in subjects with <15 BCCs at baseline (48% versus 22%; P = 0.043) and did not have an effect in subjects with ≥15 BCCs at baseline (Table 2). Celecoxib also reduced the change in BCC burden in subjects with <15 BCCs at baseline (50% versus 20%; P = 0.024).Adjustments for age and gender did not change the results.
Discussion
We have found a concordance between the effects of NSAIDs on BCC tumor development in Ptch1+/− mice and PTCH1+/− humans. In mice, celecoxib reduced microscopic BCC burden by 35%. In BCNS patients, celecoxib decreased the development of new BCCs by 50% in subjects with less severe disease (<15 BCCs at baseline). Celecoxib decreased the development of new BCCs by 30% in all subjects, but this did not reach statistical significance (P = 0.069for BCC burden).These prospective controlled results are consistent with prior observational data showing a modest effect of NSAIDs on SCCs and BCCs (30, 49). We have found that NSAID inhibition may indeed have some anti-BCC chemopreventive efficacy, but the potential cardiovascular risks associated with celecoxib would seem to preclude its widespread use. Instead, topical NSAIDs potentially might be effective chemopreventive agents against SCCs and BCCs but with fewer cardiovascular risks. Morphoeic BCCs have been reported to have elevated COX2 levels compared with other BCC subtypes and clinically are more aggressive with an increased recurrence risk (31). Hence, these subtypes might be particularly good candidates for topical NSAIDs.
With high levels in actinic keratoses and skin SCC and weak staining in murine and human BCCs, COX2 would seem to be less important in the pathogenesis of BCC than of SCC. However, COX2 expression in stromal tissue/microenvironment suggests that COX2 could be involved in the early stages of BCC development (50, 51). Therefore, the COX2 inhibitor activity in this study may be related to its effects on stromal COX2 in Ptch1+/− mice and BCNS patients; the apparent involvement of stromal COX2 in early stages of BCC development is consistent with our finding that celecoxib was most preventive in patients with less severe BCC burden at baseline. The relative importance of COX1 versus COX2 in nonmelanoma skin cancer prevention is another important issue. The preclinical data are mixed; Pentland et al. (52) have shown that COX1 deletion enhanced apoptosis but did not protect against UV-induced skin tumors.
The good correlation between the effects of NSAIDs in preventing BCCs in Ptch1+/− mice and the significant anti-BCC effects in BCNS patients supports our hypothesis that results obtained from chemoprevention studies in the Ptch1+/− mouse can predict accurately which chemopreventive agents are likely to have anti-BCC effects in humans, at least in PTCH1+/− (BCNS) patients. In fact, celecoxib was more effective in reducing BCC size/burden rather than BCC number in both mice and humans, similar to a previous report of celecoxib in reducing colorectal adenoma size rather than number (8).
Results of past attempts to preventing sporadic BCCs in patients at high risk (as assessed by having had nonmelanoma skin cancer) have included the following. (a) A reduction in new BCC formation followed reduction of dietary fat intake from the usual 40% to 20% of daily calories (53). This trial has yet to be repeated perhaps because of the difficulty in adhering to so rigorous a diet. (b) No reductions in BCCs were seen in large-scale trials of oral retinoids (3–5), whereas retinoids have been reported to have some anti-SCC efficacy (54). (c) In contrast to the retinoid trials, a recent large randomized trial found that difluoromethylornithine prevented BCC but not SCC (55). (d)Trials seeking to encourage use of sunscreens have not reduced BCCs even while diminishing SCCs (2). These and other results highlight the different etiology and molecular pathogenesis between BCCs and SCCs. In addition, we have shown previously that orally administered tea extracts are ineffective in preventing UV-induced BCC carcinogenesis in Ptch1+/− mice, whereas they inhibit the induction of SCCs in SKH-1 mice (56). These studies provide further evidence that SCCs and BCCs differ not only in their molecular pathogenesis but also in their response to chemopreventive agents.
Acknowledgments
We thank the following: members of the Scientific Advisory Committee for this Project [Mary-Margaret Chren, Stuart Gansky, Thomas Kornberg, Margaret Tempero, and Stuart Yuspa (Chair)]; members of the Data Safety Monitoring Committee [Mohammed Kashani-Sabet (Chair), Karla Kerlikowske, Alex McMillan, and Judith Walsh]; Howard Highley, Connie Northway, and associates at CCS Associates and Westat for their clinical trials monitoring expertise; clinical staff at the study Centers; Susan Fischer for the generous donation of the K14-COX2 transgenic mice; Ernie Hawk for ongoing expert advice; Wei-Yann Tsai; Juntae Yu; the PSCPs who cared for the subjects during the course of the trial (Drs. Richard Armour, Christine Avakoff, and Ken Bielinski); Betsy Billys, Shalil Busbey, Robert Bushman, Chang Cho, Armand Cognetta, Stephanie P. Diamond, Vincent DiNick, Ronald Glick, David Goldberg, Matthew Goodman, and David Gorsulowsky; Roy Grekin; Robert D. Griego, Sapna Gupta, Sharon Hrabovsky, Mark Kaufmann, Stephen Kessler, Ken Lee, Katherine Lim, Leon Lubianker, and William Lynch; Stephanie Mackey and Thomas McGovern; David Norris, Martin O’Toole, Hugo Paulson, Gary Peck, Stephen Presser, Desiree Ratner, and Paul Reicherter; Robert Rosenberg, Eli Saleeby, and Richard Salomon; Keeter Sechrist, Roberta Sengelmann, Steven Snow, Teresa Soriano, David Spott, Gloria Stevens, Edward H. Stolar, David Tashjian, Kent Taulbee, Tracie Tavel, and David Taylor; Nancy Todes-Taylor, Dana Toll, and Philip Wershler; Sherri Bale (GeneDx) and Allan Bale (Yale) for help in recruiting patients; Kristi Burr and the other members and leadership of the BCCNS Life Support network for help in recruiting subjects and constant encouragement; journals that printed our calls for subjects (Archives of Dermatology, Cutis, and the Journal of the American Academy of Dermatology); and, most importantly, all of our patients for their extreme dedication and inspiration.
Grant Support NIH grants CA81888 and CN-95116 (E.H. Epstein), NIH/National Center for Research Resources/Office of the Director University of California at San Francisco–Clinical and Translational Science Institute grant KL2 RR024130 (J.Y. Tang), Mike Rainen Family Foundation, Patricia Hughes, and Stanley Eisenberg, Merck, and Pfizer through donations of drugs for human and mouse treatments. These studies were carried out in part in the Clinical and Translational Science Institute Clinical Research Center, Mt. Zion Hospital, University of California at San Francisco, with funds provided by the National Center for Research Resources, 5 M01 RR-00079, USPHS.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Christenson LJ, Borrowman TA, Vachon CM, et al. Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA. 2005;294:681–90. doi: 10.1001/jama.294.6.681. [DOI] [PubMed] [Google Scholar]
- 2.van der Pols JC, Williams GM, Pandeya N, Logan V, Green AC. Prolonged prevention of squamous cell carcinoma of the skin by regular sunscreen use. Cancer Epidemiol Biomarkers Prev. 2006;15:2546–8. doi: 10.1158/1055-9965.EPI-06-0352. [DOI] [PubMed] [Google Scholar]
- 3.Tangrea J, Edwards B, Hartman A, et al. The ISO-BCC Study Group. Isotretinoin-basal cell carcinoma prevention trial. Design, recruitment results, and baseline characteristics of the trial participants. Control Clin Trials. 1990;11:433–50. doi: 10.1016/0197-2456(90)90020-3. [DOI] [PubMed] [Google Scholar]
- 4.Tangrea JA, Edwards BK, Taylor PR, et al. Long-term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: a multicenter clinical trial. J Natl Cancer Inst. 1992;84:328–32. doi: 10.1093/jnci/84.5.328. [DOI] [PubMed] [Google Scholar]
- 5.Greenberg ER, Baron JA, Stukel TA, et al. The Skin Cancer Prevention Study Group. A clinical trial of β carotene to prevent basal-cell and squamous-cell cancers of the skin. N Engl J Med. 1990;323:789–95. doi: 10.1056/NEJM199009203231204. [DOI] [PubMed] [Google Scholar]
- 6.Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987;66:98–113. doi: 10.1097/00005792-198703000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–52. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 8.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885–95. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 9.Matsuhashi N, Nakajima A, Shinohara K, Oka T, Yazaki Y. Rectal cancer after sulindac therapy for a sporadic adenomatous colonic polyp. Am J Gastroenterol. 1998;93:2261–6. doi: 10.1111/j.1572-0241.1998.00630.x. [DOI] [PubMed] [Google Scholar]
- 10.Niv Y, Fraser GM. Adenocarcinoma in the rectal segment in familial polyposis coli is not prevented by sulindac therapy. Gastroenterology. 1994;107:854–7. doi: 10.1016/0016-5085(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 11.Cruz-Correa M, Hylind LM, Romans KE, Booker SV, Giardiello FM. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology. 2002;122:641–5. doi: 10.1053/gast.2002.31890. [DOI] [PubMed] [Google Scholar]
- 12.Lynch HT, Thorson AG, Smyrk T. Rectal cancer after prolonged sulindac chemoprevention. A case report. Cancer. 1995;75:936–8. doi: 10.1002/1097-0142(19950215)75:4<936::aid-cncr2820750407>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 13.Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–71. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 14.Hahn H, Wicking C, Zaphiropoulos PG, et al. Mutations of the human homologue of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–51. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 15.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 16.Mancuso M, Leonardi S, Tanori M, et al. Hair cycle-dependent basal cell carcinoma tumorigenesis in Ptc1neo67/+ mice exposed to radiation. Cancer Res. 2006;66:6606–14. doi: 10.1158/0008-5472.CAN-05-3690. [DOI] [PubMed] [Google Scholar]
- 17.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743–54. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH, Jr., Scott M. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276:817–21. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 19.Swamy MV, Patlolla JM, Steele VE, Kopelovich L, Reddy BS, Rao CV. Chemoprevention of familial adenomatous polyposis by low doses of atorvastatin and celecoxib given individually and in combination to APCMin mice. Cancer Res. 2006;66:7370–7. doi: 10.1158/0008-5472.CAN-05-4619. [DOI] [PubMed] [Google Scholar]
- 20.Boolbol SK, Dannenberg AJ, Chadburn A, et al. Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res. 1996;56:2556–60. [PubMed] [Google Scholar]
- 21.Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. 2000;60:5040–4. [PubMed] [Google Scholar]
- 22.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Five-year efficacy and safety analysis of the adenoma prevention with celecoxib trial. Cancer Prev Res. 2009;2:310–21. doi: 10.1158/1940-6207.CAPR-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer SM, Conti CJ, Viner J, Aldaz CM, Lubet RA. Celecoxib and difluoromethylornithine in combination have strong therapeutic activity against UV-induced skin tumors in mice. Carcinogenesis. 2003;24:945–52. doi: 10.1093/carcin/bgg046. [DOI] [PubMed] [Google Scholar]
- 24.Fischer SM, Lo HH, Gordon GB, et al. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, and indomethacin against ultraviolet light-induced skin carcinogenesis. Mol Carcinog. 1999;25:231–40. [PubMed] [Google Scholar]
- 25.Pentland AP, Schoggins JW, Scott GA, Khan KN, Han R. Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis. 1999;20:1939–44. doi: 10.1093/carcin/20.10.1939. [DOI] [PubMed] [Google Scholar]
- 26.Orengo IF, Gerguis J, Phillips R, Guevara A, Lewis AT, Black HS. Celecoxib, a cyclooxygenase 2 inhibitor as a potential chemopreventive to UV-induced skin cancer: a study in the hairless mouse model. Arch Dermatol. 2002;138:751–5. doi: 10.1001/archderm.138.6.751. [DOI] [PubMed] [Google Scholar]
- 27.Wilgus TA, Koki AT, Zweifel BS, Rubal PA, Oberyszyn TM. Chemotherapeutic efficacy of topical celecoxib in a murine model of ultraviolet light B-induced skin cancer. Mol Carcinog. 2003;38:33–9. doi: 10.1002/mc.10142. [DOI] [PubMed] [Google Scholar]
- 28.Asgari M, White E, Chren MM. Nonsteroidal anti-inflammatory drug use in the prevention and treatment of squamous cell carcinoma. Dermatol Surg. 2004;30:1335–42. doi: 10.1111/j.1524-4725.2004.30407.x. [DOI] [PubMed] [Google Scholar]
- 29.Butler GJ, Neale R, Green AC, Pandeya N, Whiteman DC. Nonsteroidal anti-inflammatory drugs and the risk of actinic keratoses and squamous cell cancers of the skin. J Am Acad Dermatol. 2005;53:966–72. doi: 10.1016/j.jaad.2005.05.049. [DOI] [PubMed] [Google Scholar]
- 30.Grau MV, Baron JA, Langholz B, et al. Effect of NSAIDs on the recurrence of nonmelanoma skin cancer. Int J Cancer. 2006;119:682–6. doi: 10.1002/ijc.21878. [DOI] [PubMed] [Google Scholar]
- 31.Yu M, Zloty D, Cowan B, et al. Superficial, nodular, and morpheiform basal-cell carcinomas exhibit distinct gene expression profiles. J Invest Dermatol. 2008 doi: 10.1038/sj.jid.5701243. [DOI] [PubMed] [Google Scholar]
- 32.Vogel U, Christensen J, Wallin H, Friis S, Nexo BA, Tjonneland A. Polymorphisms in COX-2, NSAID use and risk of basal cell carcinoma in a prospective study of Danes. Mutat Res. 2007;617:138–46. doi: 10.1016/j.mrfmmm.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Rundhaug JE, Pavone A, Kim E, Fischer SM. The effect of cyclooxygenase-2 overexpression on skin carcinogenesis is context dependent. Mol Carcinog. 2007;46:981–92. doi: 10.1002/mc.20340. [DOI] [PubMed] [Google Scholar]
- 34.Bol DK, Rowley RB, Ho CP, et al. Cyclooxygenase-2 overexpression in the skin of transgenic mice results in suppression of tumor development. Cancer Res. 2002;62:2516–21. [PubMed] [Google Scholar]
- 35.So PL, Lee K, Hebert J, et al. Topical tazarotene chemoprevention reduces basal cell carcinoma number and size in Ptch1+/− mice exposed to ultraviolet or ionizing radiation. Cancer Res. 2004;64:4385–9. doi: 10.1158/0008-5472.CAN-03-1927. [DOI] [PubMed] [Google Scholar]
- 36.Aszterbaum M, Epstein J, Oro A, et al. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med. 1999;5:1285–91. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- 37.Dvory-Sobol H, Kazanov D, Liberman E, et al. MF tricyclic and sulindac retard tumor formation in an animal model. Int J Cancer. 2006;118:11–6. doi: 10.1002/ijc.21218. [DOI] [PubMed] [Google Scholar]
- 38.So PL, Langston AW, Daniallinia N, et al. Long-term establishment, characterization and manipulation of cell lines from mouse basal cell carcinoma tumors. Exp Dermatol. 2006;15:742–50. doi: 10.1111/j.1600-0625.2006.00465.x. [DOI] [PubMed] [Google Scholar]
- 39.Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299–308. [PubMed] [Google Scholar]
- 40.Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–80. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 41.Davison AC, Hinkley DV. Bootstrap methods and their application. Cambridge University Press; Cambridge: 1997. [Google Scholar]
- 42.Rundhaug JE, Mikulec C, Pavone A, Fischer SM. A role for cyclooxygenase-2 in ultraviolet light-induced skin carcinogenesis. Mol Carcinog. 2007;46:692–8. doi: 10.1002/mc.20329. [DOI] [PubMed] [Google Scholar]
- 43.Stoler A, Stenback F, Balmain A. The conversion of mouse skin squamous cell carcinomas to spindle cell carcinomas is a recessive event. J Cell Biol. 1993;122:1103–17. doi: 10.1083/jcb.122.5.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stempak D, Gammon J, Klein J, Koren G, Baruchel S. Single-dose and steady-state pharmacokinetics of celecoxib in children. Clin Pharmacol Ther. 2002;72:490–7. doi: 10.1067/mcp.2002.129322. [DOI] [PubMed] [Google Scholar]
- 45.Ragel BT, Jensen RL, Gillespie DL, Prescott SM, Couldwell WT. Celecoxib inhibits meningioma tumor growth in a mouse xenograft model. Cancer. 2007;109:588–97. doi: 10.1002/cncr.22441. [DOI] [PubMed] [Google Scholar]
- 46.Athar M, An KP, Tang X, et al. Photoprotective effects of sulindac against ultraviolet B-induced phototoxicity in the skin of SKH-1 hairless mice. Toxicol Appl Pharmacol. 2004;195:370–8. doi: 10.1016/j.taap.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 47.Lal G, Ash C, Hay K, et al. Suppression of intestinal polyps in Msh2-deficient and non-Msh2-deficient multiple intestinal neoplasia mice by a specific cyclooxygenase-2 inhibitor and by a dual cyclooxygenase-1/2 inhibitor. Cancer Res. 2001;61:6131–6. [PubMed] [Google Scholar]
- 48.Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 49.Clouser MC, Roe DJ, Foote JA, Harris RB. Effect of non-steroidal anti-inflammatory drugs on non-melanoma skin cancer incidence the SKICAP-AK trial. Pharmacoepidemiol Drug Saf. 2009;18:276–83. doi: 10.1002/pds.1718. [DOI] [PubMed] [Google Scholar]
- 50.An KP, Athar M, Tang X, et al. Cyclooxygenase-2 expression in murine and human nonmelanoma skin cancers: implications for therapeutic approaches. Photochem Photobiol. 2002;76:73–80. doi: 10.1562/0031-8655(2002)076<0073:ceimah>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 51.Tjiu J-W, Chen J-S, Shun C-T, et al. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J Invest Dermatol. 2009;129:1016–25. doi: 10.1038/jid.2008.310. [DOI] [PubMed] [Google Scholar]
- 52.Pentland AP, Scott G, VanBuskirk J, Tanck C, LaRossa G, Brouxhon S. Cyclooxygenase-1 deletion enhances apoptosis but does not protect against ultraviolet light-induced tumors. Cancer Res. 2004;64:5587–91. doi: 10.1158/0008-5472.CAN-04-1045. [DOI] [PubMed] [Google Scholar]
- 53.Black HS, Thornby JI, Wolf JE, Jr., et al. Evidence that a low-fat diet reduces the occurrence of non-melanoma skin cancer. Int J Cancer. 1995;62:165–9. doi: 10.1002/ijc.2910620210. [DOI] [PubMed] [Google Scholar]
- 54.Moon TE, Levine N, Cartmel B, et al. Effect of retinol in preventing squamous cell skin cancer in moderate-risk subjects: a randomized, double-blind, controlled trial. Cancer Epidemiol Biomarkers Prev. 1997;6:949–56. [PubMed] [Google Scholar]
- 55.Bailey H, Kim K, Verma A, et al. A randomized, double-blind, placebo-controlled phase 3 skin cancer prevention study of DFMO in subjects with previous history of skin cancer. Cancer Prev Res. 2010;3:35–47. doi: 10.1158/1940-6207.CAPR-09-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hebert JL, Khugyani F, Athar M, Kopelovich L, Epstein EH, Jr., Aszterbaum M. Chemoprevention of basal cell carcinomas in the ptc1+/− mouse-green and black tea. Skin Pharmacol Appl Skin Physiol. 2001;14:358–62. doi: 10.1159/000056369. [DOI] [PubMed] [Google Scholar]