

Abstract
Cancer is a complex collection of distinct genetic diseases united by common hallmarks. Here, we expand upon the classic hallmarks to include the stress phenotypes of tumorigenesis. We describe a conceptual framework of how oncogene and non-oncogene addictions contribute to these hallmarks and how they can be exploited through stress sensitization and stress overload to selectively kill cancer cells. In particular, we present evidence for a large class of non-oncogenes that are essential for cancer cell survival and present attractive drug targets. Finally, we discuss the path ahead to therapeutic discovery and provide theoretical considerations for combining orthogonal cancer therapies.
The Current State of Cancer Research
The past two decades have witnessed tremendous advances in our understanding of the pathogenesis of cancer. It is now clear that cancer arises through a multistep, mutagenic process whereby cancer cells acquire a common set of properties including unlimited proliferation potential, self-sufficiency in growth signals, and resistance to antiproliferative and apoptotic cues. Furthermore, tumors evolve to garner support from surrounding stromal cells, attract new blood vessels to bring nutrients and oxygen, evade immune detection, and ultimately metastasize to distal organs (Hanahan and Weinberg, 2000). Many of these phenotypic traits can be brought about by genetic alterations that involve the gain-of-function mutation, amplification, and/or overexpression of key oncogenes together with the loss-of-function mutation, deletion, and/or epigenetic silencing of key tumor suppressors (Hahn and Weinberg, 2002).
Cancer cells achieve these phenotypes in large part by reactivating and modifying many existing cellular programs normally used during development. These programs control coordinated processes such as cell proliferation, migration, polarity, apoptosis, and differentiation during embryogenesis and tissue homeostasis. Consistent with Darwinian principles, cancer evolves through random mutations and epigenetic changes that alter these pathways followed by the clonal selection of cells that can survive and proliferate under circumstances that would normally be deleterious.
Although a number of oncogenes and tumor suppressors, such as PI3K, Ras, p53, PTEN, Rb, and p16INK4a, are frequently mutated in cancer cells, there also appears to be a large number of low-frequency changes that can contribute to oncogenesis. Indeed, data from tumor sequencing projects reveal an astounding diversity of mutations in tumors. In one study, Stratton and colleagues estimate that individual mutations in as many as 20% of all kinases can play an active role in tumorigenesis (Greenman et al., 2007), although it remains to be seen whether mutations in 20% of other gene classes will also drive tumorigenesis. Large-scale sequencing of multiple cancers has so far failed to identify new, high-frequency mutation targets in addition to those previously identified (Cancer Genome Atlas Research Network, 2008; Ding et al., 2008; Jones et al., 2008; Parsons et al., 2008; Sjoblom et al., 2006; Wood et al., 2007). Rather, these studies found that every tumor harbors a complex combination of low-frequency mutations thought to drive the cancer phenotype. Furthermore, the repertoires of somatic mutations in different cancer types such as breast and colon cancers appear to be different. Although there is much debate with regard to the statistical requirements needed to distinguish likely driver from noncontributing passenger mutations among the large collection of mutations in tumors, it is clear that there is tremendous complexity and heterogeneity in the patterns of mutations in tumors of different origins.
The complexity of alterations in cancer presents a daunting problem with respect to treatment: how can we effectively treat cancers arising from such varied perturbations? Cancer cells have extensively rewired pathways for growth and survival that underlie the malignant phenotype. Thus, a key to successful therapy is the identification of critical, functional nodes in the oncogenic network whose inhibition will result in system failure, that is, the cessation of the tumorigenic state by apoptosis, necrosis, senescence, or differentiation. Furthermore, therapeutic agents attacking these nodes must display a sufficiently large therapeutic window with which to kill tumor cells while sparing normal cells. To borrow a term from yeast and fly genetic analyses, the therapeutic agents must constitute “synthetic lethality” with the cancer genotype/phenotype (Kaelin, 2005). In some cases, particular agents can display genotype-dependent lethality similar to synthetic lethality without directly inhibiting a particular protein. The two mainstay treatment options for cancer today—chemotherapy and radiation—are examples of agents that exploit the enhanced sensitivity of cancer cells to DNA damage. Despite all of our knowledge, however, we still do not have a clear molecular understanding of why these agents work to selectively kill tumor cells and, conversely, why they eventually fail. The advent of “targeted” therapies, which aim to attack the underlying oncogenic context of tumors, provides more sophisticated examples of synthetic lethality. When properly deployed, these therapies tend to be more effective relative to chemotherapy and radiation.
Additional Hallmarks: The Stress Phenotypes of Cancer
Although there is no simple way to predict a priori which proteins will act as nodal points to generate cancer drug targets, solutions are likely to emerge from multiple sources, including recent initiatives to understand cancer at the systems level. From a genetic point of view, it is important to appreciate that the plethora of mutations observed in the cancer genome must ultimately result in a common set of hallmarks in order to bring about the malignant phenotype. The goal of cancer therapy is, therefore, to either reverse these properties or target them as tumor-specific liabilities, preferably through the combinatorial application of a relatively small number of drugs. Thus we need a thorough understanding of the nature of these hallmarks.
In addition to the six hallmarks outlined in the seminal review by Hanahan and Weinberg (Hanahan and Weinberg, 2000) that collectively promote survival and proliferation in foreign environments (Figure 1, top), as well as the hallmark of “evading immune surveillance” proposed by Kroemer and colleagues (Kroemer and Pouyssegur, 2008) (Figure 1, left), we propose a number of additional, equally prevalent hallmarks of cancer cells based on recent analyses of cellular phenotypes. Although these cancer phenotypes are not responsible for initiating tumorigenesis, they are common characteristics of many tumor types (Figure 1, bottom). Among these additional hallmarks are DNA damage/replication stress, proteotoxic stress, mitotic stress, metabolic stress, and oxidative stress. We collectively refer to this subset as the stress phenotypes of cancers. There are often intricate functional interplays among these shared hallmarks of tumor cells, which are illustrated in Figure 1 and discussed below. Although some of these stress phenotypes are not unique to cancer cells and can be observed in other conditions such as chronic inflammation, we propose that they represent a common set of oncogenesis-associated cellular stresses that cancer cells must tolerate through stress support pathways. How these phenotypes arise is not well understood, but targeting these hallmarks and their associated vulnerabilities in a wide variety of cancers has shown promise for therapeutic intervention.
Figure 1. The Hallmarks of Cancer.

In addition to the six hallmarks originally proposed by Hanahan and Weinberg (top half, white symbols) and evasion of immune surveillance proposed by Kroemer and Pouyssegur, we propose a set of additional hallmarks that depict the stress phenotypes of cancer cells (lower half, colored symbols). These include metabolic stress, proteotoxic stress, mitotic stress, oxidative stress, and DNA damage stress. Functional interplays among these hallmarks promote the tumorigenic state and suppress oncogenic stress. For example, the utilization of glycolysis allows tumor cells to adapt to hypoxia and acidify its microenvironment to evade immune surveillance. Increased mitotic stress promotes aneuploidy, which leads to proteotoxic stress that requires compensation from the heat shock response pathway. Elevated levels of reactive oxygen species result in increased levels of DNA damage that normally elicits senescence or apoptosis but is overcome by tumor cells.
DNA Damage and DNA Replication Stress
Based on karyotypic and mutational analyses, it is clear that tumors, especially solid tumors, pass through stages of extreme genomic instability that result in the accumulation of point mutations, deletions, complex chromosomal rearrangements, and extensive aneuploidy (Hartwell and Kastan, 1994). This level of instability is due in part to a constitutive level of endogenous DNA damage, which results in activation of the DNA damage stress response (DDR) pathway (Bartkova et al., 2005; Gorgoulis et al., 2005). Elevated levels of DNA damage observed in early stage tumors are thought to be due to several factors. First, the shortening of telomeres due to replication in the absence of sufficient telomerase activity leads to the appearance of double-strand breaks (DSBs) at telomeric ends. The subsequent fusions of these deprotected ends initiate breakage-fusion-bridge cycles that result in translocations and gene amplification events (Maser and DePinho, 2002). DSBs resulting from replication stress can also lead to breakage-fusion-bridge cycles (Windle et al., 1991). Additionally, oncogene activation in precancerous lesions has been shown to increase DSBs and genomic instability (Halazonetis et al., 2008), possibly through DNA hyper-replication (Bartkova et al., 2006; Di Micco et al., 2006). Finally, mutation of genes involved in either DNA repair programs (such as excision, crosslink, or mismatch repair) or the DDR pathways (such as ATM and p53 signaling) can lead to increased DNA damage, inappropriate cell-cycle progression, and genomic instability (Harper and Elledge, 2007). In normal cells, DNA damage signals to halt proliferation, induce cellular senescence, or elicit apoptosis. Cancer cells have evolved to overcome the antiproliferative effects of DNA damage, continuing to replicate in the presence of damage (Figure 1).
Proteotoxic Stress
Tumors exhibit proteotoxic stress evidenced by their frequent constitutive activation of the heat shock response. We think this is due, in part, to the striking degree of aneuploidy (altered chromosome number) often found in solid tumors (Figure 1) (Ganem et al., 2007; Torres et al., 2008; Williams et al., 2008). Aneuploidy and gene copy-number changes can alter the relative balance of growth and survival signals, thereby promoting tumorigenesis. However, they also result in corresponding increases and decreases in transcript levels (Pollack et al., 2002; Torres et al., 2007; Tsafrir et al., 2006) that produce imbalances in the stoichiometry of protein complex subunits (Papp et al., 2003). These imbalances increase the amount of toxic, unfolded protein aggregates in the cell and place additional burdens on the protein folding and degradation machineries (Denoyelle et al., 2006). This proteotoxic stress is counteracted, in part, by the heat shock response pathway, which promotes the proper folding and/or proteolytic degradation of proteins (Whitesell and Lindquist, 2005).
Mitotic Stress
A subset of tumors display increased rates of chromosome mis-segregation, which is referred to as the CIN (chromosome instability) phenotype (Komarova et al., 2002). This instability results in a shifting chromosome distribution, thus allowing tumor cells to rapidly evolve. In principle, CIN phenotypes can result from defects in a variety of pathways involved in mitosis, including defects in mitotic proteins that execute chromosome segregation and defects in the spindle assembly checkpoint, which coordinates anaphase entry with proper alignment of chromosomes on the mitotic spindle (Cahill et al., 1998). In addition, the CIN phenotype could result from the presence of extra centrosomes in tumor cells or from stresses placed on the mitotic apparatus due to the need to segregate supernumerary chromosomes (Ganem et al., 2007). Furthermore, CIN and mitotic stress might arise indirectly as a result of DSBs and genomic instability following oncogene activation, even in lesions where the mitotic machinery is intact (Halazonetis et al., 2008). Mutations in certain oncogenes, such as Ras, and tumor suppressors, such as p53, have been suggested to contribute to the CIN phenotype (Denko et al., 1994). However, the precise cause of mitotic stress is not known for the vast majority of tumors.
Metabolic Stress
Normal cells derive the bulk of their ATP through mitochondrial oxidative phosphorylation. In what has been referred to as the Warburg effect, most cancer cells are found to predominantly produce energy by the less efficient method of glycolysis and secrete a large amount of lactic acid, even under high oxygen conditions (Warburg, 1956). Tumor cells exhibit dramatically increased glucose uptake and highly elevated rates of glycolysis (DeBerardinis et al., 2007). This provides the basis for tumor imaging by positron emission tomography (PET) using the glucose analog 18F-2-deoxyglucose. This transition to glycolysis for energy production provides several advantages to the tumor including adaptation to a low oxygen environment and the acidification of the surrounding microenvironment, which promotes tumor invasion and suppresses immune surveillance (Figure 1).
Oxidative Stress
The defining characteristic of oxidative stress is the presence of reactive oxygen species (ROS), and cancer cells typically generate more ROS than normal cells (Szatrowski and Nathan, 1991). Both oncogenic signaling (Lee et al., 1999) and the downregulation of mitochondrial function (Gogvadze et al., 2008) in tumors can contribute to ROS generation. ROS are highly reactive and likely to contribute to the increased levels of endogenous DNA damage observed in cancer cells (Figure 1). In addition, ROS are important signaling mediators, and their presence may contribute to transformation. For example, ROS promote the activation of the transcription factor HIF-1 by hypoxia (Dewhirst et al., 2008), and HIF-1 can promote the glycolytic switch and angiogenesis observed in tumors.
Attacking the Hallmarks of Cancer
Any therapy with the stated goal to treat and possibly cure cancer must show differential toxicity toward tumor cells relative to normal cells. Implicit in this statement is that some unique properties of cancer cells not shared by normal cells, such as those depicted in Figure 1, must be exploited to the specific detriment of cancer cells, i.e., the concept of synthetic lethality. In principle, cancer can be treated by inducing cancer cells to undergo apoptosis, necrosis, senescence, or differentiation. These changes can be brought about by disrupting cancer cell-autonomous processes, by interfering with autocrine/paracrine signaling within tumors, or by blocking heterotypic signaling between tumor cells and the surrounding stromal tissue or blood vessels. Enhancing immune surveillance against cancer cells expressing novel antigens is also an attractive approach that has shown efficacy in specifically killing cancer cells (Muller and Scherle, 2006).
Experiments aimed at either suppressing oncogene activity or restoring tumor suppressor function have revealed that such intervention is highly deleterious to the cancer cell. The heightened state of dependency of cancer cells on oncogenes and the loss of tumor suppressors led to the terms “oncogene addiction” (OA) and “tumor suppressor gene hypersensitivity” (Weinstein, 2002; Weinstein and Joe, 2008). These alterations are necessary for both the establishment and maintenance of the oncogenic state and therefore are logical drug targets. Indeed, much effort has been extended to pharmacologically inhibit oncoproteins. What is thought to underlie the phenomenon of oncogene addiction is the observation that oncogenes elicit strong, opposing prosurvival and proapoptotic signals in cancer cells that favor growth and survival, and the acute inhibition of oncogene function tilts this balance toward cell death (Downward, 2003; Sharma and Settleman, 2007).
To bring about their phenotypic manifestations, oncogenes rely on extensive adaptations in cellular processes that are themselves not oncogenic. In addition, cancer cells may also display an increased dependence on the normal cellular functions of certain genes that act in oncogenic pathways but are not themselves classical oncogenes. For example, mutations in many genes in a given oncogenic pathway are unable to directly promote tumor formation because, despite being required for their pathway, they cannot increase the overall activity of the pathway because they are not rate-limiting. However, a reduction in the activity of many such genes can become rate-limiting to the pathway in question, and thus, they represent potential drug targets. By this rationale, cancer cells are addicted to both oncogenes and non-oncogenes. To describe this addiction of cancer cells to the functions of non-oncogenes, we have termed this phenomenon “non-oncogene addiction,” NOA (Solimini et al., 2007). Although NOA genes, like oncogenes, are required for maintenance of the tumorigenic state, NOA genes do not undergo oncogenic mutations or functionally significant genomic alterations in tumors. The concept of non-oncogene addiction underscores the important contribution of these supporting networks to oncogenesis and highlights the potential of non-oncogenes as points of intervention for cancer therapeutics.
Whereas some gene classes and pathways fall neatly into the OA or NOA designations, others are more difficult to categorize because they exhibit characteristics of both phenomena. For example, interferon regulatory factor 4 (IRF4) (Iida et al., 1997) is oncogenic and overexpressed due to translocations in some multiple myelomas. However, it is also required for the survival of myelomas lacking IRF4 translocations or overexpression (Shaffer et al., 2008). Should it be considered as an example of OA in the latter cases? Also, should a protein that is directly activated by an oncogene and required for tumorigenesis—but is otherwise not mutated in cancer—be considered an example of NOA when it is so clearly linked to an oncogene? Both examples are clear if one adheres to a strict definition of NOA stating that NOA genes do not undergo oncogenic mutations in tumors. However, these examples often run counter to our overall intuitive sense of the different categories. Regardless, although the OA and NOA designations are not perfect, they provide a useful intellectual framework for thinking about cancer cell vulnerabilities and the principles of cancer therapies. Below, we will discuss examples of oncogene and non-oncogene addiction and describe how modern tools are being applied to identify these classes of genes for possible therapeutic exploitation.
Oncogene Addiction and Tumor Suppressor Gene Hypersensitivity
Despite the multitude of genetic and epigenetic alterations found across cancers, a given tumor is likely to be driven by only a select few changes—those that result in the gain of an oncogene or the loss of a tumor suppressor. The phrase “oncogene addiction” was coined to describe the observation that tumor maintenance often depends upon the continued activity of certain oncogenes (Weinstein, 2002). This phenomenon has been demonstrated in vivo for several oncogenes. For example, mouse models using an inducible MYC oncogene have shown that MYC-driven skin papillomas, lymphomas, and osteosarcomas can all be reversed upon MYC withdrawal (Felsher and Bishop, 1999; Jain et al., 2002; Pelengaris et al., 1999). Similarly, addictions to the HRAS or BCR-ABL oncogenes have been demonstrated in mouse models of melanoma and leukemia, respectively (Chin et al., 1999; Huettner et al., 2000). In human colorectal cancer cells bearing a KRAS mutation, somatic knockout of the KRAS oncogene results in reversion of the transformed phenotype and abrogates the ability of these cells to form tumors in nude mice (Shirasawa et al., 1993).
The subset of oncogenes whose inhibition can lead to tumor cell death, differentiation, arrest, or senescence is of great clinical interest as targets for cancer therapeutics (Table 1). This strategy has proven successful for the protein kinase oncogenes BCR-ABL (imatinib/Gleevec), EGFR (gefitinib/Iressa, erlotinib/Tarceva), and HER2 (trastuzumab/Herceptin) (Druker, 2002; Roberts and Der, 2007; Sharma et al., 2007), and efforts toward inhibition of BRAF, MDM2, and the lipid kinase PI3K are underway. Targeting non-kinase oncogenes such as RAS and MYC, however, has proven more difficult.
Table 1.
Cancer Therapies Targeting Various Hallmarks of Cancer
| Agent | Target | Addiction | Hallmarks | Potential mechanisms | References |
|---|---|---|---|---|---|
| 17AAG (small molecule) | HSP90 | NOA |

|
A geldanamycin analog that binds to the ATP-binding pocket of HSP90 and inhibits its catalytic activity | Whitesell and Lindquist, 2005 |
| 1MT, MTH-Trp (small molecule) | IDO | NOA |

|
Inhibits tryptophan catabolism in tumor microenvironment to allow T cell proliferation | Muller and Scherle, 2006 |
| 5-fluorouracil (small molecule) | DNA | NOA |

|
Inhibits pyrimidine metabolism, incorporation in to DNA and RNA causes cell-cycle arrest | Longley et al., 2003 |
| ABT-737, ABT-263 (small molecule) | BCL-XL, BCL-2 | OA |

|
Bind to the BH3 pocket of Bcl-XL and inhibit its antiapoptotic function | Stauffer, 2007 |
| Alvocidib, PD 0332991 (small molecule) | CDKs | OA |

|
Inhibit CDKs and induce cell-cycle arrest | Lee and Sicinski, 2006 |
| AP 12009 (antisense oligo) | TGFβ 2 | NOA |
|
Inhibits tumor autocrine and paracrine signaling, reverses immune suppression in the tumor microenvironment | Muller and Scherle, 2006 |
| AZD2281, AG014699 (small molecule) | PARP1 | NOA |

|
Inhibit base excision repair in homologous recombination repair-deficient cancer cells | Bryant et al., 2005; Farmer et al., 2005 |
| Bevacizumab (antibody) | VEGF | NOA |

|
Inhibits endothelial cell recruitment and tumor vasculature | Folkman, 2007 |
| BEZ235 (small molecule) | PI3K | OA |

|
Causes cell-cycle arrest in tumor cells and inhibits tumor angiogenesis | Maira et al., 2008 |
| Bortezomib (small molecule) | Proteasome | NOA |

|
Inhibits the catalytic activity of 26S proteasome and induces apoptosis | Roccaro et al., 2006 |
| Celecoxib (small molecule) | COX2 | NOA |
|
Reverses immune suppression in the tumor microenvironment, inhibits tumor autocrine and paracrine signaling | Muller and Scherle, 2006 |
| Cisplatin and analogs (small molecule) | DNA | NOA |

|
Induces DNA crosslinks | Siddik, 2003 |
| Erlotinib, Gefitinib (small molecule) | EGFR | OA |

|
Inhibit EGFR tyrosine kinase by competing with ATP binding | Sharma et al., 2007 |
| GRN163L (modified oligo) | hTERT | OA |

|
Mimics telomere sequence and inhibits the hTERT active site | Dikmen et al., 2005; Harley, 2008 |
| GRNVAC1 (cell therapy) | hTERT | OA |

|
Autologous dendritic cells transduced to express an hTERT-LAMP fusion protein to elicit T cell response to hTERT + tumor cells | Harley, 2008; Su et al., 2005 |
| GV1001 (peptide) | hTERT | OA |

|
A short immunogenic peptide from hTERT designed to elicit T cell response against hTERT + tumor cells | Harley, 2008; Nava-Parada and Emens, 2007 |
| Imatinib, Dasatinib (small molecule) | BCR-ABL, c-Kit, Src, PDGFR, other TKs | OA |

|
Tyrosine kinase inhibitor with multiple targets | Quintas-Cardama et al., 2007 |
| Mapatumumab, Lexa-tumumab (antibody) | TRAIL receptor | NOA |

|
Bind and activate TRAIL receptors to induce apoptosis | Carlo-Stella et al., 2007 |
| Methotrexate (small molecule) | DHFR | NOA |

|
Inhibits thymidine biosynthesis and induces replicative stress | McGuire, 2003 |
| Nutlin-3 (small molecule) | HDM2 | OA |

|
Binds to HDM2 and inhibits the binding and ubiquitination of p53 | Vassilev, 2007 |
| Oblimersen (antisense oligo) | BCL-2 | OA |

|
Inhibits the expression of BCL-2 by blocking translation of its mRNA | Moreira et al., 2006 |
| Paclitaxel, Vinblastine (small molecule) | Mitotic spindle | NOA |

|
Interfere with dynamics and stability of mitotic spindles, activate mitotic checkpoints, and induce chromosome mis-segregation | Weaver and Cleveland, 2005 |
| PF-00477736 (small molecule) | Chk1 | NOA |

|
Prevents activation of the DNA damage response, leading to persistent DNA damage and replication stress | Ashwell and Zabludoff, 2008 |
| PRIMA-1, MIRA-1 (small molecule) | Mutant p53 | TSGH |

|
Reactivate the function of mutant p53 | Selivanova and Wiman, 2007 |
| Rapamycin, RAD001, Temsirolimus (small molecule) | mTOR | NOA |

|
Inhibit protein synthesis | Guertin and Sabatini, 2007 |
| Retinoic acid (small molecule) | RAR, RXR | OA |

|
Induces cellular differentiation | Spira and Carducci, 2003 |
| SAHBs (stapled peptide) | BCL-XL, BCL-2 | OA |

|
Stapled BH3 domains that bind to BCL-2 family members and promote apoptosis | Verdine and Walensky, 2007 |
| Sorafenib, Sunitinib (small molecule) | Multiple kinases (VEGFR, RAF, c-Kit, PDGFR) | NOA |

|
Inhibit endothelial cell recruitment and tumor vasculature | Folkman, 2007 |
| Topotecan, Irinotecan (small molecule) | Topo-isomerase I | NOA |

|
Induce DNA breaks | Pommier, 2006 |
| Trastuzumab (antibody) | ERBB2 | OA |
|
Inhibits ERBB2 activation and induces immune destruction of cancer cells | Hynes and Lane, 2005 |
Therapeutics are selected based on the diversity of their chemical structures, the hallmarks they attack, and their cellular targets. These agents are either investigational drugs or approved for selective oncology indications. Abbreviations: OA, oncogene addiction; NOA, non-oncogene addiction; TSGH, tumor suppressor gene hypersensitivity. Symbols for each hallmark refer to those used in Figure 1.
In contrast to oncogenes, tumor suppressor genes act to provide the cellular restraints necessary to prevent aberrant growth and survival or genomic instability. Loss of tumor suppressor genes through deletion, inactivating mutation, or epigenetic silencing results in the removal of these restraints leading to tumorigenesis. Reintroduction of a tumor suppressor gene into a tumor lacking that gene can result in tumor regression. This concept has been recently demonstrated by reactivation of p53 in mouse tumor models (Martins et al., 2006; Ventura et al., 2007; Xue et al., 2007). Pharmacological exploitation of tumor suppressor mutations, however, has lagged behind efforts aimed at oncogenes because it is often difficult to use a small molecule to either restore or mimic the function of a protein that is either mutated or absent. In cases where a tumor suppressor negatively regulates the activity of a proto-oncogene, drugs targeting the corresponding proto-oncogene should prove efficacious in treating tumors lacking that tumor suppressor. For example, tumors that have lost the tumor suppressor and lipid phosphatase PTEN, which normally acts to constrain PI3K signaling, are likely to be sensitive to PI3K inhibitors. Similarly, loss of Rb, p16, p21, or p27 all result in upregulation of cyclin-dependent kinase (CDK) activity, which drives cell-cycle entry. In principle, tumors resulting from these lesions might be more sensitive to CDK inhibitors. Whether such predictions prove true will only become apparent from clinical trials employing PI3K and CDK inhibitors. In many other cases, however, such as those involving loss of the tumor suppressors p53 or ARF, there is no obvious positive signaling pathway to target, and alternative therapeutic strategies must therefore be considered.
Targeting Non-oncogene Addiction for Cancer Therapy
We proposed the concept of non-oncogene addiction (NOA) based on the understanding that the tumorigenic state depends on the activities of a wide variety of genes and pathways, many of which are not inherently oncogenic themselves (Solimini et al., 2007). Importantly, these genes and pathways are essential to support the oncogenic phenotype of cancer cells but are not required to the same degree for the viability of normal cells. From a purely genetic point of view, these dependencies should provide an ample number of drug targets that when inhibited will constitute synthetic lethality with the underlying tumor genotype. Gene interaction studies in yeast have provided precedence for this notion. For example, most mutations exhibit enhanced growth defects when paired with certain other mutations, and one study in yeast identified an average of six genetic interactions per gene (Collins et al., 2007). As a tumor contains many genetic alterations, each of these changes provides an opportunity to pair with the loss of function of a second gene to result in a severe and possibly lethal growth and survival phenotype. Furthermore, if this second gene is targeted with a drug that inhibits its protein, then a potential cancer therapy can result.
NOA genes and pathways provide important targets for antitumor therapies. In the sections below, we will discuss general classes of NOA genes with specific examples when available. NOA genes fall into two general categories, tumor intrinsic and tumor extrinsic. Whereas tumor-intrinsic NOA genes support the oncogenic state of the tumor cell in a cell-autonomous manner, tumor-extrinsic NOA genes function in stromal and vascular cells that provide heterotypic support for the tumor. An advantage of targeting these accessory cells is that, unlike tumor cells, they tend to be genetically more stable and therefore are less likely to evolve drug resistance. However, in certain circumstances tumors may be able to evolve reduced dependency on these accessory cells.
The trade-off for the tumorigenic state is that tumor cells experience numerous cellular stresses not experienced by normal cells and therefore tumor cells are more dependent on stress support pathways for their survival. In principle, there are two approaches to exploit this dependency to selectively kill tumor cells. The first approach, stress sensitization, aims to diminish the activity of the stress support pathways such that the tumor cell can no longer cope with the stress of its oncogenic state and either ceases to proliferate or initiates apoptosis or necrosis. The second approach, stress overload, aims to exacerbate existing oncogenic stress in order to overwhelm the stress support pathways in the tumor cell, leading to growth arrest or cell death. Both approaches, therefore, disrupt the balance of pro- and antisurvival signaling to the detriment of tumor cells. Examples of specific types of NOA are illustrated in Figure 2 and discussed below.
Figure 2. Examples of Non-oncogene Addictions in Cancer Cells.

The tumorigenic state results in a variety of alterations (shown on top), which are related to the hallmarks described in Figure 1. These alterations give rise to a number of potentially deleterious circumstances or vulnerabilities (detailed in the bottom half) that could be lethal to the tumor cells if left unchecked. The existence of stress support pathways (shown in red) help suppress this lethality. Many of these pathways are examples of non-oncogene addiction (NOA), and therapeutics that interfere with their functions could display synthetic lethality with the tumor genotype/phenotype.
Intrinsic Non-oncogene Addiction
DNA Damage and Replication Stress
In nearly all tumor cell types, the rate of spontaneous DNA damage and the degree of replication stress are enhanced. The presence of oncogenes can also elicit substantial DNA damage (Halazonetis et al., 2008). DNA damage stress can be exploited therapeutically through both stress sensitization and stress overload. An elaborate DNA damage response (DDR) pathway exists in the cell to ameliorate the effects of DNA damage by promoting DNA repair. Mutations in genes of this pathway result in increased sensitivity to DNA damage (Harper and Elledge, 2007). In principle, cells experiencing spontaneous DNA damage should show enhanced sensitivity to agents that interfere with this stress response pathway (Figure 2). In support of this notion, recent studies have shown that inhibitors of the DDR kinases ATM and Chk1 exhibit selective toxicity toward cancer cells (Chen et al., 2006; Kennedy et al., 2007). Although it seems counterintuitive that a pathway that normally serves to restrain proliferation and promote apoptosis would protect a cancer cell, the DNA repair and genomic stability afforded by this pathway could save a cancer cell from death caused by persistent DNA damage and replication stress.
Stress overload of the DDR pathway should also show efficacy in cancer treatment. Although it is not clear why DNA-damaging agents, such as IR and chemotherapy, are effective cancer therapies, it is possible that these are examples of stress overload, where cancer cells with already elevated levels of DNA damage and replication stress cannot repair the additional damage inflicted by these agents. An alternative explanation is that during tumorigenesis, the persistence of DNA damage selects for cells with mutations that abrogate part of the DDR pathway and therefore cannot properly sense and respond to DNA damage. These cells with a partially defective DDR might therefore be more vulnerable to the extensive DNA damage resulting from radiation or chemotherapy that is lethal without a normal DDR pathway. In this context, DNA damage exploits a stress phenotype of tumors that is analogous to non-oncogene addition.
Given the sensitivity of many cancers to DNA-damaging agents, there should exist genes whose inhibition will generate endogenous DNA damage to exacerbate this sensitivity. Exemplifying this phenomenon are cancers bearing BRCA2 mutations. These tumors are defective in homologous recombination-mediated DNA repair and are particularly sensitive to DNA crosslinkers such as cisplatin (Sakai et al., 2008). Furthermore, these tumors rely heavily on other forms of DNA repair such as base-excision repair, resulting in sensitivity to inhibitors of poly-ADP-ribose polymerase (PARP1), an enzyme that facilitates repair of single-stranded breaks (Bryant et al., 2005; Farmer et al., 2005). In normal cells, the endogenous DNA damage generated by PARP inhibition is well-tolerated because of functional compensation from homologous recombination-mediated repair. Indeed, mice deficient in PARP1 are fertile and do not exhibit increased tumor susceptibility (Conde et al., 2001). The addiction of BRCA2 mutant cells to PARP1 function is therefore an example of NOA that is exploited therapeutically. There are likely to be additional non-oncogene targets like PARP1 that can be exploited to treat these tumors. Likewise, the DNA damage sensitivity of other cancer types might also be exploited in this manner, although whether generating endogenous damage is superior to adding exogenous damage remains to be determined.
Mitotic Stress
As noted above, a variety of tumors display increased rates of chromosome mis-segregation, the CIN phenotype, because they contain mutations that alter the fidelity of mitosis. Cancer cells bearing mitotic alterations such as aneuploidy or weakened mitotic spindle regulatory machinery are likely to exhibit greater reliance on stress support pathways for proper chromosome segregation. They are therefore more sensitive to catastrophic genomic instability caused by stress overload or stress sensitization of the mitotic machinery (Weaver and Cleveland, 2005).
The spindle checkpoint can provide stress support in cells that have defects in the mitotic machinery, and in such cases inhibiting the spindle checkpoint could lead to lethality as a result of stress sensitization. Conversely, some tumors with the CIN phenotype possess mutations that weaken the spindle checkpoint (Cahill et al., 1998) and might be especially sensitive to stress overload caused by the inhibition of proteins that carryout mitosis. A notable example of stress overload is the microtubule stabilizer taxol, which interferes with proper spindle-kinetochore attachment and has shown efficacy in the treatment of breast and ovarian cancers (Weaver and Cleveland, 2005). Inhibitors of mitotic kinases such as PLK1 and Aurora-B that also promote stress overload are currently undergoing clinical trials (Carpinelli and Moll, 2008; Strebhardt and Ullrich, 2006). Aneuploidy itself might be a target of NOA as genetic screens in yeast have identified mutations that show synthetic lethality with tetraploid verses diploid cells (Storchová et al., 2006).
Proteotoxic Stress
Proteotoxic stress is a frequent occurrence in cancer cells as a result of the extreme aneuploidy, copy-number variation, and transcriptional alterations present in tumors. These changes alter the dosage balance of protein subunits in different protein complexes and consequently place increased stress on chaperone pathways to maintain normal homeostasis of the proteome. The heat shock pathway plays a major role in promoting protein folding and is activated in many tumors (Whitesell and Lindquist, 2005) and is therefore a potential NOA target (Figure 2). Studies in yeast have shown that an extra copy of a single chromosome is sufficient to activate the heat shock response (Torres et al., 2007). Pharmacological and genetic evidence supports the notion that sensitizing tumor cells toward proteotoxic stress can strongly suppress tumorigenesis. For example, the chaperone HSP90 acts to fold newly synthesized proteins and refold mis-folded proteins (Whitesell and Lindquist, 2005). The chaperone activity of HSP90 requires its ATPase activity, which can be inhibited by the anticancer drug geldanamycin. Although the antitumor activity of geldanamycin has been attributed to the destabilization of key HSP90 client proteins involved in cell proliferation such as CDK4 and HER2, it is also possible that the sensitization of tumor cells to elevated general proteotoxic stress leads to its efficacy.
Additional genetic support for NOA comes from studies of HSF1 knockout mice (Dai et al., 2007). HSF1 is the major transcription factor responsible for activating the expression of heat shock proteins, including HSP90, in response to excess unfolded proteins. Loss of HSF1 markedly reduces tumorigenesis driven by either p53 or Ras mutations (Dai et al., 2007). Given that neither HSP90 nor HSF1 have been shown to be oncogenes, they represent examples of NOA.
Unfolded proteins are generally turned over by the ubiquitin-proteasome pathway. Thus, a second key component of the proteotoxic stress response pathway is the proteasome. Genetic experiments have shown that the presence of an extra chromosome is sufficient to increase the sensitivity of yeast cells to proteasome inhibitors (Torres et al., 2007). One promising new anticancer drug bortezomib (Velcade), which has shown efficacy in treating multiple myeloma, acts to inhibit the protease function of the proteasome (Richardson et al., 2006). As proteasome inhibition is likely to stabilize many different proteins, it has been traditionally thought that the stabilization of a particular subset of proteins might be responsible for the antitumorigenic activity of Velcade. However, it is equally likely that its efficacy results from enhanced proteotoxic stress leading to stress overload and represents a prime example of NOA.
Stress sensitization and stress overload are two sides of the same coin. In principle, instead of sensitizing cancer cells toward endogenous proteotoxic stress, therapies that enhance protein unfolding should overload the stress support network in cancer cells, achieving the same result. Normal cells, on the other hand, should be less sensitive to such therapies due to their low baseline proteotoxic stress. A convenient method of inducing protein unfolding is temperature elevation. Indeed, hyperthermia is being extensively investigated as a tumor treatment for colon, breast, testicular, prostate, and liver cancers and has been the subject of ongoing clinical trials, often in combination with chemotherapy (Fiorentini and Szasz, 2006). The induction of protein unfolding by hyperthermia could provide a rationale for the efficacy of this approach. Strong support for this explanation again comes from yeast where it was shown that the presence of just one extra chromosome results in enhanced temperature sensitivity, consistent with a general sensitivity to temperature for aneuploid cells (Torres et al., 2007).
Metabolic Stress
Tumor cells exhibit altered metabolic behavior due to both cell-intrinsic properties and the tumor microenvironment. As noted above, tumor cells have increased glucose uptake and preferentially metabolize glucose through glycolysis even in the presence of oxygen. Although the reason for the Warburg effect is unclear, it has been suggested that such adaptation allows the tumor cell to divert resources toward biosynthesis, reduce the generation of ROS through mitochondrial oxidative phosphorylation, and cope with fluctuating oxygen availability in the tumor vicinity (DeBerardinis et al., 2008; Kroemer and Pouyssegur, 2008). Inhibition of the glycolytic/biosynthetic pathways in a tumor cell will lead to stress overload due to incompatibility with the persistent proliferative drive from oncogenes (Vander Heiden et al., 2001). Indeed, inhibition of ATP citrate lyase (which synthesizes acetyl-CoA from citrate), RNAi knockdown of lactate dehydrogenase A (the enzyme that converts pyruvate to lactate in the last step of glycolysis), and inhibitors against acetyl-CoA carboxylase and fatty acid synthase (which control the conversion of acetyl-CoA to manonyl-CoA to palmitate) all lead to substantial attenuation of tumor cell growth (DeBerardinis et al., 2008; Kroemer and Pouyssegur, 2008). Recently, it has been shown that the mere switching of pyruvate kinase from one splice variant (M2) commonly expressed in tumor cells to another splice variant (M1) can negatively impact the tumorigenic state, presumably by shunting pyruvate to the TCA cycle (Christofk et al., 2008). Thus targeting key metabolic enzymes that constitute NOA could effectively attenuate tumor cell proliferation.
Oxidative Stress
Tumor cells often show increased levels of intracellular ROS (Szatrowski and Nathan, 1991). Multiple causes contribute to ROS generation. Hypoxia-reperfusion in the tumor microenvironment can lead to ROS production, which in turn can initiate a viscous cycle of mitochondrial damage and further ROS generation (Gogvadze et al., 2008). In addition, oncogenes such as Ras can stimulate ROS production, a mechanism that has been implicated as an underlying cause of oncogene-induced senescence (Lee et al., 1999). ROS cause oxidative damage to DNA, proteins, lipids, and other cellular components and therefore pose a significant cellular stress (Figure 1). Cancer cells partially alleviate this stress by engaging glycolysis and downregulating mitochondrial function (Gogvadze et al., 2008). Agents that enhance ROS production, therefore, are expected to cause stress overload in cancer cells. In support of this notion, it has been shown that dichloroacetate, which inhibits pyruvate dehydrogenase kinase (PDK) and therefore stimulates mitochondrial oxidative phosphorylation and ROS production, can selectively elicit apoptosis in cancer cells but not in normal cells (Bonnet et al., 2007). Similarly, reducing the cellular ROS buffering capacity through the inhibition of glutamate-cysteine ligase (a rate-limiting enzyme in cellular glutathione synthesis) can markedly increase the radiosensitivity of cancer cells (Diehn et al., 2009).
Hypoxia and Nutrient Stress
Solid tumors, due to poor and abnormal tumor vasculature (see below), often suffer from suboptimal oxygen and nutrient supplies. This stress underlies the requirement for augmented angiogenesis (Figure 1). Consequently many tumors upregulate the transcription factor hypoxia-inducible factor HIF-1 (Pouyssegur et al., 2006). HIF-1 initiates a coordinated transcriptional program to both stimulate vessel sprouting (through the induction of VEGF-A and angiopoietin-2) and promote glycolysis (through the induction of glucose transporter 1, hexokinase, lactate dehydrogenase, and pyruvate dehydrogenase kinase 1). Furthermore, HIF-1 protects tumor cells from acidosis and promotes the extrusion of lactate by inducing carbonate anhydrases, monocarboxylate transporter MCT4, and the Na+/H+ exchanger NHE1. Drugs targeting either HIF-1 or its effector pathways will therefore sensitize tumor cells toward hypoxic stress. Indeed, anti-angiogenic therapies have been effective in various clinical settings (see below). Inhibiting the monocarboxylate transporter, which would result in intracellular lactate accumulation and acidosis, might also be a viable approach to selectively kill hypoxic tumor cells addicted to glycolysis (Pouyssegur et al., 2006).
As tumor cells grow away from the vasculature system, they experience nutrient stress and can resort to autophagy for survival. Although impaired autophagy could contribute to oncogenesis by promoting genomic instability and cytokine release (Mathew et al., 2007), autophagy is critical for the survival of apoptosis-resistant tumor cells during nutrient or growth factor restriction. Inhibiting autophagy in such cells would sensitize them toward metabolic stress and promote necrotic death. In support of this notion, the autophagy inhibitor chloroquine has been shown to synergize with DNA damage to induce tumor cell death in mice (Jin and White, 2007).
Immune Response Modulation
Most tumors express antigens that could potentially elicit an immune response due to the expression of a mutant protein that gives rise to a novel epitope. However, in the vast majority of cases tumors are able to prevent rejection by effectively suppressing an immune response in the tumor microenvironment (Muller and Scherle, 2006; Zou, 2005). Although not a stress phenotype, there are examples of NOA that can be used to reverse this hallmark. Several mechanisms protect tumor cells from immune surveillance. Tumor cells can downregulate the expression of major histocompatibility complex (MHC) molecules to reduce antigen presentation (Garrido and Algarra, 2001). Tumor cells also synthesize and release a number of immunomodulatory factors such as chemokines (e.g., CCL2), cytokines (e.g., IL6 and IL10), and prostaglandins (e.g., PGE2) to suppress the activation of cytotoxic T cells (Sharma et al., 2005). Furthermore, the depletion of key metabolites in the tumor microenvironment, such as glucose, also inhibits cytotoxic T cell expansion (Muller and Scherle, 2006).
The tumor's addiction to “immunosuppressive” NOA genes could be exploited for therapeutic gains. For example, the cycloxygenase-2 (COX2) inhibitor celecoxib, which blocks PGE2 synthesis, has been shown to enhance immune response to tumors in mice (Stolina et al., 2000). Reactivating MHC gene expression in tumor cells that have downregulated MHC, perhaps by inhibiting a histone deacetylase (HDAC) or a transcriptional repressor, is yet another potential means to enhance tumor immune surveillance. Active immunization against tumor antigens has been shown to be greatly enhanced by interfering with inhibitory T cells through the use of anti-CTLA-4 antibodies, providing another example of NOA-like therapy (Peggs et al., 2008). A major attraction of re-establishing the antitumor immune response is that one could potentially apply it in a systematic manner toward many tumor types and allow the immune system to detect and eradicate tumors at a relatively early stage.
Extrinsic Non-oncogene Addiction
Angiogenesis
One of the best examples of extrinsic NOA is the ability of solid tumors to recruit new blood vessels through the secretion of angiogenic factors. Tumor angiogenesis ensures that cells in the interior of the tumor receive sufficient nutrients and oxygen to survive. Blocking tumor angiogenesis would therefore severely restrict tumor growth (Folkman, 2007). Early experiments using mouse xenografts indicated that antibody-mediated inhibition of vascular endothelial growth factor (VEGF), which promotes the proliferation and migration of vascular endothelial cells and vessel sprouting, could severely constrain angiogenesis and tumor growth (Kim et al., 1993). These and other studies led to the development of the anti-VEGF antibody bevacizumab for therapeutic use (Ferrara et al., 2004). Additionally, the small-molecule inhibitors sorafenib and sunitinib, which target multiple receptor tyrosine kinases including VEGF receptors, have shown efficacy in treating metastatic renal cell carcinoma (Escudier et al., 2007; Motzer et al., 2006). In principle, any protein required for angiogenesis in endothelial cells is a candidate for extrinsic NOA.
Stromal Support
The tumor stroma plays a key role in supporting tumor growth (Hu and Polyak, 2008). It is increasingly clear that heterotypic interactions between the tumor and its surrounding stromal cells lead to phenotypic changes in the stroma that better support tumor growth. For example, the myofibroblasts surrounding breast tumors are distinct from normal mammary stromal fibroblasts in their ability to secrete stromal-derived factor 1 to both stimulate tumor growth and recruit endothelial progenitor cells for angiogenesis (Orimo et al., 2005). This phenotypic distinction is further supported by substantial gene expression and epigenetic changes observed in these cancer-associated stromal cells (Allinen et al., 2004). In addition, bone marrow-derived mesenchymal stem cells and hematopoietic progenitors have been found to play important roles in tumor metastasis either through cytokine-mediated paracrine signaling or through their mobilization to distal sites to help establish the metastatic niche, respectively (Kaplan et al., 2005; Karnoub et al., 2007). Although somatic mutations in tumor stromal cells are rare (Allinen et al., 2004; Qiu et al., 2008), in the case of familial neurofibromatosis 1 caused by mutations in the NF1 gene, careful modeling of the disease in mice has revealed a striking contribution of mast cells to tumor development. The contribution of NF1 heterozygous mast cells and bone marrow cells to the disease phenotype is dependent on signaling by c-kit in these cells, a receptor readily inhibited by imatinib. Indeed, imatinib was successfully applied to reduce the tumor burden of an NF1 patient (Yang et al., 2008), thus demonstrating the utility of targeting tumor-extrinsic NOA pathways for cancer therapy. We have used the stress phenotypes to illustrate NOA; however, it is important to realize that other examples of NOA pathways exist unrelated to stress that when inhibited will also be incompatible with the oncogenic state.
The Path Ahead to New Cancer Therapeutics
We are approaching a new era in cancer research as we now have the tools to mount a systematic assault on the problem of tumorigenesis in terms of both understanding the underlying pathogenic process and devising new therapies to treat cancer. Below we explore new approaches for identifying cancer drug targets and provide theoretical considerations of how therapies attacking cancer might be optimally applied.
Approaches to Target Identification
Having outlined the different classes of potential cancer targets based on their underlying cellular functions, what is the best approach that we as cancer researchers can take to quickly generate therapies that are so desperately needed? Clearly target identification and patient stratification, that is, the ability to group patients with common biomarkers to link them to the appropriate therapy, will play a major part in the evolution of new therapies. Thus, new biomarkers and means to identify therapeutic targets are needed. In a general sense, the approaches that served us so well in the first era of cancer research—the painstaking identification of individual genes and attempts to exploit them to cure cancer—will likely not suffice for the next era of cancer research. Systems-level approaches are needed to identify the vulnerabilities of cancer cells in a genome-wide and unbiased manner. Two such complementary and mutually reinforcing efforts are underway and should systematically identify candidate drug targets, providing a blueprint for cancer drug target discovery.
The first is the traditional approach to identify genetic lesions in cancer cells and exploit this knowledge for therapeutics, a tried and true method that has led to the identification of several drug targets. Such an approach is currently employed by The Cancer Genome Atlas and other similar efforts that aim to characterize, in a large number of cancer types, genomic alterations including copy-number variation, transcriptional profiles, epigenetic modifications, and DNA sequence alterations. While expensive, this approach has the potential to identify common alterations in oncogenes and tumor suppressors for further functional analysis and uncover oncogene addiction pathways that can be targeted (Cancer Genome Atlas Research Network, 2008; Jones et al., 2008; Parsons et al., 2008). It is clear that these efforts will yield important new information on human cancer and aid biomarker discovery and patient stratification. However, this approach has two limitations with respect to target discovery. First, current sequencing capacity is primarily limited to coding regions, though whole-genome sequencing will become feasible with cheaper and more highly parallel sequencing methods. The second limitation is that this approach will not discover NOA pathways that support tumorigenesis, such as those discussed above, because they are not mutated in cancers.
The second approach takes a more functional, systems-biology approach to identify cancer cell vulnerabilities (Ngo et al., 2006; Schlabach et al., 2008; Silva et al., 2008). This has been made possible by the RNA interference (RNAi) revolution. Advances in RNAi technology in mammalian cells have made it possible, for the first time, to systematically interrogate every cellular protein for potential addiction by cancer cells. A functional approach using RNAi offers several advantages in target discovery. First, RNAi-mediated protein knockdown results in either partial or complete loss of function, which in many cases closely mimics the effect of an inhibitor. Second, RNAi can be applied to any annotated gene in the human genome based on sequence information alone without the a priori knowledge of the gene's cellular function or mutational status in cancer. Therefore, this approach will uncover both known and new genes that were not previously implicated in cancer. Third, RNAi can be readily applied to animal models (Dickins et al., 2007). This provides rapid, in vivo target validation that takes advantage of the many mouse models of cancer, without the need to first develop an inhibitor. Lastly and importantly, genome-wide RNAi screening will identify genes that are not mutated in cancers and exhibit features of NOA. Systematic RNAi screens aimed to generate either “synthetic lethal signatures” against specific oncogenes or tumor suppressors or “cancer lethal signatures” using a large panel of cancer and normal cells should reveal a genetic landscape of cancer vulnerabilities that guide the rational design of therapeutic approaches.
A current limitation of the RNAi approach is the fact that many screens are being performed in cell lines in vitro and are limited to analysis of genes important for proliferation and survival. Thus, these screens will miss certain classes of genes that might function only in the proper in vivo tumor environment. This limitation can be overcome, however, if large-scale genetic screens can be approached systematically in mice with defined cancer models. Continuous RNAi library development that aims to improve gene knockdown efficiency and incorporate features (such as barcodes) that enable very high throughput screening will further enhance the power of this approach. These efforts would require a sustained commitment of resources, such as an organized consortium effort, but would result in the identification and validation of critical cancer drug targets in a relatively unbiased, comprehensive fashion. Given the long-standing success of genetics in providing unbiased insights into biology and disease mechanisms, our goal should be nothing less than identifying all the pathways upon which cancer cells depend to maintain the oncogenic state.
The identification of drug targets is only the first step in the process to develop new cancer therapeutics. The ability to identify or design small-molecule inhibitors with the appropriate pharmacological properties against any given target is the next challenge. Currently, many efforts focus only on the so-called “druggable genome,” which represents proteins with enzymatic functions that can be easily assayed or targeted. However, recent studies have shown that many proteins previously thought to be difficult to target can, in fact, be inhibited by small molecules (Wells and McClendon, 2007). New methods of identifying small molecules based on their ability to bind specific proteins and disrupt protein-protein interaction should widen the playing field and allow more validated targets that emerge from physical mapping and genetic screens to enter the drug development pipeline. Of course, it is widely hoped that RNAi itself will become a cancer therapy in the near future once a solution to its effective delivery to tumors is found. If so, this will circumvent many limitations inherent to small-molecule inhibitors and revolutionize drug development.
Mathematical Elimination of Cancer using Orthogonal Therapies
It is very likely that the oncogenes and non-oncogenes to which tumors are addicted will serve as the targets of successful cancer therapies in the future. However, it is already clear that each of even the best therapies applied alone eventually fail in the majority of cases. Each therapy can be considered to be a filter that removes a set of cancer cells with certain properties (Figure 3). However, a subset of tumor cells slip through the filter to eventually establish clones, rendering the therapy ineffective. These rare, pre-existing mutant cells contain suppressor mutations that circumvent the therapy. Examples of suppressor mutations might include an altered drug-binding site on the intended target, amplification of the drug target, activation of drug efflux pumps, or activation of an alternative pathway, such as a growth factor signaling pathway, that performs the same function as the inhibited pathway. The rate of appearance of suppressor mutations is an important parameter in cancer therapies. Unfortunately, one of the inherent properties of cancer cells is their enhanced genomic instability, which accelerates the appearance of suppressor mutations.
Figure 3. The Combinatorial Filter of Orthogonal Cancer Therapies.
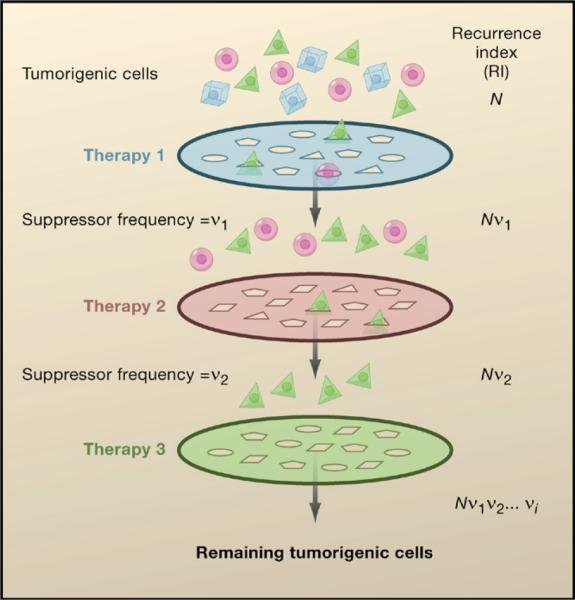
A tumor consists of genetically distinct subpopulations of cancer cells (represented by the different cell shapes), each with its own characteristic sensitivity profile to a given therapeutic agent. Each cancer therapy can be viewed as a filter that removes a subpopulation of cancer cells that are sensitive to this treatment while allowing other insensitive subpopulations to escape. This escape occurs as a result of suppressor mutations that occur at a given frequency (v) unique to each therapy and tumor type. By combining therapies with orthogonal modes of action, a combinatorial filter can be set up to minimize the recurrence index (RI) of the cancer. N represents the total number of cancer cells in the tumor. A combination of orthogonal therapies that result in RI < 1 would greatly enhance the likelihood of preventing tumor recurrence.
From a theoretical perspective, in order to overcome this problem, a combinatorial series of filters applied concurrently is needed to eliminate all of the cancer cells in a patient. How this will be accomplished remains to be determined, but we envision it to be the simultaneous application of orthogonal cancer therapies. Two therapies are considered orthogonal, and therefore act synergistically, when they attack a cancer in two different ways such that a suppressor mutation for the first therapy cannot suppress the second therapy and vice versa. It should be noted that some non-orthogonal therapies might also have efficacy in combination if only a portion of the suppressors can overcome both therapies. This leads to the simple mathematical proposition that the probability of cancer escaping the therapies, the recurrence index, RI, will be proportional to the number of cancer-initiating cells, N, times the frequency of suppressor mutations for therapy 1, v1, times the frequency of suppressor mutations for therapy 2, v2, and so on to produce RI = Nv1v2…vi. Once RI becomes much less than 1, the probability of surviving the cancer is high. Importantly, this general treatment takes full consideration of tumor cell heterogeneity such as the existence of “cancer stem cells” that might have a different property than the bulk of the tumor. Although the values of the parameters in this equation are not currently known and are likely to vary based on tumor type, it is clear that such an approach can work as it is analogous to the treatment of HIV. For HIV, which resembles tumors in that it is highly mutable, a triple cocktail of inhibitors of HIV replication can control the progression of the disease. We envision that cancer therapies will adopt this calculus and convert cancer, like AIDS, from a terminal illness to a chronic disease or even provide a cure. Given that cancer is actually a collection of distinct diseases, the orthogonal combinations will vary depending upon the tumor genotype and possibly the genotype of the patient. It is likely that the treatment regimen employed will be determined by the analysis of a series of biomarkers in each tumor that have been previously shown to predict efficacy of a particular therapy. Combinatorial therapies are currently being considered for a number of cancers. However, it will be important to understand the nature and frequency of suppressors that allow escape from each therapy when designing combinations in order to maximize the orthogonality of the combinatorial therapies. In addition, it is critical that such therapies, when tolerated by patients, be applied concurrently, not sequentially, because the latter will allow the number of cancer-initiating cells, N, to expand prior to the subsequent treatment, thereby increasing the RI. Finally, it should be noted that if one of the therapies employed is a DNA-damaging agent, it might increase the frequency of suppressor mutations for the other therapies with which it is combined.
In summary, there are many avenues through which to approach future cancer therapies. We hope these additional hallmarks depicting the stress phenotypes of cancer cells will both improve our understanding of the efficacy of existing therapies and provide rationales for new therapeutics. Cancer therapeutic targets can emerge through both the analysis of classical oncogenes and the analysis of a new class of genes that constitute NOA. New genome-wide efforts to approach cancer from a systems-biology perspective hold great promise for identifying new targets for anticancer therapies. Finally, we propose that through the proper combination of orthogonal cancer therapies, it is possible to convert cancer from a death sentence into a manageable or even curable disease.
ACKNOWLEDGMENTS
We would like to thank O. Cohen-Fix, J. Glaven, S.M. Lewis, W.G. Kaelin, and G.M. Wahl for critical reading of the manuscript. J.L. is supported by a fellowship from the American Association of Cancer Research. N.L.S. is a fellow of the Susan G. Komen for the Cure Foundation. This work is supported by grants from the NIH and the U.S. Department of Defense to S.J.E. S.J.E. is an investigator with HHMI. We dedicate this review to I. Bernard Weinstein for his contributions to cancer biology.
REFERENCES
- Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17, 32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Ashwell S, Zabludoff S. DNA damage detection and repair pathways-recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin. Cancer Res. 2008;14:4032, 4037. doi: 10.1158/1078-0432.CCR-07-5138. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864, 870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633, 637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37, 51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913, 917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300, 303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061, 1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlo-Stella C, Lavazza C, Locatelli A, Vigano L, Gianni AM, Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clin. Cancer Res. 2007;13:2313, 2317. doi: 10.1158/1078-0432.CCR-06-2774. [DOI] [PubMed] [Google Scholar]
- Carpinelli P, Moll J. Aurora kinases and their inhibitors: more than one target and one drug. Adv. Exp. Med. Biol. 2008;610:54, 73. doi: 10.1007/978-0-387-73898-7_5. [DOI] [PubMed] [Google Scholar]
- Chen Z, Xiao Z, Gu WZ, Xue J, Bui MH, Kovar P, Li G, Wang G, Tao ZF, Tong Y, et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer. 2006;119:2784, 2794. doi: 10.1002/ijc.22198. [DOI] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O'Hagan R, Pantginis J, Zhou H, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468, 472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230, 233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806, 810. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- Conde C, Mark M, Oliver FJ, Huber A, de Murcia G, Menissier-de Murcia J. Loss of poly(ADP-ribose) polymerase-1 causes increased tumour latency in p53-deficient mice. EMBO J. 2001;20:3535, 3543. doi: 10.1093/emboj/20.13.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005, 1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA. 2007;104:19345, 19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11, 20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Denko NC, Giaccia AJ, Stringer JR, Stambrook PJ. The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc. Natl. Acad. Sci. USA. 1994;91:5124, 5128. doi: 10.1073/pnas.91.11.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006;8:1053, 1063. doi: 10.1038/ncb1471. [DOI] [PubMed] [Google Scholar]
- Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer. 2008;8:425, 437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638, 642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Dickins RA, McJunkin K, Hernando E, Premsrirut PK, Krizhanovsky V, Burgess DJ, Kim SY, Cordon-Cardo C, Zender L, Hannon GJ, et al. Tissue-specific and reversible RNA interference in transgenic mice. Nat. Genet. 2007;39:914, 921. doi: 10.1038/ng2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009 doi: 10.1038/nature07733. Published online February 4, 2009. 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikmen ZG, Gellert GC, Jackson S, Gryaznov S, Tressler R, Dogan P, Wright WE, Shay JW. In vivo inhibition of lung cancer by GRN163L: a novel human telomerase inhibitor. Cancer Res. 2005;65:7866, 7873. doi: 10.1158/0008-5472.CAN-05-1215. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069, 1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 2003;3:11, 22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Druker BJ. Inhibition of the Bcr-Abl tyrosine kinase as a therapeutic strategy for CML. Oncogene. 2002;21:8541, 8546. doi: 10.1038/sj.onc.1206081. [DOI] [PubMed] [Google Scholar]
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007;356:125, 134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917, 921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell. 1999;4:199, 207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004;3:391, 400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- Fiorentini G, Szasz A. Hyperthermia today: electric energy, a new opportunity in cancer treatment. J. Cancer Res. Ther. 2006;2:41, 46. doi: 10.4103/0973-1482.25848. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007;6:273, 286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007;17:157, 162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Garrido F, Algarra I. MHC antigens and tumor escape from immune surveillance. Adv. Cancer Res. 2001;83:117, 158. doi: 10.1016/s0065-230x(01)83005-0. [DOI] [PubMed] [Google Scholar]
- Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 2008;18:165, 173. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr., Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907, 913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153, 158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9, 22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat. Rev. Cancer. 2002;2:331, 341. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352, 1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57, 70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harley CB. Telomerase and cancer therapeutics. Nat. Rev. Cancer. 2008;8:167, 179. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol. Cell. 2007;28:739, 745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821, 1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- Hu M, Polyak K. Microenvironmental regulation of cancer development. Curr. Opin. Genet. Dev. 2008;18:27, 34. doi: 10.1016/j.gde.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat. Genet. 2000;24:57, 60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer. 2005;5:341, 354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Iida S, Rao PH, Butler M, Corradini P, Boccadoro M, Klein B, Chaganti RS, Dalla-Favera R. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 1997;17:226, 230. doi: 10.1038/ng1097-226. [DOI] [PubMed] [Google Scholar]
- Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102, 104. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28, 31. doi: 10.4161/auto.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801, 1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer. 2005;5:689, 698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820, 827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557, 563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, Shimamura A, D'Andrea AD. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J. Clin. Invest. 2007;117:1440, 1449. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841, 844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- Komarova NL, Lengauer C, Vogelstein B, Nowak MA. Dynamics of genetic instability in sporadic and familial colorectal cancer. Cancer Biol. Ther. 2002;1:685, 692. doi: 10.4161/cbt.321. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13:472, 482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu ZX, Ferrans VJ, Howard BH, Finkel T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999;274:7936, 7940. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- Lee YM, Sicinski P. Targeting cyclins and cyclin-dependent kinases in cancer: lessons from mice, hopes for therapeutic applications in human. Cell Cycle. 2006;5:2110, 2114. doi: 10.4161/cc.5.18.3218. [DOI] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003;3:330, 338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brach-mann S, Chene P, De Pover A, Schoemaker K, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008;7:1851, 1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323, 1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Maser RS, DePinho RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565, 569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367, 1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JJ. Anticancer antifolates: current status and future directions. Curr. Pharm. Des. 2003;9:2593, 2613. doi: 10.2174/1381612033453712. [DOI] [PubMed] [Google Scholar]
- Moreira JN, Santos A, Simoes S. Bcl-2-targeted antisense therapy (Oblimersen sodium): towards clinical reality. Rev. Recent Clin. Trials. 2006;1:217, 235. doi: 10.2174/157488706778250050. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2006;24:16, 24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat. Rev. Cancer. 2006;6:613, 625. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- Nava-Parada P, Emens LA. GV-1001, an injectable telomerase peptide vaccine for the treatment of solid cancers. Curr. Opin. Mol. Ther. 2007;9:490, 497. [PubMed] [Google Scholar]
- Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106, 110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335, 348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Papp B, Pal C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194, 197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807, 1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggs KS, Quezada SA, Allison JP. Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol. Rev. 2008;224:141, 165. doi: 10.1111/j.1600-065X.2008.00649.x. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Littlewood T, Khan M, Elia G, Evan G. Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol. Cell. 1999;3:565, 577. doi: 10.1016/s1097-2765(00)80350-0. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl. Acad. Sci. USA. 2002;99:12963, 12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789, 802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437, 443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, Trivett M, Thompson ER, Ramakrishna M, Gorringe KL, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat. Genet. 2008;40:650, 655. doi: 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat. Rev. Drug Discov. 2007;6:834, 848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu. Rev. Med. 2006;57:33, 47. doi: 10.1146/annurev.med.57.042905.122625. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291, 3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Roccaro AM, Hideshima T, Richardson PG, Russo D, Ribatti D, Vacca A, Dammacco F, Anderson KC. Bortezomib as an antitumor agent. Curr. Pharm. Biotechnol. 2006;7:441, 448. doi: 10.2174/138920106779116865. [DOI] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116, 1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008;319:620, 624. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selivanova G, Wiman KG. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene. 2007;26:2243, 2254. doi: 10.1038/sj.onc.1210295. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454:226, 231. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, Huang M, Batra RK, Dubinett SM. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005;65:5211, 5220. doi: 10.1158/0008-5472.CAN-05-0141. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214, 3231. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 2007;7:169, 181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85, 88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265, 7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- Silva JM, Marran K, Parker JS, Silva J, Golding M, Schlabach MR, Elledge SJ, Hannon GJ, Chang K. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319:617, 620. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268, 274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986, 988. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Spira AI, Carducci MA. Differentiation therapy. Curr. Opin. Pharmacol. 2003;3:338, 343. doi: 10.1016/s1471-4892(03)00081-x. [DOI] [PubMed] [Google Scholar]
- Stauffer SR. Small molecule inhibition of the Bcl-X(L)-BH3 protein-protein interaction: proof-of-concept of an in vivo chemopotentiator ABT-737. Curr. Top. Med. Chem. 2007;7:961, 965. doi: 10.2174/156802607780906843. [DOI] [PubMed] [Google Scholar]
- Stolina M, Sharma S, Lin Y, Dohadwala M, Gardner B, Luo J, Zhu L, Kronenberg M, Miller PW, Portanova J, et al. Specific inhibition of cyclooxygenase 2 restores antitumor reactivity by altering the balance of IL-10 and IL-12 synthesis. J. Immunol. 2000;164:361, 370. doi: 10.4049/jimmunol.164.1.361. [DOI] [PubMed] [Google Scholar]
- Storchová Z, Breneman A, Cande J, Dunn J, Burbank K, O'Toole E, Pellman D. Genome-wide genetic analysis of polyploidy in yeast. Nature. 2006;443:541, 547. doi: 10.1038/nature05178. [DOI] [PubMed] [Google Scholar]
- Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer. 2006;6:321, 330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- Su Z, Dannull J, Yang BK, Dahm P, Coleman D, Yancey D, Sichi S, Niedzwiecki D, Boczkowski D, Gilboa E, et al. Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J. Immunol. 2005;174:3798, 3807. doi: 10.4049/jimmunol.174.6.3798. [DOI] [PubMed] [Google Scholar]
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794, 798. [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916, 924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Torres EM, Williams BR, Amon A. Aneuploidy: cells losing their balance. Genetics. 2008;179:737, 746. doi: 10.1534/genetics.108.090878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, Zeng Z, Liu H, Krier C, Stengel RF, Barany F, et al. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006;66:2129, 2137. doi: 10.1158/0008-5472.CAN-05-2569. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell. Biol. 2001;21:5899, 5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007;13:23, 31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661, 665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Verdine GL, Walensky LD. The challenge of drugging undrug-gable targets in cancer: lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 2007;13:7264, 7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309, 314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7, 12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes-the Achilles heal of cancer. Science. 2002;297:63, 64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077, 3080. doi: 10.1158/0008-5472.CAN-07-3293. [DOI] [PubMed] [Google Scholar]
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001, 1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer. 2005;5:761, 772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703, 709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windle B, Draper BW, Yin YX, O'Gorman S, Wahl GM. A central role for chromosome breakage in gene amplification, deletion formation, and amplicon integration. Genes Dev. 1991;5:160, 174. doi: 10.1101/gad.5.2.160. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108, 1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656, 660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/−- and c-kit-dependent bone marrow. Cell. 2008;135:437, 448. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer. 2005;5:263, 274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]