

Summary
This study is to investigate the possible role of high temperature requirement A 1 (HtrA1) in the articular cartilage degeneration. Paraffin sections were prepared from the knee and temporomandibular (TM) joints of four mouse OA models; two of the models had a genetic mutation (type IX collagen-deficient and type XI collagen-haploinsufficient) and two were surgically induced (destabilization of the medial meniscus of knee joint and discectomy of TM joint). The HtrA1 protein expression profiles of the prepared sections were examined by immunohistostaining. The level of HtrA1 mRNA in the articular cartilage taken from the knee joints of one of the genetically mutated OA models was determined by real-time PCR. Double immunohistostaining was used to examine the expression of co-localization of HtrA1 with type VI collagen and HtrA1 with discoidin domain receptor 2 (Ddr2) in the articular cartilage of knee joints from the genetically mutated OA model. The expression of HtrA1 was found to be increased in the knee and TM joints of these four models at early stages of the disease. An examination of the knee joint of a mutant mouse indicated an 8-fold increase in the level of HtrA1 mRNA, when compared to the levels observed in the knee joints of its wild-type littermates. Pericellular type VI collagen was not present in chondrocytes expressing HtrA1. Meanwhile, the expression of HtrA1 was associated with the expression of Ddr2 in the chondrocytes. Results indicate that HtrA1 may disrupt the pericellular matrix network, resulting in alteration of chondrocyte metabolisms. This eventually leads to OA.
Keywords: HtrA1, Mouse, Articular cartilage, Osteoarthritis
Introduction
Osteoarthritis (OA) is a heterogeneous disease, with variable symptoms and rates of progression (Felson et al., 2000). However, while the initial events of the disease are diverse, the pathologic progression of OA follows a consistent pattern: it begins with the gradual loss of proteoglycans and degradation of the type II collagen at the surface region of the articular cartilage, and this eventually results in the partial or complete loss of the articular cartilage (Hamerman, 1989). An understanding of the possible common molecular sequence of events underlying the progression of OA will not only broaden our knowledge of OA pathogenesis, but will also contribute invaluable information to the search in novel therapeutic targets for the prevention and treatment of OA.
Although the precise mechanism by which articular cartilage degeneration ensues is largely unknown, maintaining the integrity of the articular cartilage is key in protecting the cartilage from degradation. Chondron, consisting of the chondrocyte and its pericellular matrix and the capsule surrounding the pericellular matrix, has been considered the primary structural and functional unit involved in maintaining the integrity of the articular cartilage (Poole, 1997; Poole et al., 1988). Under normal conditions chondrocytes are well protected by their pericellular matrix, which includes type VI and IX collagens, fibronectin, decorin, fibromodulin, biglycan, cartilage oligomeric matrix protein (Comp), matrilin 3 and aggrecan. It is conceivable that excessive mechanical loading on a normal articular cartilage or normal mechanical loading on a defective articular cartilage can incite the chondrocytes to disrupt their pericellular matrix and alter certain cellular metabolic events, thus resulting in OA. In fact, mutations in type IX collagen in humans and deficiencies in type IX collagen in mice result in the early onset of OA (Fässler et al., 1994; Hu et al., 2006), as does the absence of type VI collagen in mice (Alexopoulos et al., 2009). The deletion of both fibromodulin and biglycan genes causes OA in mice (Wadhwa et al., 2005). In humans, mutations in COMP are also associated with OA (Lachman et al., 2005) and increased fibronectin fragments appear in OA cartilages (Forsyth et al., 2002; Loeser et al., 2003). These observations suggest that an enzyme/enzymes capable of degrading molecules in the pericellular matrix may play an important role in pathogenesis of OA. Results from a number of studies suggest that HtrA1, a member of the high temperature requirement family of serine proteases (Clausen et al., 2002), may be one such enzyme. Mammalian HtrA1 is a secretory enzyme. It can bind to several members of the TGF-β family, such as bone morphogenetic protein 4, TGFβ1 and TGFβ2, activin and growth differentiation factor 5, and inhibit the signaling activities that are normally initiated by the binding of these factors with their receptors (Zumbrunn and Trueb 1996). Substrates of HtrA1 have been identified, including aggrecan, decorin, biglycan, fibromodulin and fibronectin (Tsuchiya et al., 2005; Grau et al., 2006). HtrA1 has been implicated in rheumatoid arthritis (RA) and OA. Expression of HTRA1 is up-regulated in synovial fluids obtained from human RA and OA joints (Hu et al., 1998) and HTRA1 is the most abundant protease in human OA cartilages identified by proteomic analyses (Wu et al., 2007). HtrA1 is also up-regulated in experimental mouse OA articular cartilages (Oka et al., 2004). The fact that the expression of HtrA1 is increased in OA cartilages and that many of the pericellular matrix components are substrates of this enzyme suggests that HtrA1 may contribute to the pathogenesis of OA; however, the precise point in time at which the expression of HtrA1 is up-regulated and the consequence of the elevation in HtrA1 levels during the development of OA remain to be investigated.
In this study, we used two genetic mutant mouse strains that reveal OA pathology in joints similar to what is seen in humans in this study. One mouse strain is type IX collagen-deficient (Col9a1−/−) mice (Hu et al., 2006) and the other one is type XI collagen-haploinsufficient mice or heterozygous chondrodysplasia (cho/+) mice (Xu et al., 2003). Col9a1−/− mice develop normally with the exception of a flattening of knee joint surfaces and an increased distance between the knee joint condyles. The chondrodysplasia in mice is an autosomal-recessive disorder. The defect in homozygous cho/cho mice, which die at birth, is due to a single nucleotide deletion in Col11a1 leading to a frame-shift and premature termination of translation of the α1 chain of type XI collagen (Li et al., 1995). Heterozygous cho/+ mice develop normally without obvious skeletal abnormalities at birth. Histological studies show that Col9a1−/− and cho/+ mice develop OA-like changes in knee and temporomandibular (TM) joints starting at the age of 3 months and a severe OA-like pathology over 9 to 12 months (a normal life span of a laboratory mouse is about 30 months on average). We also used two surgically induced mouse OA models in this study. One is the OA-like TM joint disease induced by partial discectomy (Xu et al., 2009) and the second model is the OA-like knee joint disease induced by sectioning of the medial meniscotibial ligament (MMTL) to destabilize the medial meniscus of knee joints (Xu et al., 2007). Results from our previous studies demonstrated a typical pattern of early on-set articular cartilage degeneration in the mouse TM and knee joints following the surgery and the severity of the cartilage damage was associated with the time course following the surgery.
We examined the protein expression profile of HtrA1 in the knee and TM joints of the four mouse OA models that we mentioned above. The level of HtrA1 mRNA in the articular cartilage of knee joints, taken from one of the genetically mutated models, was also examined. This was done with the purpose of determining whether the increased expression of HtrA1 protein was paralleled by an up-regulated expression of HtrA1. Finally, we examined whether the increased expression of HtrA1 resulted in the disruption of the pericellular matrix of chondrocytes and thus led to the elevated expression of Ddr2 in the knee joints of the mutant mice.
Materials and methods
Acquisition of mouse tissues for immunohistostaining
Type IX collagen-deficient (Col9a1−/−) (Hu et al., 2006) and type XI collagen-haploinsufficient (cho/+, heterozygous chondrodysplasia) (Xu et al., 2003) mice were maintained in a virus-free animal facility at Harvard Medical School under a 12-hour lighting schedule - 12 hours with light and 12 hours without light. Knee and TM joints were harvested from Col9a1−/− and cho/+ mice at the ages of 1, 3 and 6 months. Knee and TM joints (as controls) were also harvested from C57BL/6j mice (Jackson Laboratory, Bar Harbor, ME, USA) at the ages of 1, 3 and 6 months.
Surgically induced OA mice (C57BL/6j) were generated by performing microsurgeries on knee and TM joints. The detailed microsurgical procedures have been described in our previous publications (Xu et al., 2007, 2009). OA-like mouse knee joints were induced by sectioning the medial meniscotibial ligament, in order to destabilize the medial meniscus of the knee joints. The OA-like mouse TM joints were induced by partial discectomy. Knee and TM joints were also collected from sham-surgical mice as controls.
Preparation of immunohistostaining sections
The detailed procedures for sectioning have been described in our previous publications (Xu et al., 2003; Hu et al., 2006; Lam et al., 2007). Briefly, 4 knee and 4 TM joints were isolated from 4 different mice (Col9a1−/−, cho/+ and wild-type) at each age - 1, 3 and 6 months. In addition, 4 knee joints and 4 TM joints were isolated from mice at three different time points following the surgeries - 2, 4 and 8 weeks post-surgically. All of the joints were fixed in 4% paraformaldehyde for 6 hours at room temperature. The samples were then processed for paraffin embedding. For each knee joint, a serial sectioning of 6-μm in thickness was taken. Approximately 200–250 sections represent the entirety of a mouse knee joint from its lateral to its medial side, (for the genetic mutant mice) and approximately 120–150 sections represent the entirety of a mouse knee joint from its anterior to its posterior (for the surgically induced mice). For the TM joint, it took nearly 100 sections of 6-μm in thickness to cover the entire joint, from anterior to posterior. Every 20th section was collected for immunohistostaining.
Immunohistostaining for protein expression of HtrA1
Sections were de-paraffinized and quenched for endogenous peroxidase activity. The sections were incubated with a polyclonal antibody (1:200 dilution) against HtrA1 (Cat. No. ab-38611, Abcam Inc, Cambridge, MA). After overnight incubation at 4°C, sections were washed with PBS three times and subsequently treated with a biotinylated secondary antibody (goat anti-rabbit IgG-B). Coloring was developed with the use of a peroxidase substrate (VECTOR NovaRED Substrate, Cat. No. SK-4800, Laboratories, Burlingame, CA) following treatment of the sections with a mixture of avidin and biotinylated horseradish peroxidase (VECTASTAIN ABC Kit, Cat. No. PK-4000, Vector Laboratories, Burlingame, CA). Sections were counterstained with 0.2% Fast Green solution. Staining with isotype-matched normal IgG (Vector Laboratories) and staining without primary antibody were also performed as negative controls.
Immunohistostaining for the expression of co-localization of type VI collagen, HtrA1 and Ddr2 proteins
We selected knee joints from cho/+ mice, at the age of 6 months, in order to examine the expression of co-localization of type VI collagen, HtrA1 and Ddr2 proteins. These selections were based upon the protein expression profiles of HtrA1 in the knee joints of the mutant mice. Eight sections from each knee joint were selected for immunohistostaining. There were two sample sets in this experiment. Each set included 4 knee joints from cho/+ mice and 4 knee joints from wild-type littermates. The first set of sections was incubated with a goat polyclonal antibody (1:200) against type VI collagen (Cat. No. sc-9855, Santa Cruz Biotechnology, CA) and a rabbit polyclonal antibody (1:200 dilution) against HtrA1(Cat. No. ab38611). The second set of sections was incubated with a goat polyclonal antibody (1:200) against Ddr2 (Cat. No. sc-7554, Santa Cruz Biotechnology, CA) and the polyclonal antibody (1:200 dilution) against HtrA1. After overnight incubation at 4°C, the sections were washed three times with PBS with 0.1% Tween 20. In order to detect type VI collagen, Ddr2 and HtrA1, the sections were treated with mixed secondary antibodies (1:250), Alexa Fluor488 donkey anti-rabbit IgG and Alexa Fluor594 donkey anti-goat IgG. After incubation for 30 min. at room temperature, the sections were then washed three times with PBS with 0.1% Tween 20 and examined under a fluorescence microscope (ZEISS, AXIO Imager. M1).
Real-time PCR
Total RNAs were isolated from the knee articular cartilages of 6 cho/+ and 6 wild-type littermates, at the ages of 3 and 6 months, using the Total RNA Isolation System (Promega, Madison, WI). The cDNAs were synthesized with oligo(dT) primer using Super-Script First-strand Synthesis System (BD Biosciences Clontech, CA). The concentrations of PCR primers for HtrA1 were as follows: 200 nM of forward primer 5′-GATCCGAATGATGTCGCTCAC-3′ and 200 nM of reverse primer 5′-ATTTTCCTTGAGCCCTCCGGC-3′. The t-test analysis was used to detect differences in mRNA levels between the control and experimental groups. A 5% significance level was used. PCR was performed using 25 μl of 1X PCR buffer containing 3 μM MgCl2, 200 nM dNTPs, PCR primers, 1X SYBR Green, 0.5 unit Taq DNA polymerase (Qiagen) and 0.5 μl cDNA. Real-time PCR was performed using the Icycler iQ detection system (BioRad, CA) and the PCR reaction was carried out at oC for 3 minutes followed by 50 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, with a final extension at 72°C for 4 min. At the end of the PCR cycles, a melting curve, using a temperature range from 55°C to 95°C with +0.5°C intervals, was generated to test the specificity of the PCR product. A cDNA sample in each experiment was tested in triplicate and each experiment was performed twice. We used Gapdh as the internal control. The forward primer for Gapdh was 5′-ACTGAGGACCAGGTTGTC-3′ and the reverse primer was 5′-TGCTGTAGCCGTATTCATTG-3′. The efficiency of PCR represented by a standard curve was also tested by plotting the amount of PCR product versus the known amount of a template, 0.001ng, 0.01ng, 0.1 ng, 1ng and 10ng. In this experiment, the efficiency reached 90% or higher.
Results
Increase of HtrA1 expression in the knee joints of mouse OA models
In order to determine the point in time at which the level of HtrA1 expression increases in the development of OA, we examined the protein level of the enzyme in the articular cartilages of knee joints from Col9a1−/− and cho/+ mice at the ages of 1, 3 and 6 months. For each knee joint, 10 representative paraffin sections were used for immunostaining. Thus, 40 sections (4 knees × 10) were selected for immunostaining for each age group. Brown staining (positive) cells, see figure 1, were detected in about 70% of the sections taken from Col9a1−/− mice, at the ages of 3 and 6 months, and cho/+ mice, at the age of 6 months. We noticed that the positive staining was focal and randomly scattered in the superficial layer of the entire knee joint. There were no positively stained cells in the knee joints of Col9a1−/−, cho/+ and wild-type mice at the age of 1 month (data not shown). No positively stained cells were detected in cho/+ mice at the age of 3 months and in wild-type mice at the ages of 3 and 6 months (Fig. 1).
Fig. 1.
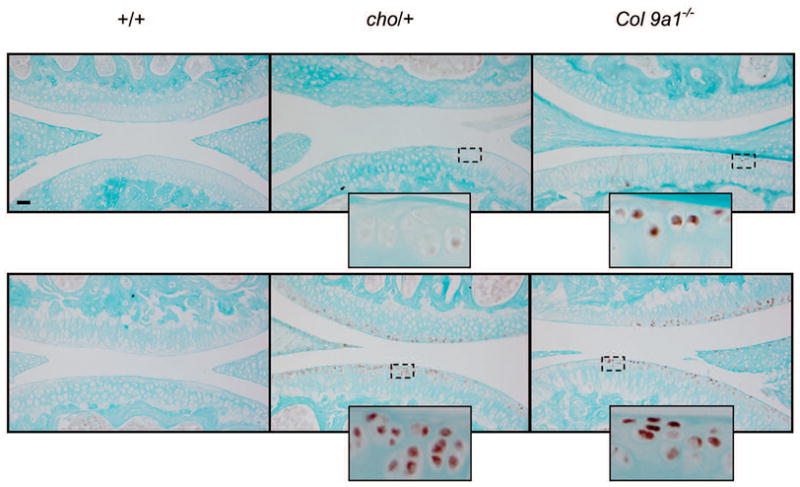
Immunostaining for HtrA1 of articular cartilage taken from the knee joints of chol+, Col9a1−/− and wild-type mice. Immunostgaining intensity of HtrA1 (see brown color staining) was increased in focal areas of the articular cartilage of Col9a1−/− mice, at 3 and 6 months of age, and of cho/+ mice, at 6 months of age, when compared to that of wild-type mice. The top panel reprsents samples taken from 3-month old mice and the bottom panel represents samples taken from 6-months old mice. The presence of positively staining cells in the 3-month old of Col9a1−/− mice and the absence of such cells in the 3-month old cho/+ mice suggests that the degenerative process in Col9a1−/− mice is much more advanced than it is in cho/− mice. This is consistent with the morphological changes observed in the articular cartilage from the knee joints of cho/+ and Col9a1−/− mice. Bar: 50 μm.
To determine whether or not the expression of HtrA1 was increased in the knee joints of mice following surgery, we examined the protein level of the enzyme in the articular cartilages of knee joints at 2, 4 and 8 weeks post-surgery. 6 representative paraffin sections were taken from each knee joint of a surgically induced mouse and used for immunostaining. Thus, 24 sections (4 joints × 6) were selected from the mice at each time point following the surgery. The results indicated that, at 4 and 8 weeks post-surgery, positively staining cells could be detected in approximately 70% of the sections taken from the surgically induced mice and hardly any such cells could be detected in the knee joints of sham-surgical mice (Fig. 2). Positively staining cells were not observed in the knee joints of both the sham and the surgically induced mice at 2 weeks following surgery (data not shown).
Fig. 2.
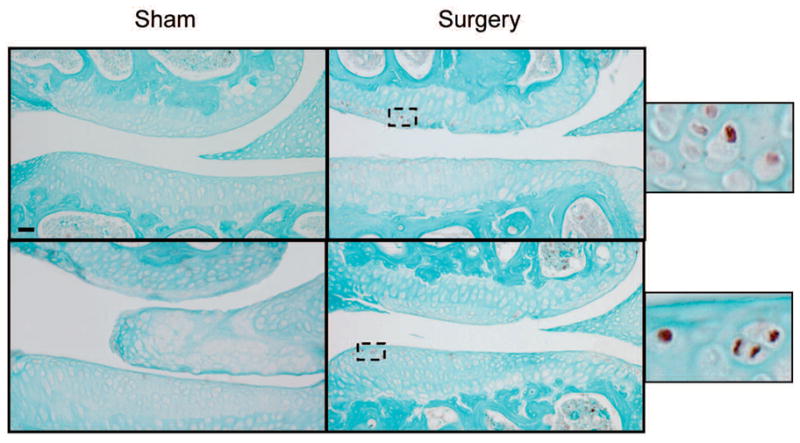
Immunostaining for HtrA1 of articular cartilage taken from surgically-induced OA knee joints. The onset of articular cartilage degeneration in the knee joints of surgically-induced mice was observed at 4 weeks post-surgery. At that point in time, focal areas of the knee joints of surgically-induced mice exhibited increased levels of immunostaining for HtrA1 (see brown color staining), when compared with those of sham-surgical mice (top panel). This relative increase in immunostaining intensity for HtrA1 was also evident in the articular cartilage of surgically-induced mouse knee joints at 8 weeks (bottom panel) post-surgery. Bar: 50 μm.
Increase of HtrA1 expression in the TM joints of mouse OA models
The articular cartilages from the TM joints of Col9a1−/−, cho/+ and wild-type mice at the ages of 1, 3 and 6 months, and from post-discectomy mice, were also examined for HtrA1 expression. For each TM joint, 5 representative paraffin sections were used for immunostaining. Therefore 20 sections (4 joints × 5) were selected from mice at each age group or time point following the surgery. Results showed that brown staining (positive) cells appeared in about 80% of the sections from Col9a1−/− and cho/+ mice, at the ages of 3 and 6 months (Fig. 3), and from post-discectomy mice, at 4 and 8 weeks following the discectomy (Fig. 4). The positive staining was focal and randomly scattered throughout the entire TM joint. There were no positively staining cells detected in the articular cartilages taken from the TM joints of wild-type mice, at the ages of 3 and 6 months (Fig. 3), and sham-surgical mice, at 4 and 8 weeks post-surgery (Fig. 4). In addition, positively stained cells were not detected in the Col9a1−/−, cho/+ and wild-type mice at 1 month of age, and in the sham-surgical and post-discectomy mice, at 2 weeks following the sham and discectomy surgeries (data not shown).
Fig. 3.

Immunostaining for HtrA1 of articular cartilage from the TM joints of cho/+, Co/9a1−/− and wild type mice. When compared with the articular cartilage from the TM joints of wild-type mice, the cartilage of Co/9a1−/− and cho/+ mice exhibited increased levels of immunostianing intensity for HtrA1 (see brown color staining), at 3 months (top panel) and 6 months (bottom panel) of age. Bar: 50 μm.
Fig. 4.
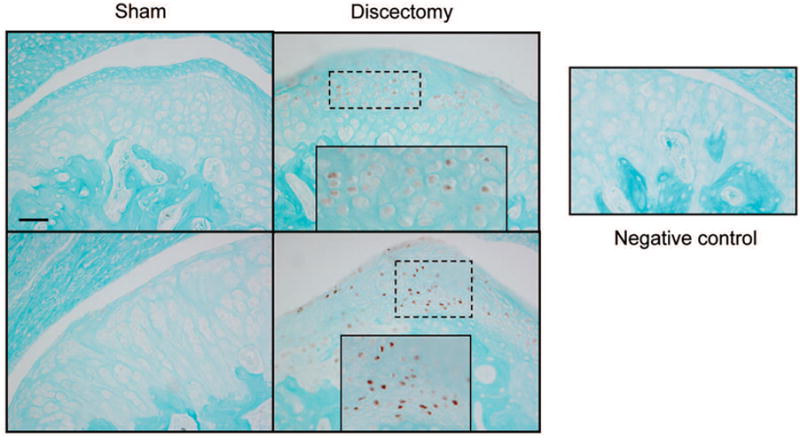
Immunohistostianig for HtrA1 of articular cartilage from surgically-induced OA TM joints. At 4 weeks (top panel) at 8 weeks (bottom panel) post-surgery, the articular cartilage from the TM joints of pot-discectomy mice exhibited increased immunostaining intensity for HtrA1 (see brom color staining) when compared to the cartilage of the sham-surgical mice. Bar: 50 μm.
In addition, positively staining cells were not detected in sections with isotype-matched normal IgG (as negative controls), see the representative section in figure 4.
Increase of HtrA1 mRNA levels in the articular cartilage of cho/+ mice at the age of 6 months
We examined the level of HtrA1 mRNA in the articular cartilage, taken from the knee joints of cho/+ mice at 6 months of age, in order to determine whether or not the evident up-regulation in the expression of HtrA1 reflected an increase in the level of HtrA1 mRNA. Real-time PCR analysis of cartilage extracts showed that the mRNA level of HtrA1 was increased 8-fold in cho/+ mice at 6 months of age, see figure 5. Hardly any HtrA1 mRNA was detected in the cho/+ mice and wild-type littermates at 3 months of age (data not shown).
Fig. 5.
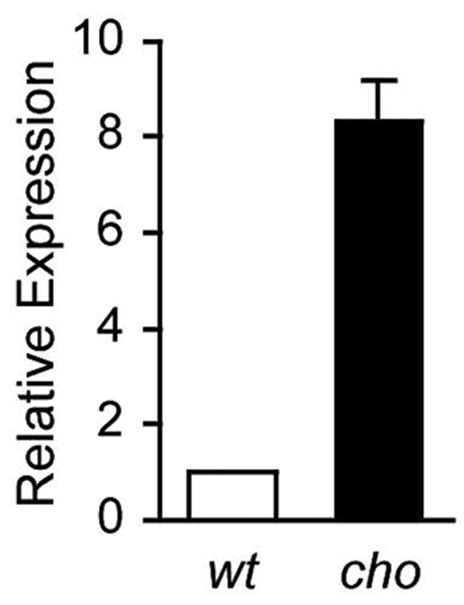
Levels of HtrA1mRNA in the articular cartilage from knee joints of cho/+ and wild-type mice at 6 months of age. When compared with the levels of HtrA1 mRNA in the articular cartilage of wild-type littermates, in the articular cartilage from the knee joints of cho/+ mice, at 6 months of age, the levels of HtrA1 mRNA exhibited an 8-fold increase. The value representing levels in wild-type mice is set at 1.0.
Absence of type VI collagen or presence of Ddr2 associated with the increased expression of HtrA1 in articular cartilage, from the knee joints of cho/+ mice at 6 months of age
In order to determine whether the increased expression of HtrA1 was associated with disruption of the pericellular matrix and an increased expression of Ddr2, we used type VI collagen as an indicator for the integrity of the pericellular matrix. Since type VI collagen is exclusively present in the pericellular matrix, disappearance of type VI collagen indicates the disruption of the pericellular matrix. Double-immunohistostaining experiments were performed using sections taken from 4 knee joints of cho/+ mice and 4 knee joints of their wild-type littermates, at 6 months of age. 8 sections from each joint were selected for staining. The results indicated that, within the articular cartilage of these knee joints, there is an apparent correlation between the expressions of these genes. When chondrocytes stained positive for HtrA1, type VI collagen was not detectable pericellularly and Ddr2 positive staining was present. To better present these results we took advantage of the fact that focal degenerative lesions can be seen in a section of a knee joint from a mouse OA model. The expression profiles of HtrA1 with type VI collagen or Ddr2 are clearly visible in the sections of knee joint taken from cho/+ mice, in which focal degenerative lesions were observed, see figure 6.
Fig. 6.

Double-immunohistostaining for HtrA1 with type VI collagen or HtrA1 with Ddr2 of articular cartilage from the knee joints of cho/+ mice at 6 months of age. Red color pericellular stianing (see arrows on A) indicates positive staining for type VI collagen. It shows that type VI collagen is present in the pericellular matrix of some chondrocytes. Green color staining (see cells in the insert on B) indicates positive staining for HtrA1. Comparison of A and B, it illustrates that tyep VI collagen is present in the pericellular matrix of chondrocytes when cells do not express HtrA1, whereas type VI collagen is absent in the pericellular matrix of chondrocytes expressing HtrA1, see the picture C. The picture C is a combination of the pictures A and B. Red color stained chondrocytes (see cells on D) indicate positive staining for Ddr2 and green color stained chondrocytes (see cells on E) indicate positive staining for HtrA1. Comparison of D and E, the figures show the co-localized expression of Ddr2 and HtrA1. The co-localization expression of these two proteins is also seen in the picture F (orange color stained cells). The picture F is a combination of the pictures D and E.
Discussion
The mechanisms responsible for the pathogenesis of OA are largely unknown. The goal of this study is to understand the molecular basis for the progression of articular cartilage degeneration that leads to OA. Based on data from both our studies and those conducted by others, we hypothesize that excessive mechanical force, due to either overloading on a normal joint or normal loading on a defective joint, can incite chondrocytes, thus resulting in increased chondrocyte activity, such as chondrocyte clustering, during the early stages of the degenerative process. As the degenerative process progress over time, the activated chondrocytes synthesize and release matrix-degrading enzymes. A consequence of the enzyme release is the disruption of the pericellular matrix of chondrocytes. This in turn enhances the exposure of the chondrocytes to type II collagen fibrils. It should be duly noted that type II collagen fibrils are present within the territorial and interterritorial locations of normal articular cartilage, and that little to no type II collagen fibrils are present close to the chondrocyte surface (Hunziker et al., 1997). Interaction of the chondrocytes with type II collagen fibrils may result in enhanced molecular signaling mediated by Ddr2, a cell membrane tyrosine kinase receptor for fibrillar type II collagens. The activation of Ddr2 then induces the expression of Mmp-13, a matrix metalloprotease, which cleaves type II collagen (Xu et al., 2005; Xu et al., 2007; Sunk et al., 2007). The resulting type II collagen fragments (CII-f) may in turn further increase the synthesis of MMP-13 through interaction with cell-surface integrins. The result is a positive feedback amplification loop that leads to the irreversible destruction of the articular cartilage. If this is the case in the majority of OA, then the following question remains: which enzyme/enzymes are responsible for the disruption of the pericellular matrix?
Results from other research groups indicate that the expression of HtrA1 is increased in human OA cartilage and in experimental mouse arthritic models. Furthermore, substrates of HtrA1 have been identified as the pericellular matrix components of chondrocytes. However, those results have left open questions of what is the precise time point of the increased expression of HtrA1 during OA progression and what is the consequence of the elevated expression of the enzyme. The data from our experiments demonstrates that the expression of HtrA1 is indeed elevated in the articular cartilages of knee and TM joints taken from genetically mutated mouse OA models, during the early stages of the disease. The increase in protein expression of HtrA1 occurred earlier in Col9a1−/− mice than in cho/+ mice. This indicates that, at any specified time point in the development of OA, the degenerative process occurring in the knee joints of Col9a1−/− mice is more advanced and aggressive than that occurring in cho/+ mice. This is consistent with the morphological changes observed in the knee joints of Col9a1−/− and cho/+ mice. Moreover, we previously found that the expression of Ddr2 was elevated in the knee joints of mutant mice at 6 months of age, and this was paralleled with an increase in the expression of HtrA1 in the mutant mice. An increase in the level of HtrA1 expression was also observed in the articular cartilage of surgically induced mouse OA knee and TM joints, at the point in time at which the articular cartilage degeneration ensued. This suggests that regardless of the nature of the initializing events, be it genetic or non-genetic that leads to cartilage degeneration, expression of this enzyme in the articular cartilage is elevated during the early stages of the degenerative process. Furthermore, in our examination of a genetically mutated mouse model the level of HtrA1 mRNA was found to be increased in the OA articular cartilage. This suggests that the increased amount of HtrA1 protein found in the OA cartilage is, at least in part, due to the increased level of HtrA1 mRNA.
The increased expression of HtrA1 in the OA-like knee joints of mice during the early stage of the disease begs the following question: what is the consequence of the up-regulated expression of this enzyme? Since the substrates of HtrA1 are components of the chondrocyte’s pericellular matrix, it is conceivable that these pericellular matrix components can be degraded by HtrA1. Type VI collagen is predominantly distributed pericellularly throughout cartilage and it is one of the major components of the pericellular matrix network. Thus, the disappearance of type VI collagen from the pericellular matrix is a good indicator that the pericellular matrix network has been disturbed. Results from our study of OA cartilage indicate that type VI collagen was absent from the pericelluar matrix of chondrocytes expressing the HtrA1 enzyme; thereby signifying that the integrity of the pericellular matrix network had been disturbed. Meanwhile, we observed that the expression of Ddr2 was elevated in those chondrocytes expressing HtrA1, indicating that the elevated expression of HtrA1 is a cellular event occurring upstream of an increase in the expression of Ddr2. This increase in the Ddr2 levels results from an interaction between chondrocytes and type II fibrillar collagen. This is consistent with our hypothesis that the disruption of the chondrocyte’s pericellular matrix is one of the early steps in the pathogenesis of OA (Fig. 7).
Fig. 7.

Molecular pathway of articular cartilage degeneration.
Taken together, these data suggest that inhibiting the activity of the HtrA1 enzyme may lead to an attenuation of the progression of OA. However, it remains to be seen whether or not side effects will result from the direct inhibition of this enzyme activity. Such side effects are particularly important when one considers the relatively broad biological function of this enzyme. Consequently, the identification of chondrocyte-specific factors that regulate the expression of HtrA1 will be central to the search in novel therapeutic targets involved in the prevention and treatment of OA.
Acknowledgments
This work was supported by NIH R01 AR-051989 (to Xu and Li).
References
- Alexopoulos LG, Youn I, Bonaldo P, Guilak F. Developmental and osteoarthritic changes in Col6a1-knockout mice: biomechanics of type VI collagen in the cartilage pericellular matrix. Arthritis Rheum. 2009;60:771–779. doi: 10.1002/art.24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T, Southan C, Ehrmann M. The HtrA family of proteases: implications for protein composition and cell fate. Mol Cell. 2002;10:443–455. doi: 10.1016/s1097-2765(02)00658-5. [DOI] [PubMed] [Google Scholar]
- Fässler R, Schnegelsberg PN, Dausman J, Shinya T, Muragaki Y, McCarthy MT, Olsen BR, Jaenisch R. Mice lacking alpha 1 (IX) collagen develop noninflammatory degenerative joint disease. PNAS. 1994;91:5070–5074. doi: 10.1073/pnas.91.11.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felson DT, Lawrence RC, Dieppe PA, Hirsch R, Helmick CG, Jordan JM, Kington RS, Lane NE, Nevitt MC, Zhang Y, Sowers M, McAlindon T, Spector TD, Poole AR, Yanovski SZ, Ateshian G, Sharma L, Buckwalter JA, Brandt KD, Fries JF. Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med. 2000;133:635–646. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- Forsyth CB, Pulai J, Loeser RF. Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002;46:2368–2376. doi: 10.1002/art.10502. [DOI] [PubMed] [Google Scholar]
- Grau S, Richards PJ, Kerr B, Hughes C, Caterson B, Williams AS, Junker U, Jones SA, Clausen T, Ehrmann M. The role of human HtrA1 in arthritic disease. J Biol Chem. 2006;281:6124–6129. doi: 10.1074/jbc.M500361200. [DOI] [PubMed] [Google Scholar]
- Hamerman D. The biology of osteoarthritis. New England J Med. 1989;320:1322–1330. doi: 10.1056/NEJM198905183202006. [DOI] [PubMed] [Google Scholar]
- Hu K, Xu L, Cao L, Flahiff CM, Brussiau J, Ho K, Setton LA, Youn I, Guilak F, Olsen BR, Li Y. Pathogenesis of Osteoarthritis-like Changes in Joints of Type IX Collagen-Deficient Mice. Arthritis Rheum. 2006;54:2891–2900. doi: 10.1002/art.22040. [DOI] [PubMed] [Google Scholar]
- Hu SI, Carozza M, Klein M, Nantermet P, Luk D, Crowl RM. Human HtrA, an evolutionarily conserved serine protease identified as a differentially expressed gene product in osteoarthritic cartilage. J Biol Chem. 1998;273:34406–34412. doi: 10.1074/jbc.273.51.34406. [DOI] [PubMed] [Google Scholar]
- Hunziker EB, Michel M, Studer D. Ultrastructure of adult human articular cartilage matrix after cryotechnical processing. Microsc Res Tech. 1997;37:271–284. doi: 10.1002/(SICI)1097-0029(19970515)37:4<271::AID-JEMT3>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Lachman RS, Krakow D, Cohn DH, Rimoin DL. MED, COMP, multilayered and NEIN: an overview of multiple epiphyseal dysplasia. Pediatr Radiol. 2005;35:116–123. doi: 10.1007/s00247-004-1323-4. [DOI] [PubMed] [Google Scholar]
- Lam NP, Li Y, Waldman AB, Brussiau J, Lee PL, Olsen BR, Xu L. Age-dependent increase of discoidin domain receptor 2 and matrix metalloproteinase 13 expression in temporomandibular joint cartilage of type IX and type XI collagen-deficient mice. Arch Oral Biol. 2007;52:579–84. doi: 10.1016/j.archoralbio.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lacerda DL, Warman ML, Beier DR, Oxford JT, Morris NP, Andrikopoulos K, Ramirez F, Wardell BB, Lifferth GD, Teuscher C, Woodward SR, Taylor BA, Seegmiller RE, Olsen BR. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. 1995;80:423–430. doi: 10.1016/0092-8674(95)90492-1. [DOI] [PubMed] [Google Scholar]
- Loeser RF, Forsyth CB, Samarel AM, Im HJ. Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem. 2003;278:24577–24585. doi: 10.1074/jbc.M304530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka C, Tsujimoto R, Kajikawa M, Koshiba-Takeuchi K, Ina J, Yano M, Tsuchiya A, Ueta Y, Soma A, Kanda H, Matsumoto M, Kawaichi M. HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development. 2004;131:1041–1053. doi: 10.1242/dev.00999. [DOI] [PubMed] [Google Scholar]
- Poole CA. Articular cartilage chondrons: form, function and failure. J Anat. 1997;191:1–13. doi: 10.1046/j.1469-7580.1997.19110001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole CA, Ayad S, Schofield JR. Chondrons from articular cartilage: I. Immunolocalization of type VI collagen in the pericellular capsule of isolated canine tibial chondrons. J Cell Sci. 1988;90:635–643. doi: 10.1242/jcs.90.4.635. [DOI] [PubMed] [Google Scholar]
- Sunk IG, Bobacz K, Hofstaetter JG, Amoyo L, Soleiman A, Smolen J, Xu L, Li Y. Increased expression of discoidin domain receptor 2 is linked to the degree of cartilage damage in human knee joints: A potential role in osteoarthritis pathogenesis. Arthritis Rheum. 2007;56:3685–3692. doi: 10.1002/art.22970. [DOI] [PubMed] [Google Scholar]
- Tsuchiya A, Yano M, Tocharus J, Kojima H, Fukumoto M, Kawaichi M, Oka C. Expression of mouse HtrA1 serine protease in normal bone and cartilage and its upregulation in joint cartilage damaged by experimental arthritis. Bone. 2005;37:323–336. doi: 10.1016/j.bone.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Wadhwa S, Embree M, Ameye L, Young MF. Mice deficient in biglycan and fibromodulin as a model for temporomandibular joint osteoarthritis. Cells Tissues Organs. 2005;181:136–143. doi: 10.1159/000091375. [DOI] [PubMed] [Google Scholar]
- Wu J, Liu W, Bemis A, Wang E, Qiu Y, Morris EA, Flannery CR, Yang Z. Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheum. 2007;56:3675–3684. doi: 10.1002/art.22876. [DOI] [PubMed] [Google Scholar]
- Xu L, Flahiff CM, Waldman BA, Wu D, Olsen BR, Setton LA, Li Y. Osteoarthritis-like changes and decreased mechanical function of articular cartilage in the joints of mice with the chondrodysplasia gene (cho) Arthritis Rheum. 2003;48:2509–2018. doi: 10.1002/art.11233. [DOI] [PubMed] [Google Scholar]
- Xu L, Peng H, Glasson S, Lee PL, Hu K, Ijiri K, Olsen BR, Goldring MB, Li Y. Increased expression of a collagen receptor discoidin domain receptor 2 in articular cartilage as a key event in the pathogenesis of osteoarthritis. Arthritis Rheum. 2007;56:2663–2673. doi: 10.1002/art.22761. [DOI] [PubMed] [Google Scholar]
- Xu L, Peng H, Wu D, Hu K, Goldring MB, Olsen BR, Li Y. Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in cho/+ mice. J Biol Chem. 2005;280:548–555. doi: 10.1074/jbc.M411036200. [DOI] [PubMed] [Google Scholar]
- Xu L, Polur I, Lim C, Servais JM, Dobeck J, Li Y, Olsen BR. Early-onset osteoarthritis of mouse temporomandibular joint induced by partial discectomy. Osteoarth Cartil. 2009;17:903–908. doi: 10.1016/j.joca.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumbrunn J, Trueb B. Primary structure of a putative serine protease specific for IGF-binding proteins. FEBS Lett. 1996;398:187–192. doi: 10.1016/s0014-5793(96)01229-x. [DOI] [PubMed] [Google Scholar]