

Abstract
A series of gramicidin S (GS) analogs have been synthesized where the Phe (i+1) and Pro (i+2) residues of the β-turn have been swapped while the respective chiralities (D-, L-) at each position are preserved, and Phe is replaced by surrogates with aromatic side chains of diverse size, orientation and flexibility. Although most analogs preserve the β-sheet structure, as assessed by NMR, their antibiotic activities turn out to be highly dependent on the bulkiness and spatial arrangement of the aromatic side chain. Significant increases in microbicidal potency against both Gram-positive and Gram-negative pathogens are observed for several analogs, resulting in improved therapeutic profiles. Data indicate that seemingly minor replacements at the GS β-turn can have significant impact on antibiotic activity, highlighting this region as a hot spot for modulating GS plasticity and activity.
Keywords: Gramidicin S, cationic antimicrobial peptides, phenylalanine analogs, NMR, β-turn, β-sheet structure
Introduction
Increasing resistance of bacteria to conventional antibiotics has posed a serious threat to human health and generated a growing need for new drugs to combat microbial infection. Cationic antimicrobial peptides (AMPs) from either microbial or eukaryotic sources have been proposed as an alternative to conventional antibiotics, since their lethal mechanism -based on disruption of the pathogen’s cytoplasmic membrane- differs from those of most conventional antibiotics in clinical use and makes induction of resistance rather unlikely.1,2
Gramicidin S (GS), one of the best-studied cationic AMPs, is synthesized non-ribosomally by Bacillus brevis3 and active against bacteria and fungi.4,5 GS is a cyclic symmetrical decapeptide, cyclo(Val-Orn-Leu-D-Phe-Pro)2 (Figure 1), that adopts a pleated β-sheet structure6–8 in which the Val, Orn and Leu residues align to form two antiparallel β-strands. Two type-II′ β-turns centered at the D-Phe-Pro sequences flank the β-strands, stabilized by four cross-strand hydrogen bonds (Figure 1). Amphipaticity of the β-sheet structure is achieved by positioning of the hydrophobic side chains of Leu and Val facing one side of the molecule whereas the two basic Orn residues project to the opposite side.5,9
Figure 1.

Structure of gramicidin S (top) and the analog generated (1) by sequence inversion at the β-turn (bottom). In both cases the peptide adopts aβ-sheet structure.
Although the mechanism of action of GS is not completely understood,10 it is generally accepted that the peptide kills bacterial cells through destabilization and permeabilization of their cytoplasmic membranes.11 Unfortunately, GS displays poor selectivity between microbial and mammalian cells, a fact that restricts its use to topical applications.5 In recent years, considerable effort has been devoted to developing GS analogs with improved therapeutical index where the antimicrobial and cytotoxic (e.g., hemolytic) activities are dissociated. In this quest, both the β-strand12–14 and the β-turn15–25 regions have been extensively modified in SAR studies that have shed light on factors governing GS bioactivity, such as cationic nature,11,24 amphipathic character,26,27 β-sheet structure,24,26,27 ring size28 and global hydrophobicity.26,27 The latter property results not only from the hydrophobic domain defined by the Val and Leu side chains at the β-strands, but also from the presence of an aromatic side chain at the two β-turn regions that is regarded as essential for bioactivity.15–17,24,29–32 Indeed, replacement of D-Phe by either Gly, D-Ala, or D-cyclohexylalanine leads to less active or inactive compounds,29,30 thus evidencing that an aromatic group at this particular position is needed for full bactericidal potency. Moreover, by replacing the D-Phe residues in GS by a variety of non-coded aromatic amino acids, we have recently shown25 that the pharmacological profile changes dramatically with the size and spatial arrangement of the aromatic side chain at the i+1 position of the β-turns.
These latter findings have led us to question whether an aromatic moiety is strictly necessary at the i+1 position of the β-turn. In line with this, we hypothesized that the i+1 and i+2 positions could be swapped without dramatic alteration of GS overall architecture (i.e., the β-turns and the whole β-sheet structure) as long as the chirality of the said i+1 and i+2 positions (D- and L-, respectively) remained unaltered, as in peptide 1 (Figure 1). The hypothesis is supported on the known requirement for a D-amino acid (or glycine) at the i+1 position33 of type II′ β-turns such as that in GS, and on the fact that Pro is the amino acid residue with the highest propensity to occupy an i+1 position at a type-II β-turn;33–35 therefore, a D-Pro at the i+1 position would appear to be an optimal choice for nucleating a type II′ β-turn. Herein we report on a family of GS analogs bearing double modifications based on the aforementioned considerations. Specifically, in the lead analog 1, cyclo(Val-Orn-Leu-D-Pro-Phe)2 (Figure 1), both D-Phe-Pro sequences in GS have been replaced by D-Pro-Phe. In the remaining analogs of the series, Phe is replaced by a variety of non-proteinogenic counterparts bearing aromatic side chains of different size, orientation, and flexibility. The structure of these GS analogues has been analyzed by NMR spectroscopy and related to their antibiotic and hemolytic activities.
Results
Peptide Design and Synthesis
The amino acids selected as Phe replacements (Phe*) in the GS analog 1 (Figure 1) are shown in Figure 2. β, β-Diphenylalanine (Dip), fluorenylglycine (Flg), dibenzylglycine (Dbg), 1, 2, 3, 4-tetrahydroisoquinoline-3-carboxylic acid (Tic), and the two enantiomers of the cyclopropane amino acid c3diPhe were initially considered. Most of them incorporate two phenyl rings exhibiting diverse orientations and degrees of conformational freedom. In Dbg, the aromatic substituents can freely rotate, whereas in c3diPhe they are tightly held. Both Dip and Flg bear two phenyl groups on the same β carbon that in the latter case are forced to be coplanar. The orientation of the aromatic moiety in Tic is fixed by the methylene unit linking it to the backbone. In addition, it should be noted that Dbg is achiral while c3diPhe is characterized by an achiral α carbon and two chiral β carbons. In order to evaluate separately the structural effect associated exclusively to the strained three-membered cycle present in c3diPhe, the unsubstituted cyclopropane amino acid Ac3c was also considered for this study. Cyclopropane amino acids have been shown to exert a strong stabilizing effect on β-turns when incorporated into Pro-Xaa dipeptides36 and are therefore expected to stabilize the β-sheet structure of GS, provided no steric conflict between the cyclopropane substituents and the contiguous residues arises.
Figure 2.

Amino acids selected as L-Phe replacements (Phe*) in 1 (left). Sequence and numbering of the corresponding gramicidin S analogs generated (2–13) (right). For amino acids containing two chiral centers, configurations are given for the 2, 3 (in c3diPhe) or 1, 2 (in c3Phe) carbon atoms.
The different antibiotic properties found for the peptides incorporating the two c3diPhe enantiomers (see below) prompted us to evaluate the effect produced by the cyclopropane amino acid bearing a single phenyl substituent, c3Phe (Figure 2). In this case, four stereoisomeric forms are possible, each of them with a different orientation of the aromatic ring. It is worth noting that each c3diPhe residue can be regarded as formally containing the aromatic side chain of two c3Phe stereoisomers. Additionally, α-methylphenylalanine [(αMe)Phe] was included in this study as the open-chain counterpart of c3Phe, in the same way as Dbg can be viewed as the acyclic equivalent of c3diPhe. Both (αMe)Phe and Dbg keep the α-tetrasubstituted character of c3Phe and c3diPhe but not the restriction imposed by the three-membered ring. As an α-methylated amino acid, (αMe)Phe is also particularly suitable to occupy the i+2 position of a β-turn.37
Solid-phase Boc chemistry protocols19,23 were applied to the synthesis of peptides 1–13 (Figures 1,2), as done for GS and the first series of analogues in our previous work.25 All stereoisomers of c3diPhe, c3Phe, and Flg shown in Figure 2 were obtained as N-Boc derivatives in enantiomerically pure form following previously reported procedures.38–40 Achiral Boc-Dbg-OH was synthesized according to the protocol described by Kotha.41 Boc-(αMe)Phe-OH was obtained from commercially available H-(αMe)Phe-OH, and the side chain of N-Boc ornithine was protected as a formamide as described by Kitagawa.42 Each peptide was synthesized following the general route presented in Scheme 1. Unsuccessful coupling of the Dbg residue prevented the synthesis of the linear precursor of peptide 4. For the remaining peptides, the solid-phase synthetic protocols were completed satisfactorily. The linear decapeptides were released from the polymeric support by treatment with TFMSA and then cyclized in solution under high-dilution conditions. Finally, deprotection of the Orn side chains by acid hydrolysis followed by reversed-phase HPLC purification furnished the target peptides (1–3, 5–13), in good overall yields and high purity (≥99%, see Supporting Information). All peptides were satisfactorily characterized by MS and NMR.
Scheme 1.
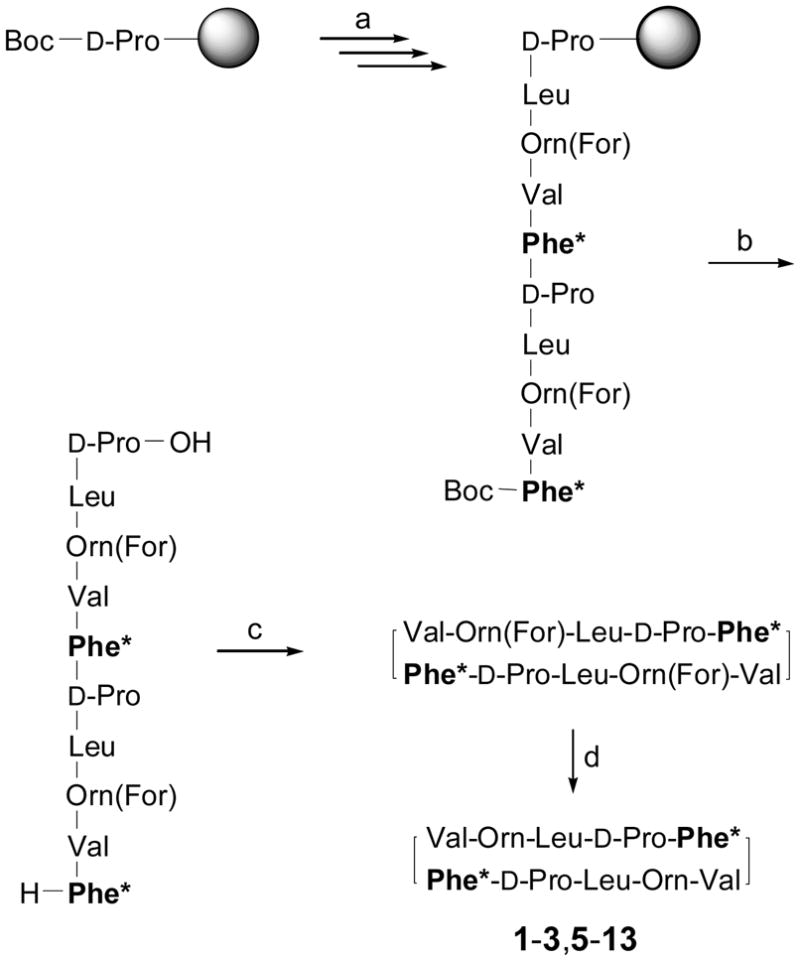
Synthetic Strategy for the GS Analogsa
aReagents and conditions: (a) standard solid-phase peptide synthesis (Boc chemistry); (b) TFA/TFMSA/TIS 10:1:1, rt, 90 min; (c) HATU/HOAt/DIEA (3:3:5 equiv), DMF, rt, 1 h; (d) 20% HCl in MeOH, 37 °C, 21 h. Phe* stands for Phe or the corresponding substitute.
NMR Structural Studies
The structure of GS analogs 1–3 and 5–13 was evaluated in aqueous solution by NMR as previously done for GS and a first series of analogs.25 A set of minor NMR signals coexisting with those of the major species were present in the spectra of all analogs, except for peptide 5 (Figure 3, see also Supporting Information, Figure SF2, and Table ST1). In all cases, the major conformer was found to contain both Leu-D-Pro bonds in a trans arrangement, as inferred from the presence of an NOE between the Leu Hα proton and the Pro Hδδ′ protons (Figure 3 and Table ST2, Supporting Information), as well as from the 13Cβ–13Cγ chemical shift differences in the 2.9–5.2 ppm range observed for the Pro residues (ΔδPro = δCβ–δCγ ppm; see Supporting Information, Table ST2).44 For peptides 1–3, as expected for Pro-containing peptides, the minor species were assigned to the cis rotamer of at least one Leu-D-Pro bond, as deduced from the presence of an NOE between the Hα protons of Leu and Pro residues and from a ΔδPro value near 10 ppm.44 The most populated of the two minor species is an aymmetric conformer in which one Leu-D-Pro bond corresponds to the trans rotamer and the other to the cis rotamer, and the least populated is a symmetric conformer with the two Leu-D-Pro bonds in cis (Figure 3A, and Supporting Information, Table ST3). In contrast, analogs 6–13 showed broad NMR signals and, except for peptides 11 and 13, exchange crosspeaks between signals belonging to the major and minor species, as well as between signals of different minor species, were observed in the 150 ms NOESY spectra (Supporting Information, Figure SF2 and Table ST1). This indicates that the conformational equilibrium in peptides 6–13 is faster than that usually associated to the isomerization of the Leu-D-Pro bonds. More strikingly, the minor species observed in peptides 6, 7 and 9 could not be attributed to the cis-trans isomerism of the amide bonds preceding Pro residues, as they exhibit NOEs between the Leu Hαproton and the Pro Hδ and Hδ ′ protons, and ΔδPro values below 5.0 ppm (Supporting Information, Figure SF2 and Table ST4), both typical of trans rotamers. Hence, the minor species observed for peptides 6–13 could not be attributed to the cis-trans isomerism of the amide bonds preceding Pro. Since these compounds (6–13) incorporate an α-tetrasubstituted amino acid, the observed conformational equilibrium could be somehow related to the presence of such non-proteinogenic residues. It should be noted that peptides 6, 7, and 9–13 contain Phe surrogates that are not only α-tetrasubstituted but also bear one (or more) bulky, rigidly held phenyl group(s) that could further hamper rotation about the bonds in the neighborhood of the α carbon. The presence of minor species, broad signals and exchange NOE cross-peaks made the spectral analysis of some of these peptides rather complex (see Supporting Information, Tables ST2 and ST4). Results described below correspond, in all cases, to the major trans-trans species.
Figure 3.

Selected NOESY spectral regions for peptides 1 (A) and 5 (B) in D2O (top panels) and in H2O/D2O 9:1 v/v (all other panels) at pH 3 and 5 °C. Green, blue and red labels correspond, respectively, to symmetrical major, asymmetrical minor and symmetrical minor species. The top panel for peptide 1 shows the sequential NOEs between the Leu Hα proton and the Pro Hδ and Hδ′ protons indicative of the trans rotamery for the two Leu-Pro bonds in the major species, and for one Leu-Pro bond in the aymmetrical minor species, as well as the sequential NOE between the Leu and Pro Hα protons demonstrating the cis rotamery for one Leu-Pro bond in the aymmetrical minor species. Non-sequential NOEs, as those between the amide protons of Val1 and Leu3′, and Val1′ and Leu3 (Figure 1 and Table 2), are boxed.
Chemical shifts of CαH protons and Cα carbons are dependent on the φ, ψ torsion angles and are, therefore, good indicators of the secondary structure adopted by the peptide backbone. Lys δrandom coil values43 were used to calculate the Δδ values for the Orn residues. For all compounds investigated (Figure 4), Orn2/2′ and Leu3/3′ exhibited positive ΔδCαH conformational shifts (ΔδCαH = δCαHobserved–δCαHrandom coil, ppm) and negative ΔδCαvalues (ΔδCα= δCαobserved–δCαrandom coil, ppm), as expected for β-strand residues (ΔδCαH = +0.40 ppm and ΔδCα= −1.6 ppm on average in β-sheets).45,46 The Orn2/2′ residues in 2 were the sole exception to this pattern. The ΔδCαH and ΔδCα profiles observed for Val1/1′ do not seem to follow a particular trend, and adopt either positive or negative values. This anomalous behavior may be explained by the ring current effects generated by the aromatic groups in the contiguous position (Phe*5/5′), as observed in our previous work on GS analogs25 and some β-hairpin peptides.47 Indeed, in the absence of aromatic substituents (peptide 8), Val1/1′ shows conformational shifts appropriate for a β-strand residue.
Figure 4.

ΔδCαH and ΔδCαprofiles (Δδ = δobserved–δrandom coil, ppm) for GS analogs in aqueous solution at pH 3.0 and 5 °C. Lys δrandom coil values43 were used to calculate the Δδ values for the Orn residues. The δCαH and δCα values for the Orn residues in 12 could not be assigned.
The large 3JCαH–NH coupling constants observed for Val1/1′, Orn2/2′ and Leu3/3′ (8.0–9.8 Hz, Supporting Information, Table ST5) confirm that these residues adopt a β-sheet conformation. Again, in analog 2 the Orn2/2′ residues deviate from the expected behavior (3JCαH–NH = 5.7 Hz). On the other hand, in the (R, S)c3Phe derivative (12) the 3JCαH–NH values of Val1/1′ and Leu3/3′ could not be determined because of signal broadening.
The involvement of the NH amide protons in intramolecular hydrogen bonds was deduced from the temperature coefficients (Δδ/ΔT) (Table 1). The amide protons of Orn2/2′ and Phe*5/5′ displayed large negative Δδ/ΔT values (below −7.0 ppb K−1) in all peptides, except again for analog 2, in agreement with their non-hydrogen bonded, solvent-exposed character in the GS β-sheet (Figure 1). In comparison, and with the exception of compound 12, Δδ/ΔT values above −5.1 ppb K−1 were observed for the NH protons of Val1/1′ and Leu3/3′, which are involved in intramolecular hydrogen bonds in the GS structure (Figure 1).
Table 1.
Temperature Coefficientsa (Δδ/ΔT, ppb.K−1) for the Amide Protons.
| Peptide | Phe*5,5′b | Val1,1′ (NH) | Orn2,2′ (NH) | Leu3,3′ (NH) | Phe*5,5′ (NH) |
|---|---|---|---|---|---|
| 1 | Phe | −2.2 | −9.1 | −1.8 | −8.7 |
| 2 | Dip | −2.9 | −4.1 | +0.9 | −7.4 |
| 3 | Flg | −0.7 | −9.9 | +2.6 | −7.3 |
| 5 | Tic | −2.0 | −8.5 | −3.8 | — |
| 6 | (R, R)c3diPhe | −3.2 | −10.5 | −4.2 | −7.1 |
| 7 | (S, S)c3diPhe | −1.0 | −9.0 | −1.5 | −9.0 |
| 8 | Ac3c | −1.9 | −9.0 | −5.1 | −11.8 |
| 9 | (S, S)c3Phe | −1.6 | −9.8 | −4.0 | Ovc |
| 10 | (R, R)c3Phe | −2.1 | −9.0 | −3.9 | −7.1 |
| 11 | (S, R)c3Phe | −3.1 | −10.0 | −3.7 | −8.1 |
| 12 | (R, S)c3Phe | −7.5 | −8.5 | −10.0 | −15.1 |
| 13 | (αMe)Phe | −1.6 | −7.0 | −3.0 | −9.9 |
Measured in H2O/D2O 9:1 (v/v) at pH 3.0 and temperature range of 5–25 °C.
Phe* stands for Phe or the corresponding substitute.
Not determined because of signal overlapping.
The above structural data suggest that most analogs of this study fit into a β-sheet conformation, as does GS.6–8,25 It is worth noting that in almost all peptides of the series the Val1/1′ NH exhibits the typical behavior of a hydrogen-bonded amide proton, which is not the case for either GS or the analogs in our previous work.25 This may suggest that the peptides (with the probable exception of 2 and 12) adopt a more regular β-sheet structure, which could be attributed to the presence of the D-Pro-Phe* instead of the D-Phe*-Pro sequence.
NOE data provided further structural information on the current set of analogs. The GS β-sheet structure (Figure 1) should give rise to five characteristic backbone proton NOEs: CαH Orn2–CαH Orn2′ (distance in a canonical antiparallel β-sheet: 2.2 Å), CαH Orn2–NH Leu3′, CαH Orn2′–NH Leu3 (distance in a canonical antiparallel β-sheet: 3.2 Å), NH Val1–NH Leu3′, and NH Val1′–NHLeu3 (distance in a canonical antiparallel β-sheet: 3.3 Å). Given the molecule symmetry, the first NOE would not be observable, the second and third ones will be undistinguisable from the sequential CαH Orn2/2′–NH Leu3/3′ NOEs, while the last two must overlap into a single cross-peak. Such a cross-peak is indeed observed in the NOESY spectra of all peptides investigated (Table 2, Figure 3) with the exception of 2 and 12, which once again deviate from the expected behavior. NOE data are also informative on the layout of the side chains. The backbone β-sheet conformation orients the Val and Leu side chains towards the same side of the molecule (Figure 1) and, indeed, NOE correlations between hydrogens of these side chains are observed for all peptides (Table 2, Figure 3) except 7, for which spectrum complexity precluded analysis. The presence of such NOE cross-peaks in analogs 2 and 12 suggests that the Val and Leu side chains are proximal. This could translate into a certain amphiphilic character despite the fact that these GS analogs do not seem to adopt a canonical β-sheet structure.
Table 2.
Summary of NOEs Observed.a
| Peptideb | Phe*5,5′c | backbone NOEd | side-chain NOEe | |||
|---|---|---|---|---|---|---|
| NHVal1–NHLeu3′/NHVal1′–NHLeu3 | Val1,1′–Leu3,3′ | Orn2,2′–Phe*5,5′ | Val1,1′–Phe*5,5′ | D-Pro4,4′–Phe*5,5′ | ||
| 1 | Phe | + | + | + | − | − |
| 2 | Dip | − | + | + | + | + |
| 3 | Flg | −f | + | − | + | + |
| 5 | Tic | + | + | − | + | − |
| 6 | (R, R)c3diPhe | + | + | +g | + | + |
| 8 | Ac3c | + | + | +h | − | +h |
| 9 | (S, S)c3Phe | + | + | +h | + | + |
| 10 | (R, R)c3Phe | + | + | +h | − | + |
| 11 | (S, R)c3Phe | + | + | − | + | +h |
| 12 | (R, S)c3Phe | − | +h | + | + | − |
| 13 | (αMe)Phe | + | + | +h | + | + |
Observation of one or more NOEs is shown by a “+”, and non-detected NOEs by a “–”.
Non-ambiguous assignment of NOESY cross-peaks was precluded in peptide 7 because of its very broad signals and the presence of numerous exchange cross-peaks.
Phe* stands for Phe or the corresponding surrogate.
NOE involving backbone protons.
NOE involving at least one side chain proton of the residues indicated.
Not observed due to closeness to diagonal.
Weak peak intensities.
Only protons of the cyclopropane ring or theα-methyl group are involved.
Biological Activity
The 1–3 and 5–13 analogs (peptide 4 was not synthetically viable, see above) were tested along with GS against two Gram-positive (Staphylococcus aureus and Listeria monocytogenes) and two Gram-negative bacterial strains [Acinetobacter baummanii ATCC 19606 and an isogenic strain resistant to colistin (polymyxin E)]. Although GS is mostly active against Gram-positives, the last two strains were included in view of the growing incidence of A. baumannii infections, particularly those caused by strains resistant to polymyxin E, which until recently has been the last universally active drug against these pathogens.48 Microbicidal activities were determined in solution, as in solid media they tend to be underestimated, especially for Gram-negatives.4 Pseudomonas aeruginosa, another Gram-negative, was included earlier on in the assays, but as none of the analogs showed growth inhibitions above 30% at 50 μM, the highest concentration assayed, it was disregarded. Results are given in Table 3 and show that the replacement of the D-Phe-Pro sequences in GS by D-Pro-Phe in 1 has a negative impact on activity. However, further modification of the Phe residue allows the recovery and even an enhancement of the microbicidal potency of the natural peptide. Thus, the Dip (2), Flg (3), Tic (5) and (R, R)c3diPhe (6) analogs turned out to be more active than GS against Gram-positive bacteria, while 3 and 5 were also superior against Gram-negatives. The improved microbicidal activity of the Tic analog (5) against the colistin-resistant strain is also worth mentioning. On the other hand, the Ac3c derivative (8), lacking an aromatic moiety at the β-turn region was, perhaps not surprisingly, practically devoid of any antibiotic activity.
Table 3.
Hemolytic and Antimicrobial Activitiesa (μM) of Gramicidin S (GS) Analogs with Sequence Inversion at the β-Turn
| Peptide | Phe*5,5′b | Erythrocytes | S. aureus | L. monocytogenes | A. baumanni S | A.baumannii Rd | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| HC50 | MIC50 | TIc | MIC50 | TI | MIC50 | TI | MIC50 | TI | ||
| GS | — | 21.1 (± 2.6) | 7.9 (± 0.8) | 2.7 (1.0) | 8.0 (± 0.0) | 2.6 (1.0) | 11.0 (± 0.4) | 1.9 (1.0) | 13.1 (± 0.0) | 1.6 (1.0) |
| 1 | Phe | 78.7 (± 4.2) | 15.8 (± 0.0) | 4.9 (1.8) | 23.6 (± 0.0) | 3.3 (1.3) | 24.8 (± 1.2) | 3.1 (1.7) | >40 | n.d. |
| 2 | Dip | 34.0 (± 3.7) | 3.8 (± 0.8) | 8.9 (3.3) | 3.4 (± 0.5) | 10.0 (3.8) | 20.3 (± 0.3) | 1.7 (0.9) | 23.9 (± 2.1) | 1.4 (0.9) |
| 3 | Flg | 2.1 (± 1.4) | 2.2 (± 0.1) | 0.9 (0.3) | 2.2 (± 0.2) | 0.9 (0.3) | 5.0 (± 0.3) | 0.4 (0.2) | 14.3 (± 1.9) | 0.1 (0.1) |
| 5 | Tic | 7.7 (± 1.5) | 2.4 (± 0.2) | 3.2 (1.2) | 2.4 (± 0.1) | 3.2 (1.2) | 5.1 (± 0.1) | 1.5 (0.8) | 8.5 (± 2.1) | 0.9 (0.6) |
| 6 | (R, R)c3diPhe | 26.0 (± 2.4) | 3.2 (± 0.5) | 8.1 (3.0) | 5.1 (± 0.5) | 5.1 (1.9) | >40 | n.d.e | >40 | n.d. |
| 7 | (S, S)c3diPhe | >60 | 25.8 (± 0.0) | 2.3 (0.8) | >30 | n.d. | >40 | n.d. | >40 | n.d. |
| 8 | Ac3c | >60 | >30 | n.d. | >30 | n.d. | >40 | n.d. | >40 | n.d. |
| 9 | (S, S)c3Phe | >60 | >30 | n.d. | >30 | n.d. | >40 | n.d. | >40 | n.d. |
| 10 | (R, R)c3Phe | >60 | >30 | n.d. | >30 | n.d. | >40 | n.d. | 41.0 (± 11) | n.d. |
| 11 | (S, R)c3Phe | 57.0 (± 3.8) | >30 | n.d. | >30 | n.d. | >40 | n.d. | >40 | n.d. |
| 12 | (R, S)c3Phe | >60 | >30 | n.d. | >30 | n.d. | >40 | n.d. | >40 | n.d. |
| 13 | (αMe)Phe | 56.5 (± 3.9) | 11.9 (± 1.0) | 4.7 (1.7) | 6.8 (± 0.3) | 8.3 (3.2) | 9.6 (± 0.4) | 5.9 (3.1) | 7.0 (± 1.4) | 8.0 (5.0) |
HC50: peptide concentration required for 50% lysis of sheep erythrocytes; MIC50: peptide concentration required for 50% inhibition of bacterial growth after 24 h, relative to a control culture; standard deviations for HC50 and MIC50 are shown in parentheses.
Phe*5,5′ stands for Phe or the corresponding surrogate (see Figures 1 and 2); GS contains D-Phe4,4′.
TI= therapeutic index = HC50/MIC50; values relative to TI of GS in parentheses.
An A. baumannii strain resistant to colistin (polymyxin E).
n.d. = not determined.
The contrasting activity of the analogs incorporating the c3diPhe enantiomers (6, 7) deserves some comment. Thus, while 7 had the lowest activity against all bacterial strains among the 1–7 subset, 6 was unique in its highest selectivity for Gram-positives. These differences, which might be ascribed to the rigid orientation imposed by the cyclopropane ring on the aromatic substituents, prompted us to expand the analog series with the four c3Phe stereoisomers bearing a single phenyl group (Figure 2). Surprisingly enough, all analogs containing these Phe surrogates (9–12) showed marginal activity, if at all, against all organisms. In contrast, the analog with the closely related (αMe)Phe residue (13) not only retained the activity of GS but displayed the best therapeutic index (TI, see below) against both A. baumannii strains.
The hemolytic effect of the peptides was evaluated on sheep erythrocytes as an indicator of toxicity against mammalian cells. Results are summarized in Table 3, where the therapeutic index (TI, defined as HC50/MIC50) is also given as a combined measure of both effects. As a general rule, the antibiotic and hemolytic activities were found to run parallel. Thus, peptides with very low microbicidal power (1, 7–12) were hardly harmful to erythrocytes, whereas analogs of high antibiotic activity (3, 5) turned out to be also fairly toxic. However, some notable deviations to this general behavior were observed. For instance, the Dip analog 2 was more active against Gram-positives yet slightly less cytotoxic than GS, which translated into a modest (about 3-fold) but significant TI increase vs. GS. A similar improvement in TI was observed for the (R, R)c3diPhe-containing analog (6) regarding the S. aureus strain. The peptide incorporating (αMe)Phe (13) was found to be essentially harmless to erythrocytes while exhibiting an antibiotic potency similar or even superior to that of GS, which resulted in an enhanced TI for all pathogens considered. Particularly noteworthy is the fact that the most significant increase in TI for this analog (near 5-fold that of the natural peptide) was observed for the Gram-negative A. baumannii strand resistant to conventional antibiotics. It should also be noted that improved TIs reported in the literature for GS analogs most often stem from lower cytotoxicity,24,25,27,49,50 whereas the most significant increases in TI relative to GS found in this work (≥ 3-fold) are also associated to a higher antibiotic potency.
Discussion
Several groups, including our own,25,29–32 have shown that the β-turn of GS is particularly suited to modulation of the TI by subtle substitutions. In this work we have explored the impact of replacing Phe by residues bearing one or two phenyl rings that impart different degrees of hydrophobicity, bulkiness and rotational freedom to the GS molecule. In order to carry out these substitutions with minimal perturbation of the β-turn architecture, the native D-Phe-L-Pro sequence of GS has been changed to D-Pro-L-Phe, since it is known that simple residue permutation (i.e., to L-Pro-D-Phe) results in practically full loss of activity.22 This distinguishing feature of the present series compares favorably with our earlier work,25 where the canonical D-Phe-L-Pro β-turn was used. In absolute terms, sequence inversion to D-Pro-L-Phe, as exemplified by analog 1, entails a certain loss of antimicrobial potency relative to the parent GS. However, the change also involves a substantial decrease in cytotoxicity (determined as hemolysis) that, when factored into the therapeutic index (TI), actually results in an almost 2-fold improvement against the Gram-positive S. aureus as well as the colistin-sensitive strain of the Gram-negative A. baumannii. A similarly improved antimicrobial profile can be found for the 2 (Dip) and 6 [(R, R)c3diPhe] analogs against Gram-positives, as well as for the (αMe)Phe replacement (13) against both Gram-positives and -negatives. The Tic replacement, which in the previous series proved to be very advantageous,25 is now (analog 5) of less consequence, as the enhanced antimicrobial potency entails a considerable penalty in cytotoxicity. In the canonical D-Phe-L-Pro series,25 the proximity between Orn and the aromatic side chain exclusive of the Tic analog was invoked to explain the TI improvement. Such interaction is not observed in 5, neither in 3, the most hemolytic analog (Tables 2–3). Interestingly, however, Orn-Phe* NOEs are observed for analogs less hemolytic than GS, such as 1 and 2 (Tables 2–3).
Among several factors known to influence the interaction of AMPs with bacterial or mammalian membranes, hydrophobicity is viewed as crucial. In general, and regardless of the anionic or zwitterionic nature of the bilayer, a hydrophobic patch of size above a minimal threshold is required for a peptide to partition into the membrane. In this scenario, hydrophobicity correlates relatively well with AMP activity against systems such as eukaryotes (e.g., hemolysis) or Gram-positive bacteria, where the peptide can diffuse relatively easily through the peptidoglycan layer. For GS and the twelve analogs of the present series, however, hydrophobicity does not provide a straightforward interpretation to antimicrobial action. RP-HPLC retention times (Figure 5), widely accepted as a criterion of relative hydrophobicity for structurally related compounds, clearly show that while the least (8) and most (3) hydrophobic analogs respectively show low and high activities against Gram-positives and erythrocytes, analogs of intermediate hydrophobicity follow no discernible general trend in this regard. For instance, the four c3Phe derivatives (9–12) differ significantly in hydrophobicity but coincide in (a very poor) activity. Again, comparing GS with 2 or 5, of similar hydrophobicity but higher potency against Gram-positives, reveals similar discrepancies, albeit in the opposite sense. The fact that the antimicrobial performance of the current set of analogs cannot be explained simply on the basis of hydrophobicity agrees with recent data on AMPs like lactoferricin51 or again GS,52 for which additional parameters (e.g., bulkiness) have been invoked as crucial for activity.
Figure 5.

Retention times (Rt) of GS analogs on RP-HPLC. Elution conditions: see experimental section. The retention time of GS under the same conditions is also shown for comparison.
Against Gram-negatives, the situation is more complex due to the existence of an outer membrane that AMPs must traverse before interacting with the plasma membrane. In this context hydrophobicity can be regarded as a two-edged sword since, above a certain limit, it will induce aggregation and poor water solubility, causing the AMP to be sequestered within the outer membrane, unable to reach and disrupt the plasma membrane. Thus, a balanced combination of hydrophobicity with additional parameters such as amphipathicity, molecular flexibility or cationic character becomes essential for AMP interaction with the LPS of the outer membrane and efficacy against Gram-negatives. In the present work, improved performance against Gram-negatives is observed for analogs 3, 5 and 13, all of them with hydrophobicities (i.e., retention times) equal or superior to GS and with substantial structural diversity as Phe surrogates (i.e., Flg in 3, Tic in 5, (αMe)Phe in 13). The failure of analogs 6–12 against Gram-negatives may be explained because, in these analogs, the bulkiness and rigidity of the cyclopropane system may dramatically impair AMP transit through the membrane, regardless of its potential to disrupt the plasma membrane final target.
Changes in antimicrobial activity patterns were evident not only between Gram-positive and Gram-negative organisms but also at the species or even the strain level. The case of the Gram-negative pathogen Acinetobacter baumannii, of which both colistin-susceptible and -resistant strains that reflect differences in outer membrane composition were tested, is illustrative. Against this organism, a substantially improved TI was found for analog 13 (3-fold relative to the parent GS for the colistin-sensitive strain), making this peptide an optimized lead within the current D-Pro-L-Phe series, with the added incentive that the TI optimization applies also to the resistant strain, which constitutes a serious clinical concern53 given that colistin is the last drug universally active on this pathogen. It has been proposed that the self-uptake mechanism used by GS to traverse the outer membrane of Gram-negatives differs from the more general one of other membrane-active AMPs,54 for which high-affinity binding to LPS (mediated by both hydrophobicity and cationic character) entails disruption of the outer membrane and access to the periplasmic space. For GS, instead, the mechanism would involve diffuse binding to LPS and further passage driven by hydrophobic interactions. This alternative mechanism would not seem to apply to the current set of analogs, since the resistant strain, with a highly modified LPS, is significantly more insensitive to analogs such as 3 or 5, which incorporate Phe* surrogates equally or more hydrophobic than Phe. Also, recent experiments have shown the permeability of the outer membrane to be quite compromised in the resistant strain.55
In summary, although inversion of the β-turn sequence of GS has a detrimental impact on antibiotic potency, this can be productively offset by the incorporation of Phe surrogates containing an additional aromatic group or a single phenyl substituent with the adequate orientation. Since the NMR data, with the probable exception of analogs 2 and 12 (see Results), do not reveal substantial alterations in conformation, it is fair to assume that, as long as a robust global β-sheet structure is preserved, sequence inversion at the β-turn of GS is a productive source of antimicrobial leads, provided that a modicum of hydrophobicity is preserved and that the conformational restriction involved is not inadequate (as in the cyclopropane analogs except 6). As is often the case in SAR studies of peptides, subtle changes such as the addition of a methyl group (compare analogs 1 and 13) can have a dramatic impact on the biological profile. Thus, as shown by analogs 7 and 9–12, structural constraints imposing inadequate orientation of the aromatic substituent at the β-turn of GS can be highly detrimental for antibiotic potency. In contrast, analogs 2, 6 and 13 show a remarkable 3-fold boost in TI against Gram-positives relative to GS. Of these, 6 is selective for Gram-positives, and, more interestingly, analog 13 is quite effective against Gram-negatives, including an interesting 5-fold increase in TI against the colistin-resistant Acinetobacter strain.
Experimental Section
Chemicals and Instrumentation for Peptide Synthesis
The different stereoisomers of c3diPhe38, c3Phe39 and fluorenylglycine40 were prepared as previously reported. Dibenzylglycine was obtained following the methodology described by Kotha.41 N-Boc ornithine was purchased from Bachem (Bubendorf, Switzerland) and its side-chain amino group was protected as a formamide by reaction with formic acid and 1,1′-oxalyldiimidazole.42 All other amino acids were purchased from Senn Chemicals (Dielsdorf, Switzerland), NeoMPS (Strasbourg, France) or Fluka (Buchs, Switzerland). Chloromethylated Merrifield resin and TFMSA were from Fluka, HATU from GenScript (Piscataway, NJ), and HBTU from Matrix Innovation (Montreal, Quebec). Solvents and other chemicals were from SDS (Peypin, France). Mass determination was done by the ESI technique in a MicrOTOF-Q spectrometer (Bruker Daltonics, Billerica, MA). Analytical RP-HPLC was performed on an LC-2010A workstation (Shimadzu Corporation, Kyoto, Japan) with a Luna C8 (3 μm, 50 × 4.6 mm) column (Phenomenex B.V., Utrecht, The Netherlands) eluted with a linear 5–95% gradient of CH3CN (+0.036% TFA, v/v) into H2O (+0.045% TFA, v/v) over 15 min at 1 mL/min flow rate, with UV detection at 220 nm. Preparative RP-HPLC purification was done on a Phenomenex Luna C8 column (10 μm, 250 × 10 mm) running linear gradients of CH3CN (+0.1% TFA, v/v) into H2O (+0.1% TFA, v/v) as indicated for each peptide, at a flow rate of 5 mL/min. The preparative system included two Shimadzu LC-8A pumps, a Shimadzu SPD-10A detector and a Foxy Jr. fraction collector (Teledyne Isco, Lincoln, NE).
General Procedure for Peptide Synthesis
All peptides were synthesized manually by solid-phase methods on a Boc-D-Pro-Merrifield resin (0.1 mmol) using Boc chemistry. Boc-amino acids (0.3 mmol) were coupled by HBTU/DIEA (0.3 and 0.6 mmol, respectively) for 30–45 min in DMF. HATU was used instead of HBTU for the coupling reactions involving the 5/5′ residues. To avoid diketopiperazine formation, the third amino acid of each sequence was incorporated by the in situ neutralization method.56 Coupling and deprotection reactions were monitored by the nynhidrin57 or p-nitrophenylester58 colorimetric tests. The linear decapeptide was cleaved from the resin by treatment with TFA/TFMSA/TIS 10:1:1 (v/v/v) (4 mL) at room temperature for 90 min and then precipitated with cold tert-butyl methyl ether. The peptide was redissolved in glacial acetic acid, filtered off the resin, and lyophilized. Cyclization was performed by dissolving the peptide in DMF to a final concentration of 2 mg/mL and stirring for 1 h at room temperature in the presence of HATU/HOAt/DIEA (3:3:5 equiv). After solvent removal and lyophilization, the cyclized peptide was deformylated by treatment with 20% hydrochloric acid in methanol at 37 °C for 21 h. The solvent was evaporated under reduced pressure, and the residue was taken up in glacial acetic acid and lyophilized. Final purification was carried out by preparative reversed-phase HPLC as indicated in each case. HPLC-homogeneous fractions were combined and lyophilized to give white powders of ≥99% HPLC purity (see Supporting Information). Variations to this general protocol are indicated for each peptide.
cyclo(Val-Orn-Leu-D-Pro-Phe)2 (1)
RP-HPLC, linear 30–70% CH3CN gradient into H2O for 30 min (66 mg, 0.063 mmol, 47% overall yield). HRMS (ESI) C60H93N12O10 [M+H]+: calcd. 1141.7132, found 1141.7088; C60H92N12O10Na [M+Na]+: calcd. 1163.6952, found 1163.6924.
cyclo(Val-Orn-Leu-D-Pro-Dip)2 (2)
RP-HPLC, linear 45–75% CH3CN gradient into H2O for 30 min (55 mg, 0.042 mmol, 34% overall yield). HRMS (ESI) C72H101N12O10 [M+H]+: calcd. 1293.7758, found 1293.7682; C72H100N12O10Na [M+Na]+: calcd. 1315.7578, found 1315.7521.
cyclo(Val-Orn-Leu-D-Pro-Flg)2 (3)
RP-HPLC, linear 35–65% CH3CN gradient into H2O for 30 min (57 mg, 0.044 mmol, 38% overall yield). HRMS (ESI) C72H97N12O10 [M+H]+: calcd. 1289.7445, found 1289.7420; C72H96N12O10Na [M+Na]+: calcd. 1311.7265, found 1311.7228.
cyclo(Val-Orn-Leu-D-Pro-Tic)2 (5)
The cyclized peptide was purified by RP-HPLC (linear 60–90% CH3CN gradient into H2O for 30 min) prior to deformylation. Final RP-HPLC, linear 40–70% CH3CN gradient into H2O for 30 min (52 mg, 0.045 mmol, 39% overall yield). HRMS (ESI) C62H93N12O10 [M+H]+: calcd. 1165.7132, found 1165.7088; C62H92N12O10Na [M+Na]+: calcd. 1187.6952, found 1187.6895.
cyclo[Val-Orn-Leu-D-Pro-(R, R)c3diPhe]2 (6)
Precipitation of the linear decapeptide by addition of cold tert-butyl methyl ether proved unsuccessful. The organic solvent was then evaporated and the crude was redissolved in CH3CN. The peptide precipitated upon addition of H2O. The cyclization mixture was allowed to react overnight. The cyclized peptide was purified by RP-HPLC (linear 65–95% CH3CN gradient into H2O for 30 min) prior to deformylation. Final RP-HPLC, linear 40–75% CH3CN gradient into H2O for 30 min (24 mg, 0.018 mmol, 15% overall yield). HRMS (ESI) C74H101N12O10 [M+H]+: calcd. 1317.7758, found 1317.7737; C74H100N12O10Na [M+Na]+: calcd. 1339.7578, found 1339.7557.
cyclo[Val-Orn-Leu-D-Pro-(S, S)c3diPhe]2 (7)
The linear decapeptide was precipitated with H2O, as described above for 6. Final RP-HPLC, linear 40–75% CH3CN gradient into H2O for 30 min (64 mg, 0.049 mmol, 41% overall yield). HRMS (ESI) C74H101N12O10 [M+H]+: calcd. 1317.7758, found 1317.7717; C74H100N12O10Na [M+Na]+: calcd. 1339.7578, found 1339.7559.
cyclo(Val-Orn-Leu-D-Pro-Ac3c)2 (8)
RP-HPLC, linear 30–60% CH3CN gradient into H2O for 30 min (68 mg, 0.067 mmol, 59% overall yield). HRMS (ESI) C50H85N12O10 [M+H]+: calcd. 1013.6506, found 1013.6494; C50H84N12O10Na [M+Na]+: calcd. 1035.6326, found 1035.6309.
cyclo[Val-Orn-Leu-D-Pro-(S, S)c3Phe]2 (9)
The linear decapeptide was precipitated with H2O, as described above for 6, and purified by RP-HPLC (linear 40–70% CH3CN gradient into H2O for 30 min) before cyclization. The cyclized peptide was purified by RP-HPLC (linear 50–80% CH3CN gradient into H2O for 30 min) prior to deformylation. Final RP-HPLC, linear 40–70% CH3CN gradient into H2O for 30 min (61 mg, 0.052 mmol, 45% overall yield). HRMS (ESI) C62H93N12O10 [M+H]+: calcd. 1165.7132, found 1165.7088; C62H92N12O10Na [M+Na]+: calcd. 1187.6952, found 1187.6912.
cyclo[Val-Orn-Leu-D-Pro-(R, R)c3Phe]2 (10)
The linear decapeptide was precipitated with H2O, as described above for 6, and purified by RP-HPLC (linear 45–75% CH3CN gradient into H2O for 30 min) before cyclization. The cyclized peptide was purified by RP-HPLC (linear 50–80% CH3CN gradient into H2O for 30 min) prior to deformylation. Final RP-HPLC, linear 35–65% CH3CN gradient into H2O for 30 min (49 mg, 0.042 mmol, 36% overall yield). HRMS (ESI) C62H93N12O10 [M+H]+: calcd. 1165.7132, found 1165.7102; C62H92N12O10Na [M+Na]+: calcd. 1187.6952, found 1187.6928.
cyclo[Val-Orn-Leu-D-Pro-(S, R)c3Phe]2 (11)
RP-HPLC, linear 45–65% CH3CN gradient into H2O for 30 min (26 mg, 0.021 mmol, 18% overall yield). HRMS (ESI) C62H93N12O10 [M+H]+: calcd. 1165.7132, found 1165.7117; C62H92N12O10Na [M+Na]+: calcd. 1187.6952, found 1187.6947.
cyclo[Val-Orn-Leu-D-Pro-(R, S)c3Phe]2 (12)
The cyclization step required an additional amount of the HATU/HOAt/DIEA (0.6:0.6:1.0 equiv) coupling mixture. RP-HPLC, linear 35–60% CH3CN gradient into H2O for 30 min (67 mg, 0.057 mmol, 50% overall yield). HRMS (ESI) C62H93N12O10 [M+H]+: calcd. 1165.7132, found 1165.7116; C62H92N12O10Na [M+Na]+: calcd. 1187.6952, found 1187.6939.
cyclo[Val-Orn-Leu-D-Pro-(αMe)Phe]2 (13)
RP-HPLC, linear 35–65% CH3CN gradient into H2O for 30 min (76 mg, 0.065 mmol, 56% overall yield). HRMS (ESI) C62H97N12O10 [M+H]+: calcd. 1169.7445, found 1169.7458; C62H96N12O10Na [M+Na]+: calcd. 1191.7265, found 1191.7261.
NMR spectroscopy
Samples for NMR experiments were prepared at 1–2 mM peptide concentration in 0.5 mL of H2O/D2O 9:1 (v/v) or pure D2O at pH 3.0. pH was measured with a glass microelectrode and was not corrected for isotope effects. NMR spectra were acquired on a Bruker AV 600 MHz spectrometer equipped with a z-gradient cryoprobe. A methanol sample was used to calibrate the temperature of the NMR probe. One-dimensional (1D) and two-dimensional (2D) spectra were acquired by standard pulse sequences using presaturation of the water signal. Mixing times for 2D TOCSY and NOESY were 60 and 150 ms, respectively. The 1H–13C and 1H–15 N HSQC spectra59 at natural 13C and 15N abundance were recorded in D2O and H2O/D2O 9:1 (v/v), respectively. Data were processed using TOPSPIN (Bruker Biospin, Rheinstetten, Germany) software. Sodium 2,2-dimethyl-2-silapentane-5-sulphonate (DSS) was used as an internal reference for 1H chemical shifts. The 13C and 15N chemical shifts were indirectly calibrated by multiplying the spectrometer frequency that corresponds to 0 ppm in the 1H spectrum, assigned to internal DSS reference, by 0.25144954 and 0.101329118, respectively.60 The 1H-NMR signals of the peptides were assigned by sequential assignment methods.61 The 13C and 15N resonances were then assigned following the cross-correlations observed in the HSQC spectra between the proton and the hetero-nucleus to which it is bonded.
Structure Calculation
Structure calculations were performed by using the CYANA program62 and an annealing strategy. Since the non-proteinogenic amino acids were not included in the standard CYANA libraries, we built them using MOLMOL63 and manual optimization (these libraries are available upon request from the authors). For this purpose, X-ray data of compounds containing these amino acids were obtained from our laboratories or retrieved from the Cambridge Structural Database.64 Theoretical constraints for the GS β-sheet incorporated for structure calculation included φ and ψ angle restraints, lower and upper-limit distance restraints for the four characteristic cross-strand hydrogen-bonds, and upper-limit distance restraints for the backbone atoms of strand residues (see Supporting Information). Experimental distance constraints for peptides 1 and 5 were derived from 2D NOESY spectra recorded in H2O/D2O 9:1 (v/v). The NOE cross-peaks were integrated by using the automatic integration subroutine of the Sparky program (T.D. Goddard & S.G. Kneller, University of California at San Francisco) and then calibrated and converted to upper-limit distance constraints with CYANA.62 Given the symmetrical nature of the peptides, for structure calculations, residues were re-numbered from 1 to 10 starting at Leu3 (Figure 1). Lower and upper limit restraints required for peptide backbone cyclization were introduced with CYANA (see Supporting Information). For each peptide, a total of 50 conformers were generated and the 20 conformers with the lowest target function were analyzed. Model structures were examined with MOLMOL.63 A side chain torsion angle was considered as well defined when its root mean square deviation between values in the 20 best calculated structures was less than ±30°.
Antimicrobial Activity
Stocks of Staphylococcus aureus CECT 240, Listeria monocytogenes CECT 4032, Acinetobacter baumannii ATCC 19606 and its isogenic colistin-resistant strain 19606R (obtained by continuous growing under increasing colistin concentration) were maintained at −80 °C in freezing medium (65% glycerol, 0.1 M MgSO4, 25 mM Tris-HCl, pH 8.0). Two days prior to the assay for microbicidal activity, they were thawed and grown in MBH medium (Mueller-Hinton II Broth Cation Adjusted (Becton-Dickinson, Cockeysville, MD) at 37 °C; for 19606R, 64 μg/mL colistin sulfate (Sigma, Madrid, Spain) was included.47 Bacterial cells were harvested at exponential growth phase, washed twice with phosphate buffered saline (PBS, 10 mM Na2HPO4, 1 mM KH2PO4, 140 mM NaCl, 3 mM KCl, pH 7.0) and resuspended in MBH, at 5 × 105 colony forming units/mL. Aliquots (100 μL) from this suspension were transferred into a polypropylene 96 well plate, and bacteria were allowed to proliferate for 24 h at 37 °C in the presence of the corresponding peptide concentration. Afterwards, growth was measured by turbidimetry at 600 nm in a model 680 microplate reader (Bio-Rad Laboratories, Hercules. CA). Determinations were carried out twice on triplicated samples. MIC50 was defined as the lowest peptide concentration inhibiting bacterial growth by 50%, relative to untreated control, and was calculated using the SigmaPlot (Systat Software, San Jose, CA) software, v. 9.0.
Hemolytic Activity Assay
As above, hemolytic activity of the peptides was also determined twice on triplicate samples. Defibrinated sheep blood (Biomedics, Madrid, Spain) was centrifuged and washed twice with Hank’s medium (136 mM NaCl; 4.2 mM Na2HPO4; 4.4 mM KH2PO4; 5.4 mM KCl; 4.1 mM NaHCO3, pH 7.2), supplemented with 20 mM D-glucose (Hank’s-Glc). Erythrocytes were resuspended in the same buffer at 2×107 erythrocytes/mL and 100 μL-aliquots of the suspension were incubated with the peptides (4 h, 37 °C). The remaining erythrocytes were harvested in a Micro 200 microfuge (A. Hettich GmbH & Co KG, Germany) (14,000 rpm, 5 min, 4 °C), 80 μL of the supernatant were transferred into a 96-well culture microplate, and released hemoglobin was determined at 550 nm in a Bio Rad 680 (Hercules, CA) microplate reader. The asymptotic ordinate of the GS supernatant was taken as 100% hemolysis. HC50 values were calculated using SigmaPlot, version 9.0.
Supplementary Material
Acknowledgments
This work was supported by Ministerio de Ciencia e Innovación (BIO2008-04487-CO3-02 to D.A., CTQ2008-00080/BQU to M.A.J., CTQ2007-62245 to C.C.), Fondo de Investigaciones Sanitarias (PI061125 and RD06/0021/0006 to L.R.), by the regional governments of Aragón (research group E40), Catalonia (SGR2008-492), and Madrid (COMBACT S-BIO-0260/2006). C.S. and C.M.S. thank Ministerio de Educación y Ciencia and Consejo Superior de Investigaciones Científicas-European Social Fund for FPU and I3P fellowships, respectively. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the view of the policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- Ac3c
1-aminocyclopropanecarboxylic acid
- AMP
antimicrobial peptide
- ATCC
American Type Culture Collection
- (αMe)Phe
α-methylphenylalanine
- Boc
tert-butoxycarbonyl
- CECT
Spanish Type Culture Collection
- COSY
correlated spectroscopy
- c3diPhe
1-amino-c-2, t-3-diphenylcyclopropane-r-1-carboxylic acid
- c3Phe
1-amino-2-phenylcyclopropanecarboxylic acid
- Dbg
dibenzylglycine
- DIEA
N, N-diisopropylethylamine
- Dip
β, β-diphenylalanine
- DMF
N, N-dimethylformamide
- DSS
sodium 2,2-dimethyl-2-silapentane-5-sulphonate
- ESI
electrospray ionization
- Flg
fluorenylglycine
- For
formyl
- GS
gramicidin S
- HATU
2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HC50
50% hemolytic concentration
- HOAt
N-7-aza-hydroxybenzotriazole
- HRMS
high resolution mass spectrometry
- HSQC
heteronuclear single quantum coherence spectra
- LPS
lipopolysacharide
- MIC50
minimal inhibitory concentration
- NOESY
nuclear Overhauser enhancement spectroscopy
- Orn
ornithine
- RMSD
root mean square deviation
- SAR
structure-activity relationship
- TFA
trifluoroacetic acid
- TFMSA
trifluoromethanesulfonic acid
- TI
therapeutic index (HC50/MIC50)
- Tic
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
- TIS
triisopropylsilane
- TOCSY
total correlated spectroscopy
Footnotes
Supporting Information Available
NMR data, details on structure calculations, and analytical data on the peptides synthesized. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hirsch T, Jacobsen F, Steinau HU, Steinstraesser L. Host defense peptides and the new line of defence against multiresistant infections. Protein Pept Lett. 2008;15:238–243. doi: 10.2174/092986608783744252. [DOI] [PubMed] [Google Scholar]
- 2.Parisien A, Allain B, Zhang J, Mandeville R, Lan CQ. Novel alternatives to antibiotics: bacteriophages, bacterial cell wall hydrolases, and antimicrobial peptides. J Appl Microbiol. 2008;104:1–13. doi: 10.1111/j.1365-2672.2007.03498.x. [DOI] [PubMed] [Google Scholar]
- 3.Gause GF. Gramicidin S and its use in the treatment of infected wounds. Nature. 1944;154:703. [Google Scholar]
- 4.Kondejewski LH, Farmer SW, Wishart DS, Hancock RE, Hodges RS. Gramicidin S is active against both gram-positive and gram-negative bacteria. Int J Pept Protein Res. 1996;47:460–466. doi: 10.1111/j.1399-3011.1996.tb01096.x. [DOI] [PubMed] [Google Scholar]
- 5.Prenner EJ, Lewis RN, McElhaney RN. The interaction of the antimicrobial peptide gramicidin S with lipid bilayer model and biological membranes. Biochim Biophys Acta. 1999;1462:201–221. doi: 10.1016/s0005-2736(99)00207-2. [DOI] [PubMed] [Google Scholar]
- 6.Hull SE, Karlsson R, Main P, Woolfson MM, Dodson EJ. The crystal structure of a hydrated gramicidin S-urea complex. Nature. 1978;275:206–207. [Google Scholar]
- 7.Schmidt GM, Hodgkin DC, Oughton BM. A crystallographic study of some derivatives of gramicidin S. Biochem J. 1957;65:744–750. doi: 10.1042/bj0650744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tishchenko GN, Andrianov VI, Vainstein BK, Woolfson MM, Dodson E. Channels in the gramicidin S-with-urea structure and their possible relation to transmembrane ion transport. Acta Crystallogr D: Biol Crystallogr. 1997;53:151–159. doi: 10.1107/S0907444995000916. [DOI] [PubMed] [Google Scholar]
- 9.Ovchinnikov YA, Ivanov VT. Conformational states and biological activity of cyclic peptides. Tetrahedron. 1975;31:2177–2209. [Google Scholar]
- 10.Jelokhani-Niaraki M, Hodges RS, Meissner JE, Hassenstein UE, Wheaton L. Interaction of gramicidin S and its aromatic amino-acid analogs with phospholipid membranes. Biophys J. 2008;95:3306–3321. doi: 10.1529/biophysj.108.137471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katsu T, Kobayashi H, Fujita Y. Mode of action of gramicidin S on Escherichia coli membrane. Biochim Biophys Acta. 1986;860:608–619. doi: 10.1016/0005-2736(86)90560-2. [DOI] [PubMed] [Google Scholar]
- 12.Afonin S, Glaser RW, Berditchevskaia M, Wadhwani P, Guhrs KH, Mollmann U, Perner A, Ulrich AS. 4-fluorophenylglycine as a label for 19F NMR structure analysis of membrane-associated peptides. ChemBioChem. 2003;4:1151–1163. doi: 10.1002/cbic.200300568. [DOI] [PubMed] [Google Scholar]
- 13.Arai T, Imachi T, Kato T, Ogawa HI, Fujimoto T, Nishino N. Synthesis of [hexafluorovalyl1,1′]gramicidin S. Bull Chem Soc Jpn. 1996;69:1383–1389. [Google Scholar]
- 14.Waki M, Abe O, Okawa R, Kato T, Makisumi S, Izumiya N. Studies of peptide antibiotics. XII. Syntheses of [2,2′-α, γ-diaminobutyric acid]-and [2,2′-lysine]-gramicidin S. Bull Chem Soc Jpn. 1967;40:2904–2909. doi: 10.1246/bcsj.40.2904. [DOI] [PubMed] [Google Scholar]
- 15.Aimoto S. The synthesis of a heavy-atom derivative of gramicidin S (GS), [D-Phe(4-Br)4,4′]-GS, by a novel method. Bull Chem Soc Jpn. 1988;61:2220–2222. [Google Scholar]
- 16.Andreu D, Ruiz S, Carreño C, Alsina J, Albericio F, Jiménez MA, de la Figuera N, Herranz R, García-López MT, González-Muñiz R. IBTM-containing gramicidin S analogs: Evidence for IBTM as a suitable type II′ β-turn mimetic. J Am Chem Soc. 1997;119:10579–10586. [Google Scholar]
- 17.Grotenbreg GM, Buizert AE, Llamas-Saiz AL, Spalburg E, van Hooft PA, de Neeling AJ, Noort D, van Raaij MJ, van der Marel GA, Overkleeft HS, Overhand M. βTurn modified gramicidin S analogs containing arylated sugar amino acids display antimicrobial and hemolytic activity comparable to the natural product. J Am Chem Soc. 2006;128:7559–7565. doi: 10.1021/ja0588510. [DOI] [PubMed] [Google Scholar]
- 18.Kawai M, Yamamura H, Tanaka R, Umemoto H, Ohmizo C, Higuchi S, Katsu T. Proline residue-modified polycationic analogs of gramicidin S with high antibacterial activity against both Gram-positive and Gram-negative bacteria and low hemolytic activity. J Pept Res. 2005;65:98–104. doi: 10.1111/j.1399-3011.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee DL, Hodges RS. Structure-activity relationships of de novo designed cyclic antimicrobial peptides based on gramicidin S. Biopolymers. 2003;71:28–48. doi: 10.1002/bip.10374. [DOI] [PubMed] [Google Scholar]
- 20.Ripka WC, Delucca GV, Bach AC, Pottorf RS, Blaney JM. Protein β-turn mimetics II: Design, synthesis and evaluation in the cyclic peptide gramicidin S. Tetrahedron. 1993;49:3609–3628. [Google Scholar]
- 21.Sato K, Kato R, Nagai U. Studies on β-turn of peptides. XII. Synthetic conformation of weak activity of [D-Pro5,5′]-Gramicidin S predicted from β-turn preference of its partial sequence. Bull Chem Soc Jpn. 1986;59:535–538. [Google Scholar]
- 22.Tamaki M, Okitsu T, Araki M, Sakamoto H, Takimoto M, Muramatsu I. Synthesis and properties of gramicidin S analogs containing Pro-D-Phe sequence in place of D-Phe-Pro sequence in the β-turn part of the antibiotic. Bull Chem Soc Jpn. 1985;58:531–535. [Google Scholar]
- 23.Wishart DS, Kondejewski LH, Semchuk PD, Sykes BD, Hodges RS. A method for the facile solid-phase synthesis of gramicidin S and its analogs. Lett Pept Sci. 1996;3:53–60. [Google Scholar]
- 24.Yamada K, Shinoda SS, Oku H, Komagoe K, Katsu T, Katakai R. Synthesis of low-hemolytic antimicrobial dehydropeptides based on gramicidin S. J Med Chem. 2006;49:7592–7595. doi: 10.1021/jm061051v. [DOI] [PubMed] [Google Scholar]
- 25.Solanas C, de la Torre BG, Fernández-Reyes M, Santiveri CM, Jiménez MA, Rivas L, Jiménez AI, Andreu D, Cativiela C. Therapeutic index of gramicidin S is strongly modulated by D-phenylalanine analogs at the β-turn. J Med Chem. 2009;52:664–674. doi: 10.1021/jm800886n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jelokhani-Niaraki M, Kondejewski LH, Farmer SW, Hancock RE, Kay CM, Hodges RS. Diastereoisomeric analogs of gramicidin S: structure, biological activity and interaction with lipid bilayers. Biochem J. 2000;349:747–755. doi: 10.1042/bj3490747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondejewski LH, Jelokhani-Niaraki M, Farmer SW, Lix B, Kay CM, Sykes BD, Hancock RE, Hodges RS. Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alterations in amphipathicity. J Biol Chem. 1999;274:13181–13192. doi: 10.1074/jbc.274.19.13181. [DOI] [PubMed] [Google Scholar]
- 28.Abraham T, Marwaha S, Kobewka DM, Lewis RN, Prenner EJ, Hodges RS, McElhaney RN. The relationship between the binding to and permeabilization of phospholipid bilayer membranes by GS14dK4, a designed analog of the antimicrobial peptide gramicidin S. Biochim Biophys Acta. 2007;1768:2089–2098. doi: 10.1016/j.bbamem.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawai M, Nagai U. Comparison of conformation and antimicrobial activity of synthetic analogs of gramicidin S: Stereochemical considerations of the role of D-phenylalanine in the antibiotic. Biopolymers. 1978;17:1549–1565. doi: 10.1002/bip.1978.360170613. [DOI] [PubMed] [Google Scholar]
- 30.Higashijima T, Miyazawa T, Kawai M, Nagai U. Gramicidin S analogs with a D-Ala, Gly, or L-Ala residue in place of the D-Phe residue: molecular conformations and interactions with phospholipid membrane. Biopolymers. 1986;25:2295–2307. doi: 10.1002/bip.360251207. [DOI] [PubMed] [Google Scholar]
- 31.Ando S, Aoyagi H, Waki M, Kato T, Izumiya N. Studies of peptide antibiotics. XLIII. Syntheses of gramicidin S analogs containing D-serine or dehydroalanine in place of D-phenylalanine and asymmetric hydrogenation of the dehydroalanine residue. Tetrahedron Lett. 1982;23:2195–2198. [PubMed] [Google Scholar]
- 32.Grotenbreg GM, Spalburg E, de Neeling AJ, van der Marel GA, Overkleeft HS, van Boom JH, Overhand M. Synthesis and biological evaluation of novel turn-modified gramicidin S analogs. Bioorg Med Chem. 2003;11:2835–2841. doi: 10.1016/s0968-0896(03)00219-0. [DOI] [PubMed] [Google Scholar]
- 33.Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Protein Chem. 1985;37:1–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 34.Marraud M, Aubry A. Crystal structures of peptides and modified peptides. Biopolymers (Pept Sci) 1996;40:45–83. doi: 10.1002/(sici)1097-0282(1996)40:1<45::aid-bip3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Hutchinson EG, Thornton JM. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiménez AI, Cativiela C, Aubry A, Marraud M. β-turn preferences induced by 2,3-methanophenylalanine chirality. J Am Chem Soc. 1998;120:9452–9459. [Google Scholar]
- 37.Toniolo C, Crisma M, Formaggio F, Peggion C. Control of peptide conformation by the Thorpe-Ingold effect (Cα-tetrasubstitution) Biopolymers (Pept Sci) 2001;60:396–419. doi: 10.1002/1097-0282(2001)60:6<396::AID-BIP10184>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 38.Jiménez AI, López P, Oliveros L, Cativiela C. Facile synthesis and highly efficient resolution of a constrained cyclopropane analog of phenylalanine. Tetrahedron. 2001;57:6019–6026. [Google Scholar]
- 39.Cativiela C, Díaz-de-Villegas MD, Jiménez AI, López P, Marraud M, Oliveros L. Efficient access to all four stereoisomers of phenylalanine cyclopropane analogs by chiral HPLC. Chirality. 1999;11:583–590. doi: 10.1002/(SICI)1520-636X(1999)11:7<583::AID-CHIR11>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 40.Royo S, Jiménez AI, Cativiela C. Synthesis of enantiomerically pure β, β-diphenylalanine (Dip) and fluorenylglycine (Flg) Tetrahedron: Asymmetry. 2006;17:2393–2400. [Google Scholar]
- 41.Kotha S, Behera M. Synthesis and modification of dibenzylglycine derivatives via the Suzuki-Miyaura coupling reaction. J Pept Res. 2004;64:72–85. doi: 10.1111/j.1399-3011.2004.00171.x. [DOI] [PubMed] [Google Scholar]
- 42.Kitagawa T, Arita J, Nogahata A. Convenient one-pot method for formylation of amines and alcohols using formic acid and 1,1′-oxalyldiimidazole. Chem Pharm Bull (Tokyo) 1994;42:1655–1657. [Google Scholar]
- 43.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of the nearest-neighbor effects. J Biomol NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 44.Schubert M, Labudde D, Oschkinat H, Schmieder P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. J Biomol NMR. 2002;24:149–154. doi: 10.1023/a:1020997118364. [DOI] [PubMed] [Google Scholar]
- 45.Santiveri CM, Rico M, Jiménez MA. 13Cαand 13Cβchemical shifts as a tool to delineate β-hairpin structures in peptides. J Biomol NMR. 2001;19:331–345. doi: 10.1023/a:1011224625129. [DOI] [PubMed] [Google Scholar]
- 46.Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 47.Santiveri CM, Rico M, Jiménez MA. Position effect of cross-strand side-chain interactions on beta-hairpin formation. Protein Sci. 2000;9:2151–2160. doi: 10.1110/ps.9.11.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saugar JM, Rodríguez-Hernández MJ, de la Torre BG, Pachón-Ibáñez ME, Fernández-Reyes M, Andreu D, Pachón J, Rivas L. Activity of cecropin A-melittin hybrid peptides against colistin-resistant clinical strains of Acinetobacter baumannii: molecular basis for the differential mechanisms of action. Antimicrob Agents Chemother. 2006;50:1251–1256. doi: 10.1128/AAC.50.4.1251-1256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kondejewski LH, Lee DL, Jelokhani-Niaraki M, Farmer SW, Hancock REW, Hodges RS. Optimization of microbial specificity in cyclic peptides by modulation of hydrophobicity within a defined structural framework. J Biomol Chem. 2002;1:67–74. doi: 10.1074/jbc.M107825200. [DOI] [PubMed] [Google Scholar]
- 50.McInnes C, Kondejewski LH, Hodges RS, Sykes BD. Development of the structural basis for antimicrobial and hemolytic activities of peptides based on gramicidin S and design of novel analogs using NMR spectroscopy. J Biol Chem. 2000;275:14287–14294. doi: 10.1074/jbc.275.19.14287. [DOI] [PubMed] [Google Scholar]
- 51.Haug BE, Strøm MB, Svendsen JS. The medicinal chemistry of short lactoferricin-based antibacterial peptides. Curr Med Chem. 2007;14:1–18. doi: 10.2174/092986707779313435. [DOI] [PubMed] [Google Scholar]
- 52.Van der Knaap M, Engels E, Busscher HJ, Otero JM, Llamas-Saiz AL, van Raaij MJ, Mars-Groenendijk RH, Noort D, van der Marel GA, Overkleeft HS, Overhand M. Synthesis and biological evaluation of asymmetric gramicidin S analogues containing modified D-phenylalanine residues. Bioorg Med Chem. 2009;17:6318–6328. doi: 10.1016/j.bmc.2009.07.042. [DOI] [PubMed] [Google Scholar]
- 53.Valencia R, Arroyo LA, Conde M, Aldana JM, Torres MJ, Fernández-Cuenca F, Garnacho-Montero J, Cisneros JM, Ortiz C, Pachón J, Aznar J. Nosocomial outbreak of infection with pan-drug-resistant Acinetobacter baumannii in a tertiary care university hospital. Infect Control Hosp Epidemiol. 2009;30:257–263. doi: 10.1086/595977. [DOI] [PubMed] [Google Scholar]
- 54.Zhang L, Dhillon P, Yan H, Farmer S, Hancock RE. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2000;44:3317–3321. doi: 10.1128/aac.44.12.3317-3321.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernández-Reyes M, Rodríguez-Falcón M, Chiva C, Pachón J, Andreu D, Rivas L. The cost of resistance to colistin in Acinetobacter baumannii: a proteomic perspective. Proteomics. 2009;9:1632–1645. doi: 10.1002/pmic.200800434. [DOI] [PubMed] [Google Scholar]
- 56.Schnölzer M, Alewood P, Jones A, Alewood D, Kent SB. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int J Pept Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 57.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in solid-phase synthesis of peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 58.Madder A, Farcy N, Hosten NGC, De Muynck H, De Clercq PJ, Barry J, Davies AP. A novel sensitive colorimetric assay for visual detection of solid phase bound amines. Eur J Org Chem. 1999:2787–2791. [Google Scholar]
- 59.Bax A, Lerner L. Two-dimensional nuclear magnetic resonance spectroscopy. Science. 1986;232:960–967. doi: 10.1126/science.3518060. [DOI] [PubMed] [Google Scholar]
- 60.Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wüthrich K. Recommendations for the presentation of NMR structures of proteins and nucleic acids--IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. J Biomol NMR. 1998;12:1–23. doi: 10.1023/a:1008290618449. [DOI] [PubMed] [Google Scholar]
- 61.Wüthrich K, Billeter M, Braun W. Polypeptide secondary structure determination by nuclear magnetic resonance observation of short proton-proton distances. J Mol Biol. 1984;180:715–740. doi: 10.1016/0022-2836(84)90034-2. [DOI] [PubMed] [Google Scholar]
- 62.Güntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 63.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 64.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.