

SUMMARY
Porphyromonas gingivalis, a self-limiting oral pathogen, can colonize and replicate in gingival epithelial cells (GECs). P. gingivalis-infected GECs are protected from mitochondrion-dependent apoptosis, partially through activation of phosphatidyl inositol 3-kinase/Akt signaling. Biochemical events associated with P. gingivalisinduced inhibition of apoptosis include the blocking of mitochondrial membrane permeability and cytochrome-c release. We studied functional importance of Akt and the status of associated key mitochondrial molecules, pro-apoptotic Bad and caspase-9, during infection of GECs. We found that P. gingivalis infection caused significant phosphorylation of Bad progressively, while messenger RNA levels for Bad slowly decreased. Fluorescence microscopy showed translocation of the mitochondrial Bad to the cytosol post-infection. Conversely, P. gingivalis lost the ability to promote phosphorylation and translocation of Bad in Akt-deficient GECs. Caspase-9 activation induced by a chemical inducer of apoptosis was significantly inhibited by infection over time. However, Akt depletion by small interfering RNA did not reverse inhibition of caspase-9 activation by infection. Hence, P. gingivalis inactivates pro-apoptotic Bad through Akt. The inhibition of caspase-9 activation appears to be independent of Akt. Overall, our findings suggest that Akt is a key component of anti-apoptotic pathways stimulated by P. gingivalis. The P. gingivalis uses other mitochondrial pathways to protect host cells from cell-death and to ensure its survival in gingival epithelium.
Keywords: apoptosis, cell survival, mitochondria, persistence, Porphyromonas
INTRODUCTION
The human oral mucosa maintains barrier and signaling functions in the presence of a diverse microbial consortium indicating highly sophisticated co-evolved relationships between the epithelium and its specialized microbial colonizers. While these interactions often result in commensal harmony within the oral cavity, harmful interactions between the invading oral microbes and the host can lead to debilitating acute or chronic diseases (Jenkinson & Lamont, 2005). The latter outcome has been found to be frequently associated with ‘potential pathogens’ that have evolved a variety of complex strategies that allow successful encounters with the immune defense system and adaptation to host metabolic circumstances enabling co-existence with their host (Finlay & Falkow, 1989).
Within the gingival compartment, gingival epithelial cells are among the first host cells encountered by the colonizing oral bacteria. Porphyromonas gingivalis, a gram-negative, opportunistic anaerobe, and a prominent etiological agent in severe forms of periodontal disease, is a successful colonizer of oral epithelial tissues (Socransky et al., 1998; Lamont & Yilmaz, 2002). The organism is highly invasive, and can swiftly enter human primary cultures of gingival epithelial cells (GECs) (Belton et al., 1999). Interestingly, P. gingivalis can exist in the host epithelium without the presence of overt disease as determined using fluorescently labeled 16S ribosomal RNA probes in conjunction with confocal microscopy from in vivo samples (Rudney et al., 2005; Colombo et al., 2007). The P. gingivalis can replicate to high levels intracellularly, maintain viability for extended periods in primary GECs, and spread from cell-to-cell through actin-based membrane projections later in the infection (Yilmaz et al., 2006). Infected GECs harboring large numbers of intracellular P. gingivalis do not undergo apoptotic or necrotic death and are resistant to apoptosis. The infection induces an anti-apoptotic phenotype in primary GECs by rendering the host cells resistant to cell death from various potent pro-apoptotic agents including staurosporine (STS), camptothecin, and extracellular ATP (Nakhjiri et al., 2001; Yilmaz et al., 2004, 2008; Mao et al., 2007). Furthermore, P. gingivalis-infected GECs undergo successful mitosis and the infection accelerates the host-cell cycle progression (Kuboniwa et al., 2008). The organism impacts multiple anti-apoptotic/survival host pathways and prevents host-cell death by partially blocking mitochondrion-dependent apoptosis. This includes inhibition of mitochondrial membrane depolarization and cytochrome-c release, upregulation of anti-apoptotic Bcl-2 and downregulation of pro-apoptotic Bax, and inhibition of caspase-3 activation through dual JAK/Stat and Akt signaling (Nakhjiri et al., 2001; Yilmaz et al., 2004; Mao et al., 2007). Also, P. gingivalis infection activates the pro-survival phosphatidyl inositol 3 (PI3) kinase/Akt pathway in primary GECs, and a specific PI3 kinase inhibitor, LY294002, substantially abolishes the infected cells’ resistance to apoptosis stimulated by STS. Phosphorylation of Akt on Ser473 by P. gingivalis infection exerts a strong anti-apoptotic effect, possibly through the inhibition of mitochondrial permeability changes as the suppression of Akt activation by the PI3 kinase inhibitor results in the loss of mitochondrial membrane potential, cytochrome-c release, and DNA fragmentation (Yilmaz et al., 2004). While the previous studies revealed that P. gingivalis infection can modulate several mitochondrial cell death pathways, the complete range and nature of mitochondrial molecules associated with infection have yet to be fully understood.
In mammalian cells mitochondrial integrity, which is central to both caspase-dependent and independent cell death, is tightly regulated by Bcl-2 family proteins (Chao & Korsmeyer, 1998; Cosulich et al., 1999). Bad, a pro-apoptotic member of the Bcl-2 family, can mediate mitochondrial release of cytochrome-c and promote apoptotic death. The pro-apoptotic activity of Bad can be abrogated through phosphorylation by Akt kinase (Datta et al., 1999; Chen et al., 2005). Akt can also concurrently interact and inhibit the initiator apoptotic enzyme, caspase-9, an upstream regulator of the executioner caspases including caspase-3 and caspase-6 (Thornberry & Lazebnik, 1998; Zhang et al., 2004). Hence, Akt can modulate mitochondrion-associated cell death and its associated key molecules, promoting enhanced host-cell survival and protection against apoptosis. The goal of this study was, therefore, to further determine the functional significance of Akt signaling on P. gingivalis-induced survival of primary GECs and simultaneously to characterize the role of some key mitochondrial molecules in this process. We found that Bad phosphorylation levels significantly increased over the course of infection while Bad messenger RNA (mRNA) levels slowly decreased. Conversely, knock-down of Akt by small interfering RNA (siRNA) prevented P. gingivalis-dependent phosphorylation and translocation of Bad. Caspase-9 activation induced by STS treatment was significantly inhibited by P. gingivalis infection over time. However, depletion of Akt did not reverse the inhibition of caspase-9 activation by infection. Hence, P. gingivalis, an opportunistic oral pathogen, successfully uses multiple mitochondrial survival pathways to ensure effective colonization in the gingival epithelium.
METHODS
Bacteria and cell culture
The P. gingivalis ATCC 33277 was cultured anaerobically for 24 h at 37°C in trypticase soy broth supplemented with yeast extract (1 μg/ml), hemin (5 μg/ml), and menadione (1 μg/ml). Bacteria were grown for 24 h, harvested by centrifugation at 6000 g and 4°C for 10 min, washed twice, and resuspended in Dulbecco’s phosphate-buffered saline (PBS), pH 7.3, before incubation with host cells. Bacteria were quantified using a Klett–Summerson photometer.
Primary GECs were obtained after oral surgery in the clinics of the University of Florida from healthy gingival tissue as previously described (Lamont et al., 1995). Cells were cultured as monolayers in serum-free keratinocyte growth medium (Lonza, Walkersville, MD) at 37°C in 5% CO2. GECs were used for experimentation at 80% confluence and cultured for 48 h before infection with bacterial cells or exposure to other test reagents in keratinocyte growth medium.
Infection of cells with P. gingivalis and treatment with STS and PI3 kinase inhibitor
GECs were infected at a multiplicity of infection of 100 with P. gingivalis 33277 for 30 min, 60 min, 2, 6, 12 or 24 h at 37°C in a CO2 incubator. All time points for the infections were carried out backwards; i.e. instead of beginning all infections at the same time, infections were initiated at the indicated times before time zero so that all incubations could be stopped at the same time. For induction of apoptosis studies, GECs were treated with a potent apoptosis inducer, STS, 2 μM or 4 μM (Sigma, St. Louis, MO), for 3 h after 21 h infection with or without the bacteria. Additionally, after 3 h infection with P. gingivalis 33277, GECs were treated with the PI3 kinase inhibitor, LY294002 (LY), 20 μM (Sigma), for 18 h (3 h post-infection) before the 3-h STS treatment. All treatments were performed in GEC culture media at 37°C in the CO2 incubator.
Analysis of apoptosis by annexin-V and propidium iodide staining
Early apoptotic changes were identified by using fluorescein isothiocyanate-conjugated Annexin-V-fluos (green fluorescence) (Roche Applied Science, Indianapolis, IN), which binds to phosphatidylserine exposed on the outer leaflet of apoptotic cell membranes. Propidium iodide (PI) (red fluorescence) (Sigma) was used for the discrimination of necrotic cells from the Annexin-V-positively stained cells. Briefly, GECs were grown on four-well chambered slides (Nalge-Nunc International, Rochester, NY), infected with P. gingivalis for 24 h and incubated with various agents as described above. The slides were washed with ice cold PBS and immediately treated with 100 μl Annexin-V-Fluos binding solution containing 10 μl Annexin-V-Fluos labeling reagent per 1000 μl HEPES buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 5 mM CaCl2) and 1 μg/ml PI. After 15 min incubation in the dark at room temperature, the slides were washed with ice-cold PBS and fixed in 10% neutral buffered formalin for 20 min. Slides were mounted in Vectashield Mounting Medium (Vector Laboratories, Burlingame, CA) containing 4′,6-diamidino-2-phenylindole (DAPI) for nuclear staining and examined using a fluorescence microscope (Zeiss Axio imager A1) equipped with bandpass optical filter sets appropriate for imaging of dyes. The images were captured with a cooled CCD camera (Qimaging, Surrey, Canada) controlled by Qcapture software. Cells that were untreated and incubated in the binding buffer with Annexin-V and PI or only with Annexin-V or PI separately served as controls for determining the threshold of fluorescence intensity. Cells that were treated with only STS or ethanol served as positive staining for Annexin-V and PI, respectively. Approximately 1000 cells per condition from three separate experiments were analysed to determine the percentage of cells positively stained for Annexin-V and PI. The same microscopy settings were employed throughout all experiments.
Depletion of Akt by RNA interference
Primary cultures of GECs at 50% confluence were transfected in GEC growth medium using 100 nM of siRNA duplexes in 5 μl siRNA Akt DharmaFECT1 agent (Dharmacon, Lafayette, CO). Briefly, 5 μl transfection agent was added drop-wise into 195 μl of GEC growth medium and the incubation was performed for 10 min at room temperature. The 100 nM siRNA Akt sequences (Dharmacon) were added to diluted transfection agent, mixed gently, and incubated for 10 min at room temperature. Finally, 50 μl of this mixture was added to each well, the plate was rocked gently, and further incubated for 48 h at 37°C 5% CO2. Non-target pool siRNA (Dharmacon) and transfection agent alone were used as negative controls.
Confirmation of Akt knockdown by Western immunoblotting
Western blot analysis was performed up to 48 h post-transfection using equal amount of protein in each sample determined by a bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL), and the samples were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Proteins were blotted onto nitrocellulose membrane, blocked in 5% dried milk solution diluted in Tris-buffered saline containing 0.01% Tween 20 (TBS/T), incubated with Akt (1/2/3) antibody at a dilution of 1: 2000 (Cell Signaling, Danvers, MA), and treated with horseradish peroxidase (HRP) -conjugated secondary antibody at 1: 5000 (Cell Signaling). The blot was then stripped and probed with anti-β-actin antibody 1: 1000 and HRP-conjugated secondary antibody (Cell Signaling) at 1: 2000 used as control. Results were visualized using an enhanced chemiluminescence detection kit (Amersham Biosciences, Piscataway, NJ). Densitometric scanning was quantified using NIH-ImageJ (Bethesda, MD) analysis. The knockdown experiments were repeated at least three separate times.
Assay of Bad activation by immunoprecipitation
GECs were infected with P. gingivalis 33277 for 6, 12, or 24 h. Cells were washed twice with cold PBS and solubilized in lysis buffer (50 mM Tris–HCI pH 7.2, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mM EDTA with phosphatase inhibitor and protease cocktail inhibitor (Sigma). Lysates were clarified by centrifugation at 10,000 g for 15 min at 4°C and protein concentration was determined by the bicinchoninic acid protein assay (Pierce Company). Bad was precipitated from the cell lysates with anti-Bad-specific antibody (Cell Signaling) overnight at 4°C. The protein –antibody complexes were collected with Protein A–Sepharose beads at 1: 10 by volume and washed three times with lysis buffer. Samples were resolved by 12% SDS–PAGE and transferred to nitrocellulose membranes. Ser136 phosphorylation of Bad was assessed by reaction at 4°C overnight with a 1: 500 dilution of specific anti-phosphoBad antibody (Cell Signaling) followed by a 1: 2000 dilution of HRP-conjugated secondary antibody (Cell Signaling). Results were visualized by the enhanced chemiluminescence detection system (Amersham Pharmacia, Piscataway, NJ), analysed by scanning densitometry and quantified using NIH-ImageJ. Blots were then stripped and reprobed with a 1: 1000 dilution of anti-Bad and 1: 2000 dilution of HRP-conjugated secondary antibody (Cell Signaling) to determine total Bad in the samples.
Bad localization assay by fluorescence microscopy
GECs were grown on four-well chambered slides (Nalge-Nunc International), washed with ice-cold PBS, and fixed in 10% neutral buffered formalin for 20 min. The cells were permeabilized for 10 min with 0.1% Triton X-100 at 4°C and the same slides were incubated with anti-Bad monoclonal antibody 1: 50 (Santa Cruz) and anti-P. gingivalis 33277 rabbit polyclonal antibody at 1: 1000 in PBS containing 0.1% Tween and 3% bovine serum albumin for 1 h. After washing with PBS twice, samples were stained with Oregon Green 488 goat anti-mouse secondary antibody and Alexa-Fluor 594 anti-rabbit secondary antibody, respectively, for 1 h at room temperature (Invitrogen, Carlsbad, CA). Samples with no primary antibody incubation were included as control. Slides were mounted in Vectashield Mounting Medium (Vector Laboratories) containing DAPI for nuclear staining and examined using a fluorescence microscope (Zeiss Axio imager A1) as described previously (Yilmaz et al., 2006). The images were captured by multiple exposures using a cooled CCD camera controlled by Qcapture software (Surrey, Canada). The images are representative of 100 cells studied per sample from at least two separate experiments performed.
Real-time quantitative polymerase chain reaction
Total RNA was isolated from triplicate independent control and P. gingivalis-infected GECs using RNeasy Mini Kit (Qiagen, Germantown, MD). The genomic DNA was removed by DNase 1 treatment (Ambion, Austin, TX). Total RNA (1 μg) from each sample was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI). Real time quantitative polymerase chain reaction (PCR) was conducted in triplicate for each complementary DNA (cDNA) sample with the iCycler iQ real-time PCR detection system using iQ™ SYBR Green Supermix (Bio-Rad, Hercules, CA). Two microlitres template cDNA was added to a final volume of 25 μl with 1XSYBR Green Supermix and 1 μl (20 μM) of the following primer pairs: Bad: forward 5′-GGGCACAGCAACGCAGATG-3′, reverse 5′-TGGGAACGGGTGGAGTTTCG-3′; 18s rRNA forward 5′-CGCCGCTAGAGGTGAAATTC-3′ reverse 5′-TCTTGGCAAATGCTTTCGCT-3′.
18s rRNA was used as an endogenous control. Real-time results were analysed using iCycler™ iQ Optical System software (Bio-Rad). The melt curve profile was analysed to verify a single peak for each sample, indicating primer specificity.
Preparation of standards
Specific DNA products for each gene under investigation were synthesized from chromosomal DNA using standard PCR methods and were visualized by gel electrophoresis to verify that a single specific product had been generated. Each product was purified using the QIAquick PCR Purification Kit (Qiagen), and quantified using the GeneQuant spectrophotometer. DNA product copy number was calculated using the formula (Yin et al., 2001):
A 10-fold dilution series of each DNA standard was prepared for starting quantities of 108−101 copies/μl. These were used at least in duplicate in each real-time PCR assay to allow the real-time PCR software to estimate starting quantity of that gene in the cDNA samples.
Measurement of caspase-9 activity
GECs or Akt-deficient GECs were treated with STS (4 μM) for 3 h, or control buffer, or infected with P. gingivalis 33277 for 30, 60 min, 2, 6, 12, and 24 h with or without STS. Caspase-9 activity was measured by incubating cells with caspase-9-specific substrate, FAM-LEHD-FMK at 1: 30 dilution (Immunochemistry Technologies, Bloomington, MN). The fluorescence increase in cells with activated caspase-9 was measured with a 96-well fluorescence plate reader (excitation at 488 nm, emission at 520 nm with black microtiter plates). Caspase-9 activation was quantified as the amount of green fluorescence emitted from the FLICA (fluorescent labeled inhibitors of caspases) probes bound to the enzyme. The relative fluorescence intensities were obtained by subtracting the value of the sample with the smallest fluorescence intensity from each sample. The results were then expressed as percentages of untreated control sample with fluorescent intensity normalized to 100.
RESULTS
Depletion of Akt by siRNA reverses P. gingivalis-induced protection of GECs against cell death
Signaling through Akt plays a critical role in protection of cells from a variety of severe stresses, as may develop during infection by intracellular pathogens (Datta et al., 1999; Verbeke et al., 2006). Our previous findings through the pharmacological inhibition and phosphorylation assays revealed a potentially important role for Akt signaling in P. gingivalis-mediated protection against cell death by exogenous apoptotic stimuli targeting mitochondria (e.g. STS). Therefore, we further examined the involvement of Akt in this process by RNA interference. Forty-eight hours after transfection with Akt siRNA, approximately 85% of the protein levels of Akt were reduced in primary GECs as determined by Western blotting (Fig. 1). We confirmed that Akt siRNA by itself did not induce apoptosis of uninfected cells, and Akt-deficient cells had infection levels comparable to siRNA-untreated cells, indicating that Akt deficiency did not impact the internalization efficiency of P. gingivalis (not shown). We first measured 24-h infected and uninfected GECs treated with the two concentrations (2 and 4 μM) of the apoptosis inducer STS for 3 h and analysed cell death by immunofluorescence microscopy using Annexin-V-PI double-staining (Fig. 2A,B). In addition, we used LY294002 (20 μM), a specific inhibitor of PI3 kinase an immediate upstream mediator of Akt, as described previously (Yilmaz et al., 2004). Following treatment with LY294002, the infected and uninfected GECs were incubated with the varying concentrations of STS (Fig. 2A). Simultaneously, we measured the cell death in the Akt-deficient cells. The GECs were first transfected with Akt siRNA, infected with P. gingivalis, and then treated with STS and/or LY294002 similar to the treatment of normal cells (Fig. 2C,D). Consistent with previous findings, P. gingivalis did not promote cell death. Approximately 83 and 74% of normal cells that were infected with P. gingivalis and treated with 2 μM or 4 μM STS, respectively, were resistant to apoptosis, whereas approximately 45 and 53% of normal cells without infection became severely apoptotic in a concentration-dependent manner following treatment with 2 μM or 4 μM STS (Fig. 2A). In the absence of Akt, the level of apoptosis in the infected cells and the cells infected and treated with STS displayed approximately three- to fourfold increases. These results were statistically significant (P < 0.05 and P < 0.01, respectively) by Student’s t-test (Fig. 2C,D). Treatment of both normal GECs and the Akt-deficient cells with LY294002 without infection and STS did not induce any significant level of apoptosis (not shown). In addition, the treatment of the infected deficient cells with LY294002 followed by the STS treatments did not promote further increase in the level of apoptosis, proposing a specific role for Akt (Fig. 2C,D) as LY294002 will also inhibit PI3 kinase and downstream pathways parallel to Akt. Taken together, these results suggest that Akt plays an essential role in P. gingivalis-mediated protection against mitochondrion-dependent cell death in primary GECs because knock-down of Akt by siRNA significantly reversed the pro-survival phenotype of host cells induced during the infection.
Figure 1.
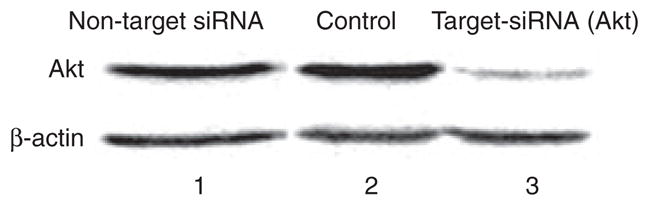
Knockdown of Akt by small interfering RNA (siRNA) in primary gingival epithelial cells. A target-specific Akt antibody was used to confirm the inhibition of Akt expression by Western blotting. A non-target antibody (β-actin) was used to control proper loading and specificity of Akt siRNA. Column 1 is non-target siRNA (control), column 2 is transfection agent alone (control), and column 3 is target siRNA with transfection agent. Densitometric scanning of the products from the 48-h post-transfection samples (column 3, Akt lane) displayed a approximately 85% decrease in the level of Akt demonstrating a successful inhibition of the protein in the primary cultures of gingival epithelial cells.
Figure 2.

Quantitative analysis of cell death by Annexin-V and propidium iodide (PI). The percentage of dying cells was determined by dual staining with fluorescein isothiocyanate-conjugated Annexin-V and PI using fluorescence microscopy. The threshold of fluorescence intensity was determined with samples that were uninfected and untreated. Staurosporine (STS) treated cells and ethanol-treated cells (not shown), respectively, were used to evaluate the positively Annexin-V and/or PI-stained cells. At least 10 separate fields containing an average of 50 cells were studied quantitatively from three independent experiments performed in duplicate (A). Primary gingival epithelial cell (GEC) monolayers grown on microscopic chambers were incubated in the binding buffer containing Annexin-V (green fluorescence) and PI (red fluorescence). PI was used for the discrimination of necrotic or late apoptotic cells from the Annexin-V-positively stained cells. The nuclear stain DAPI (blue fluorescence) was used to visualize the number of cells in the field. (B) The simultaneous quantitative analysis and microscopic examinations were employed in the Akt-deficient GECs for similar conditions to those employed with normal GECs (C, D). Asterisks denote statistical significance (*P = 0.006, **P = 0.001, and ***P = 0.01; Student’s t-test) for 24-h uninfected + STS-treated (2 μM) GECs versus 24-h infected + STS-treated (2 μM) GECs, for 24-h uninfected + STS-treated (4 μM) GECs versus 24-h infected + STS-treated (4 μM) GECs, and for 24-h infected GECs versus 24-h infected + STS (2 μM) + LY-treated GECs, respectively (A). Asterisks denote statistical significance (*P = 0.03 and **P = 0.005; Student’s t-test) for 24-h uninfected + siRNA-treated GECs versus 24-h infected + siRNA-treated GECs and 24-h infected + siRNA-treated GECs versus 24-h infected + STS + siRNA-treated GECs, respectively (C).
Akt mediates pro-apoptotic Bad phosphorylation in GECs infected by P. gingivalis
Our previous study showed that P. gingivalis infection results in phosphorylation/activation of Akt. Akt has been shown to promote cell survival through its ability to phosphorylate Bad at the Ser136 residue (Datta et al., 1997; Seo et al., 2004; Yilmaz et al., 2004). Phosphorylated Bad dissociates from a heterodimeric complex formed with anti-apoptotic Bcl-2 and/or Bcl-xL proteins, thereby, increasing anti-apoptotic effects of the Bcl-2 family. In light of our new results, we investigated next whether activated Akt can phosphorylate pro-apoptotic Bad. We therefore measured phosphorylation from the samples immunoprecipitated with specific Bad antibody over the course of infection in normal versus Akt-deficient GECs by immunoblots using antibodies against a phosphorylation site of Bad Ser136 residue.
We found that phosphorylation of Bad noticeably increased (1.6-fold) at 6 h infection, was raised more than threefold at 12 h and peaked to approximately 4.5-fold after a 24-h infection in the ratio of phosphorylated: total Bad, as determined by densitometry analysis of Western blotting products (Fig. 3A,B). There was no change detected after 2 h of infection (not shown). In contrast, the level of phosphorylated Bad remained unchanged in Akt siRNA-transfected cells over the course of infection (Fig. 3A) as verified by the ratio of phosphorylated: total Bad (Fig. 3C). The protection of P. gingivalis-infected GECs against cell death therefore appears partially to be the result of inactivation of pro-apoptotic Bad, which appears to be directly associated with activation of Akt signaling. This conclusion is further strengthened by the analysis of Akt phosphorylation on the Ser473 residue by a quantitative immunofluorescence assay, which revealed enhanced activation kinetics (maximal 2.5-fold increase) for Akt for the similar time points of P. gingivalis infection in GECs (data not shown).
Figure 3.

Porphyromonas gingivalis infection induces a large gradual increase in Bad phosphorylation. Primary gingival epithelial cells (GECs) and the Akt-deficient GECs were infected with P. gingivalis for 0 min (control), 6, 12, and 24 h. Cell lysates immunoprecipitated with anti-Bad-specific antibody analysed by immunoblotting with antibodies against phosphorylated Bad (Ser136) (A). Blots were analysed by scanning densitometry and ratios of phosphorylated:total Bad were determined relative to ratios in control cells. The values show relative fold change calculated for a representative experiment and represent results obtained from at least three experiments (B,C).
Effect of P. gingivalis infection on Bad at the mRNA level
A previous study by Nakhjiri et al. (2001) showed that the P. gingivalis infection in primary GECs also modulates the expression of Bcl-2 family members transcriptionally. This includes downregulation of expression of the pro-apoptotic molecule Bax and upregulation in the expression of the anti-apoptotic molecule Bcl-2. Similarly, we wanted to further characterize the effect of infection on Bad at the mRNA level both in normal and Akt-deficient GECs. Although our protein phosphorylation assays (Fig. 3A) illustrated a major increase in the levels of Bad phosphorylation, quantitative (real-time) reverse transcription-PCR was performed on mRNA extracted from primary GECs infected with P. gingivalis and showed gradual (approximately 60%) decrease in Bad mRNA levels following the 24-h incubation (Fig. 4A). Interestingly, there was no change in the levels of Bad mRNA from the uninfected samples deficient in Akt, but there was an approximately 20% increase in the samples treated with P. gingivalis for 24 h (Fig. 4B) pointing to a potentially additional role for the Akt pathway in suppressing pro-apoptotic Bad at the transcriptional level.
Figure 4.

Bad messenger RNA (mRNA) levels slowly decreased by Porphyromonas gingivalis infection. Gene expression was measured by quantitative reverse transcription–polymerase chain reaction on mRNA from primary gingival epithelial cells (GECs) (A) and Akt-deficient GECs (B) infected with P. gingivalis for 0 min (control), 6, 12, and 24 h. Relative fold change was calculated by dividing the copy number of the gene transcript in P. gingivalis-infected cells by the copy number in control cells. Data are representative of three independent experiments performed in triplicate. *P = 0.05; Student’s t-test) for 24-h uninfected GECs versus 24-h infected GECs.
Porphyromonas gingivalis infection sequesters Bad in cytosol of GECs through Akt
Since we observed a dramatic increase in the levels of phosphorylated Bad during the 24-h infection of P. gingivalis in GECs and phosphorylated Bad is normally maintained in cytosol in an inactive form, we wanted to next determine whether 24-h infection alters the intracellular distribution of Bad in GECs using immunofluorescent microscopy. Akt-deficient GECs were also analysed to further verify the importance of Akt in this interaction. Uninfected control cells had a large proportion of Bad localized in the cytosol in the absence of apoptotic stimuli (Fig. 5A). Addition of STS to the uninfected cells resulted in strong staining of Bad around the nuclei, where typically mitochondria are clustered in the cell, indicating a large amount of Bad translocated to mitochondria (Fig. 5A). However, the cells infected with P. gingivalis showed abundant Bad accumulated in cytosol. On the other hand, GECs that were lacking Akt displayed similarly strong staining of Bad around the nucleus both in infected and STS-treated uninfected cells (Fig. 5B). Overall results further confirmed the role of Akt-associated signaling in sequestration of pro-apoptotic Bad in cytosol, during P. gingivalis infection, thereby, increasing anti-apoptotic effects of the organism in primary GECs.
Figure 5.

Porphyromonas gingivalis infection redistributes Bad localization in primary gingival epithelial cells (GECs) through Akt. Intracellular Bad localization was detected by immunofluorescence using antibodies against Bad (green). The samples were also stained with P. gingivalis antibody (red) and DAPI (blue) to visualize the nuclei. (A) Uninfected cells displayed large proportions of Bad localized in the cytosol. However, incubation with the apoptosis inducer staurosporine (STS) caused strong staining in the perinuclear area, indicating translocation of Bad to mitochondria. Infection with P. gingivalis produced sequestration of Bad in the cytosol. (B) Akt knockdown cells were prepared as in (A). The localization of Bad showed intense staining around the nuclei, where the mitochondria are. This was similar in the uninfected STS-treated cells (control). The images were captured with a fluorescence microscope equipped with a cooled CCD.
Inhibition of pro-apoptotic caspase-9 by P. gingivalis is independent of Akt
Akt can independently interact and block the activation of initiator apoptotic enzyme, caspase-9, localized in mitochondria, an upstream regulator of the executioner caspases including caspase-3 and caspase-6 (Kennedy et al., 1997; Nicholson & Thornberry, 1997). A recent study has shown that P. gingivalis infection in primary GECs can inhibit caspase-3 activation through dual JAK/Stat and Akt signaling (Mao et al., 2007). To determine whether caspase-9 could be involved in the resistance to apoptosis associated with mitochondrial death pathways by P. gingivalis infection, we first examined the caspase-9 activation kinetics throughout the infection in GECs with and without STS using the caspase-9 substrate, FAMLEHD-FMK, whose relative fluorescence increases inside cells with activated caspase-9 (Fig. 6A).
Figure 6.

Inhibition of caspase-9 by Porphyromonas gingivalis infection is independent of Akt. Caspase-9 activity was determined by a fluorescent substrate assay and data are expressed as per cent of uninfected treated cells (control). Staurosporine (STS) -treated uninfected cells were used as control for the caspase activation. Primary gingival epithelial cells (GECs) were infected with P. gingivalis for the indicated times or were infected and treated with STS (4 μM) as described in Methods. P. gingivalis infection had no effect on caspase-9 activity in the initial incubation periods, yet it produced a noticeable decrease in enzyme activity at 6, 12, and 24 h (A). Akt-deficient GECs also displayed similar caspase-9 activity with normal primary GECs (B). The values show per cent averages and SD for two samples obtained from at least two experiments performed in duplicate. Asterisks denote statistical significance (*P = 0.05, **P = 0.08, ***P = 0.04; Student’s t-test) for 24-h infected GECs versus 24-h uninfected GECs, for 24-h infected + STS-treated GECs versus 24-h uninfected + STS-treated GECs, and for 24-h uninfected GECs versus 24-h uninfected + STS-treated GECs, respectively (A). Asterisks denote statistical significance (*P = 0.02, **P = 0.03, and ***P = 0.01; Student’s t-test) for 6 -h infected + siRNA-treated GECs versus uninfected + siRNA-treated GECs, 24-h infected + siRNA-treated GECs versus uninfected + siRNA-treated GECs, and for 24-h infected + STS + siRNA-treated GECs versus uninfected + STS + siRNA-treated GECs (B).
Consistent with our previous findings, caspase-9 displayed a small increase in its enzymatic activity within the first hours of infection (30 min and 1 h) compared with untreated controls, and was significantly decreased (approximately 50% lower than the baseline levels of caspase-9 activity) after 2, 6, 12, and 24 h of infection (Fig. 6A). After the addition of STS (4 μM), within the early time periods of infection (30 min, 1 and 2 h), there was a large increase in the levels of caspase-9 activation. However, the enzymatic activity of caspase-9 significantly started to reduce around 6 h and declined to the control levels following 24 h of infection. Unexpectedly, the simultaneous experiments performed with GECs transfected with Akt siRNA revealed similar levels of caspase-9 activity compared with the normal GECs infected with P. gingivalis or treated with STS (Fig. 6B). Also, the treatments of Akt-deficient cells with a specific inhibitor of JAK-1 (Pyridone 6), (5 μM), which was shown to be involved in the inhibition of camptothecin-induced caspase-3 activation by P. gingivalis, did not have any effect on the suppression of caspase-9 activation during the infection (unpublished data). Hence, these findings suggest that caspase-9, a key pro-apoptotic molecule in mitochondria-associated cell death pathways, may represent an alternate anti-apoptotic pathway in addition to Akt and JAK/Stat signaling mechanisms.
DISCUSSION
The cell biology of infections by the intracellular pathogens illustrates that there is a great level of control on host-cell apoptotic pathways (Collins, 1995; Danelishvili et al., 2003; Byrne & Ojcius, 2004). Apoptosis is a critical process in host–pathogen dynamics that can be advantageous to the host by contributing to pathogen removal. Induction of apoptosis can also be used by the pathogen as a virulence strategy facilitating dissemination of infection. For microorganisms that favor or strictly require life inside a eukaryotic cell, a viable host is essential for replication and survival. Consequently, inhibition of apoptosis can provide a safe haven for microbes, and make intracellular organisms invisible to the immune system (Hacker et al., 2006). The sites of entry for the potential pathogens frequently include mucosal regions lined by epithelial tissues, which function as an important part of innate immunity. As a result, the epithelial cells can serve as effectual colonizing niches for these opportunistic species. A common finding for an increasing number of successful host-adapted bacteria is that they delay or inhibit apoptotic pathways in epithelial cells while the same bacteria act as pro-apoptotic stimuli in other tissue types (Byrne & Ojcius, 2004; Hacker et al., 2006). For example, Salmonella typhimurium infection is shown to cause apoptosis in macrophages but not epithelial cells. Similar to Salmonella, Shigella flexneri, which kills macrophages rapidly through apoptosis, also does not induce cell death in epithelial cells (Zychlinsky et al., 1992; Monack et al., 1996; Knodler & Finlay, 2001).
Porphyromonas gingivalis, one of the major constituents of the subgingival polymicrobial consortium, is predominantly identified in severe forms of periodontal disease and possesses the ability to effectively colonize, adapt, and persist in its target gingival epithelial cells without being destructive until the environment becomes favorable for causing disease (Yilmaz, 2008). The suppression of cell death and promotion of cell survival in GECs by P. gingivalis appears to trigger the bacterium’s capacity to disseminate intercellularly. GECs harboring high level of intracellular P. gingivalis also demonstrate increased host-cell cycle progression and successful mitosis following 1 day of infection (Yilmaz et al., 2006; Kuboniwa et al., 2008). In parallel with these findings, a recent study showed that P. gingivalis infection can interfere with the P2X7 receptor-dependent apoptosis of primary GECs by consuming ATPe by way of its secreted putative nucleoside diphosphate kinase and preventing activation of P2X7 receptors by ATP ligation (Yilmaz et al., 2008). In this way, P. gingivalis can use distinct host-survival strategies to perpetually colonize oral epithelial tissues.
Previous studies showed that P. gingivalis promotes the survival of the gingival epithelial host-cell, at least partially, by activating PI3 kinase/Akt pathway, blocking caspase-3 activation through JAK/Stat signaling, and targeting the members of the Bcl-2 family proteins (Nakhjiri et al., 2001; Yilmaz et al., 2004; Mao et al., 2007). Our study verified the predicted functional role of PI3 kinase/Akt signaling in promoting epithelial cell survival stimulated by P. gingivalis infection. Knock-down of Akt by siRNA demonstrated that Akt is probably central to the success of the organism in limiting mitochondrion-dependent host cell death and in colonizing gingival epithelial tissues as a successful persistent opportunistic pathogen.
The biochemical features that are associated with inhibition of apoptosis during P. gingivalis infection include blocking of mitochondrial permeability transition and inhibition of cytochrome-c release from mitochondria (Yilmaz et al., 2004). Anti-apoptotic Bcl-2 and Bcl-xL proteins of the Bcl-2 family are located at the outer membrane of mitochondria and can inhibit the release of cytochrome-c. In the presence of apoptotic stimuli, pro-apoptotic Bad translocates to mitochondria to associate with Bcl-2 and Bcl-xL. This induces membrane depolarization of mitochondria and subsequent release of cytochrome-c (Chao & Korsmeyer, 1998). On the other hand, the apoptotic activity of Bad can be abrogated through phosphorylation by Akt (Datta et al., 1997). Phosphorylated Bad dissociates from a heterodimeric complex formed with anti-apoptotic Bcl-2 and/or Bcl-xL proteins and is retained in the cytosol in an inactive form, thereby increasing anti-apoptotic effects of the Bcl-2 family. Hence, the phosphorylation status and intracellular localization of Bad are critically important biochemical features in cell-death pathways. Accordingly, our results showed P. gingivalis infection causes a significant level of Bad phosphorylation in GECs through Akt. This results in sequestering of Bad away from mitochondria and so its pro-apoptotic action is blocked.
The results also suggested that the inactivation of Bad mediated by Akt during the infection occurred gradually and was a time-dependent process. Interestingly, the dramatic decrease in Bad mRNA levels following the 24 h infection proposed a potentially complementary mechanism for the Bad inactivation. Recent studies examining the quantitative proteomics of intracellular P. gingivalis in gingival epithelial cells showed the secretion of a large number of distinctive P. gingivalis proteins that could be important for adaptation and survival (Xia et al., 2007; Yilmaz et al., 2008). It is tempting to hypothesize that secretion of effector molecules by the intracellular bacterium may provide an additional strategy for the inhibition of pro-apoptotic Bcl-2 family proteins.
In an alternative process, Akt can phosphorylate caspase-9, an initiator caspase of the apoptotic caspase family. Akt phosphorylation of caspase-9 significantly reduces the caspase proteolytic activity and diminishes the following sequential caspase activation including caspase-6 and caspase-3. Caspase-3 activation, which significantly increases the mitochondrial permeability, is shown to be inhibited by P. gingivalis infection through dual Akt and JAK/Stat signaling (Mao et al., 2007). Similarly, our results demonstrated that caspase-9 activation is significantly impaired by P. gingivalis in a time-dependent manner. Yet, the inhibition of caspase-9 was independent of Akt and JAK, as indicated by the joint siRNA and the pharmacological inhibition assays. The early and short-lived detection of caspase-9 activation was not surprising because our previous study showed that P. gingivalis triggers activation of caspases at early time points of infection in GECs determined by the inhibition of rapid phosphatidylserine exposure by the broad-spectrum caspase inhibitor zVAD-fmk. However, neither caspase activation nor phosphatidylserine externalization leads to host-cell death (Yilmaz et al., 2004). Further, caspase-9 activation is also required for transcription factor p53-dependent cell death. Infection of GECs with P. gingivalis causes a large reduction in p53 levels and this is consistent with the caspase-9 inactivation observed here (Kuboniwa et al., 2008). Nevertheless, our results showed that Akt is not responsible for the caspase-9 inhibition stimulated by the infection, and future investigations are required to characterize the exact molecular mechanisms mediating this inhibition.
Overall, this study identified novel processes between the intracellular molecules, Akt and Bad, in response to P. gingivalis infection that may be critical for the organism’s survival and colonization in the epithelial tissues. Together, these findings indicate a high degree of complexity that culminates in the modulation of mitochondrion-dependent death during the infection of P. gingivalis in GECs and may represent potential therapeutic targets for removal of cells infected by this prevalent opportunistic pathogen.
Acknowledgments
This study was supported by NIDCR grant R01DE016593.
References
- Belton CM, Izutsu KT, Goodwin PG, Park Y, Lamont RJ. Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol. 1999;1:215–223. doi: 10.1046/j.1462-5822.1999.00022.x. [DOI] [PubMed] [Google Scholar]
- Byrne GI, Ojcius DM. Chlamydia and apoptosis: life and death decisions of an intracellular pathogen. Nat Rev Microbiol. 2004;2:802–808. doi: 10.1038/nrmicro1007. [DOI] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- Chen YL, Law PY, Loh HH. Inhibition of PI3K/Akt signaling: an emerging paradigm for targeted cancer therapy. Curr Med Chem Anticancer Agents. 2005;5:575–589. doi: 10.2174/156801105774574649. [DOI] [PubMed] [Google Scholar]
- Collins M. Potential roles of apoptosis in viral pathogenesis. Am J Respir Crit Care Med. 1995;152:S20–S24. doi: 10.1164/ajrccm/152.4_Pt_2.S20. [DOI] [PubMed] [Google Scholar]
- Colombo AV, da Silva CM, Haffajee A, Colombo AP. Identification of intracellular oral species within human crevicular epithelial cells from subjects with chronic periodontitis by fluorescence in situ hybridization. J Periodontal Res. 2007;42:236–243. doi: 10.1111/j.1600-0765.2006.00938.x. [DOI] [PubMed] [Google Scholar]
- Cosulich SC, Savory PJ, Clarke PR. Bcl-2 regulates amplification of caspase activation by cytochrome c. Curr Biol. 1999;9:147–150. doi: 10.1016/s0960-9822(99)80068-2. [DOI] [PubMed] [Google Scholar]
- Danelishvili L, McGarvey J, Li YJ, Bermudez LE. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol. 2003;5:649–660. doi: 10.1046/j.1462-5822.2003.00312.x. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dukek H, Tao X, et al. Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Finlay BB, Falkow S. Common themes in microbial pathogenecity. Microbiol Rev. 1989;53:210–230. doi: 10.1128/mr.53.2.210-230.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker G, Kirschnek S, Fischer SF. Apoptosis in infectious disease: how bacteria interfere with the apoptotic apparatus. Med Microbiol Immunol (Berlin) 2006;195:11–19. doi: 10.1007/s00430-005-0239-4. [DOI] [PubMed] [Google Scholar]
- Jenkinson HF, Lamont RJ. Oral microbial communities in sickness and in health. Trends Microbiol. 2005;13:589–595. doi: 10.1016/j.tim.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Wagner AJ, Conzen SD, et al. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- Knodler LA, Finlay BB. Salmonella and apoptosis: to live or let die? Microbes Infect. 2001;3:1321–1326. doi: 10.1016/s1286-4579(01)01493-9. [DOI] [PubMed] [Google Scholar]
- Kuboniwa M, Hasegawa Y, Mao S, et al. P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes Infect. 2008;10:122–128. doi: 10.1016/j.micinf.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont RJ, Yilmaz O. In or out: the invasiveness of oral bacteria. Periodontol 2000. 2002;30:61–69. doi: 10.1034/j.1600-0757.2002.03006.x. [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel DJ, Weinberg A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63:3878–3885. doi: 10.1128/iai.63.10.3878-3885.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao S, Park Y, Hasegawa Y, et al. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 2007;9:1997–2007. doi: 10.1111/j.1462-5822.2007.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci USA. 1996;93:9833–9838. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhjiri SF, Park Y, Yilmaz O, et al. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol Lett. 2001;200:145–149. doi: 10.1111/j.1574-6968.2001.tb10706.x. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Rudney JD, Chen R, Sedgewick GJ. Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, and Tannerella forsythensis are components of a polymicrobial intracellular flora within human buccal cells. J Dent Res. 2005;84:59–63. doi: 10.1177/154405910508400110. [DOI] [PubMed] [Google Scholar]
- Seo YS, Chen Y-b, Ivanovska I, et al. BAD is a pro-survival factor prior to activation of its pro-apoptotic function. J Biol Chem. 2004;279:42240–42249. doi: 10.1074/jbc.M406775200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Verbeke P, Welter-Stahl L, Ying S, et al. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog. 2006;2:e45. doi: 10.1371/journal.ppat.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Wang T, Taub F, et al. Quantitative proteomics of intracellular Porphyromonas gingivalis. Proteomics. 2007;7:4323–4337. doi: 10.1002/pmic.200700543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology. 2008;154:2897–2903. doi: 10.1099/mic.0.2008/021220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751. doi: 10.1128/IAI.72.7.3743-3751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Verbeke P, Lamont RJ, Ojcius DM. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect Immun. 2006;74:703–710. doi: 10.1128/IAI.74.1.703-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Yao L, Maeda K, et al. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X(7)-mediated host-cell apoptosis. Cell Microbiol. 2008;10:863–875. doi: 10.1111/j.1462-5822.2007.01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JL, Shackel NA, Zekry A, et al. Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) for measurement of cytokine and growth factor mRNA expression with fluorogenic probes or SYBR Green I. Immunol Cell Biol. 2001;79:213–221. doi: 10.1046/j.1440-1711.2001.01002.x. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Rao JN, Guo X, et al. Akt kinase activation blocks apoptosis in intestinal epithelial cells by inhibiting caspase-3 after polyamine depletion. J Biol Chem. 2004;279:22539–22547. doi: 10.1074/jbc.M314337200. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–169. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]