

Abstract
Purpose of review
A significant proportion of hearing loss and deafness is caused by defects in the structure or function of cells within the organ of Corti. Identification of the molecular factors that regulate the development of this structure should provide valuable insights regarding inner ear formation and the signaling pathways that underlie congenital auditory deficits. In addition, targeted modulation of these same factors could be developed as therapies for hair cell regeneration.
Recent findings
Results from experiments using transgenic and mutant mice, as well as in vitro techniques, have identified genes and signaling pathways that are required to either specify unique auditory cell types, such as hair cells or supporting cells, or to generate the highly ordered cellular pattern that is characteristic for the organ of Corti. In particular, the hedgehog and fibroblast growth factor signaling pathways modulate the formation of the progenitor cells that will give rise to the organ of Corti. Sox2, a transcription factor that is required for the formation of the cochlear progenitor cell population, has paradoxically been shown to also act as an inhibitor of hair cell development. Finally, the motor protein Myosin II regulates extension of the organ of Corti and the alignment of hair cells and supporting cells into ordered rows.
Summary
A better understanding of the signaling pathways that direct different aspects of cochlear development, such as specific of cell fates or cellular patterning, offers the potential to identify new pathways or molecules that could be targeted for therapeutic interventions.
Keywords: hair cell, pillar cell, atoh1, organ of Corti, Sox2
Introduction
The organ of Corti contains inner and outer hair cells that are arranged into four or five ordered rows, depending on species, which extend along the length of the cochlear spiral [1]. In addition, the organ also contains several types of supporting cells, including inner phalangeal cells, border cells, pillar cells, Deiters' cells, Hensen's cells and Claudius' cells that surround the hair cells to form a highly stereotyped and invariant cellular pattern (Fig. 1). Previous results have demonstrated that both hair cells and supporting cells are required for normal auditory function [2,3]. While a similar requirement for cellular patterning has not been directly demonstrated, the conservation of this pattern in all known therian mammals strongly suggests that it plays a crucial role in cochlear function.
Figure 1.
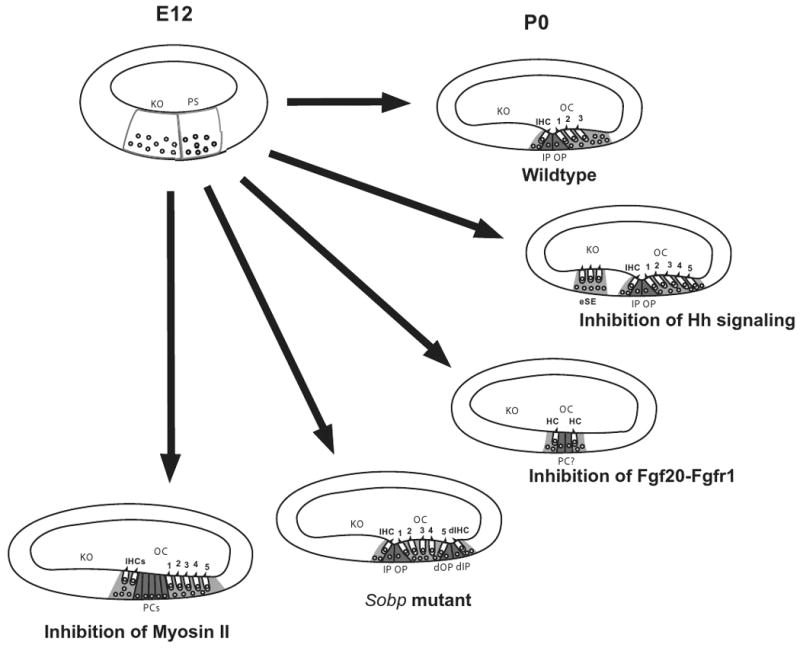
Phenotypic changes in the organ of Corti in response to changes in specific signaling pathways. At E12, the floor of the developing cochlear duct is comprised of undifferentiated cells that can be classified as part of the prosensory domain (PS) or Kolliker's organ (KO) based on relative position. By P0, the normal cellular pattern of the organ of Corti (OC), including a single inner hair cell (IHC), three outer hair cells (numbered), inner and outer pillar cells (IP and OP) and supporting cells (light gray), is present. Inhibition of hedgehog signaling leads to the formation of ectopic hair cells and supporting cells (eSE) in Kolliker's organ as well as an overproduction of outer hair cells. In contrast, inhibition of Fgf20-Fgfr1 interactions leads to the formation of small patches of unpatterned hair cells and supporting cells including some cells that may be pillar cells. In Sobp mutants, mirror-image duplications of the organ of Corti are present including a duplicated inner hair cells (dIHC) and duplicated pillar cells (dIP and dOP). Finally, inhibition of Myosin II leads to defects in extension of the cochlear duct. In a cross-section this appears as additional rows of all cell types.
All of the cells that will ultimately develop as hair cells or supporting cells within the organ of Corti are derived from progenitor cells initially located in the otocyst, placodally-derived invaginations that form adjacent to the developing hindbrain [4,5]. Once formed, intrinsic and extrinsic factors act to direct cells within the otocyst to one of three primary fates: neuroblasts that will delaminate from the otocyst to form the neurons of the statoacoustic (VIII cranial) ganglion, prosensory cells that will go on to form the sensory regions of the auditory and vestibular systems, and non-sensory cells that comprise the remaining epithelial regions of the inner ear (Fig. 2). Within the prosensory lineage, individual cells will subsequently become specified as either hair cells or supporting cells. The factors that direct cells along each of these lineages remain poorly understood, however recent results have identified new signaling pathways that are important for specific aspects of lineage restriction or cell fate specification.
Figure 2.

Lineage relationships of cells originating in the otocyst. Cells within the otocyst have three broad fates, Prosensory Cell, Non-sensory cell, or Neuroblast. As discussed in the text, Sox2 actually plays a role in the specification of both the Prosensory and Neuroblast lineages. Prosensory cells then make a secondary choice between Hair Cell and Supporting Cell fates, with activation of Fgfr1 by Fgf20 positively influencing the hair cell fate, leading to activation of Atoh1. In contrast, Sox2 acts as an inhibitor of the hair cell fate at this decision point, probably at least in some cells through activation of Prox1. For the particular case of pillar cells, developing inner hair cells express Fgf8 which activates Fgfr3 in adjacent progenitors, leading to an inhibition of the hair cell fate and an induction of the pillar cell fate in those cells. Finally, the hedgehog signaling pathway acts to prevent at least some Non-Sensory Cells from spontaneously switching into the Prosensory lineage.
Hedgehog signaling inhibits prosensory development
The hedgehog signaling pathway is a highly conserved signaling pathway that has been shown to regulate diverse aspects of animal development and disease [6]. Previous results had demonstrated that hedgehog signaling at the stage of early otocyst development is required for formation of ventral structures within the inner ear, including the entire cochlear duct [7]. However, the absence of a cochlea in hedgehog mutants precluded an examination of its role in sensory development. Recently, cochlear patterning was examined in a mouse model (Gli3Δ699) for Pallister-Hall syndrome, a disorder that arises through truncating mutations in GLI3, a downstream mediator of hedgehog signaling [8]. Because of partial functional redundancy with another hedgehog effector, GLI2/Gli2, truncating mutations in GLI3/Gli3 result in an incomplete loss of hedgehog function. Gli3Δ699/Δ699 mice survive until birth, and analysis of these mice indicated the presence of shortened cochleae, expanded sensory regions, and ectopic sensory patches in Kölliker's organ, a region of non-sensory otocyst-derived cells located adjacent to the organ of Corti and comprising the lateral half of the cochlear duct at embryonic time points (Fig 1) [9*]. The truncated GLI3 protein is thought to act as a repressor of hedgehog signaling, so the expansion of sensory cells within the cochlea suggested that hedgehog signaling normally acts to restrict the size of the prosensory domain. This hypothesis was supported by in vitro experiments that demonstrated that increased activation of the hedgehog pathway acts to inhibit sensory formation while inhibition of hedgehog signaling mimics the phenotype seen in Gli3Δ699/Δ699 mice. Finally, audiologic assessment of a limited number of Pallister-Hall patients indicated variable hearing loss across multiple frequencies.
These results suggest that activation of the hedgehog pathway regulates the size and position of the organ of Corti by inhibiting non-sensory cells within the cochlear duct from developing as prosensory cells (Fig. 2). Activation of hedgehog signaling is dependent on the presence of one or more of the known hedgehog ligands, sonic hedghehog (SHH), desert hedgehog (DHH) or Indian hedgehog (IHH). An initial screen for expression of these ligands in the cochlea indicated that the developing spiral ganglion neurons strongly express Shh during the period of cochlear development. Since the ganglion is located adjacent to the lateral half of the cochlear duct, this result is consistent with a higher concentration of SHH in Kolliker's organ where it could act to prevent sensory formation.
Sox2 regulates multiple events in cochlear development
As discussed, cells within the otocyst are initially specified along one of three primary lineages. Previous results had demonstrated that the HMG box transcription factor Sox2 is required for the formation of the prosensory lineage [10,11]. However, subsequent studies demonstrated that Sox2 expression is not limited to the period of prosensory formation. Instead Sox2 is maintained in cells within the developing organ of Corti through at least the early postnatal period, although it is downregulated in developing hair cells [12-14]. This pattern of expression suggested that following a role in formation of prosensory cells, Sox2 might act to inhibit hair cell development. This hypothesis was supported by the demonstration that cochleae from mice that only expressed 20% of the normal level of SOX2 actually contained an increased number of hair cells [14*]. Concomitant in vitro studies demonstrated that Sox2 can induce prosensory formation in non-sensory cells but actually inhibits hair cell formation when expressed in cells that have already been specified as prosensory cells. Finally, Prox1, a homeobox-containing transcription factor, that was known to be expressed in a subset of Sox2-positive cells within the developing organ of Corti [15], was shown to be a Sox2 target [14]. Forced expression of Prox1 in prosensory cells also acts to inhibit hair cell development, suggesting that Prox1 may be a major effecter of the inhibitory role of Sox2 on hair cell formation (Fig 2) [14,16].
Fibroblast growth factor signaling is required for formation of the organ of Corti
In contrast with hedgehog signaling, the fibroblast growth factor signaling pathway has recently been shown to be required for formation of the organ of Corti. Previous studies had demonstrated a crucial role for Fibroblast growth factor receptor 1 (Fgfr1) in the formation of the sensory epithelium however the specific ligand responsible for receptor activation had not been determined [17]. A recent paper demonstrated that Fgf20 is specifically expressed in the developing organ of Corti in a pattern that partially overlaps with Fgfr1 [18*]. Inhibition of FGF20 in vitro using a function blocking antibody recapitulates the phenotype of Fgfr1 mutants (Fig 1). While definitive proof awaits the generation of an Fgf20 mutant mouse, the results strongly suggest that Fgf20 acts through Fgfr1 to induce formation of the organ of Corti.
Atoh1 induces functional hair cells in vivo
The basic Helix-Loop-Helix transcription factor Atoh1 is expressed in developing hair cells and was shown to be required for hair cell formation in Atoh1 mutant mice [19,20]. In addition, both in vivo and in vitro approaches have demonstrated that forced expression of Atoh1 is sufficient to induce cells that are morphologically consistent with hair cells and express many hair cell-specific markers (Fig 2) [21-25]. However, a definitive test of these cells using electrophysiological techniques had not been completed.
This issue was addressed in an elegant paper from the laboratories of John Brigande and Tony Ricci in which a new in utero electroporation technique was used to transfect otocyst cells with an Atoh1 expression vector at E11 [26*]. Following electroporation, the embryos were allowed to develop through birth and up to post-natal day 4 to 6, at which point cochleae were removed and Atoh1-transfected cells were identified based on co-expression of the marker green fluorescent protein. The mechanotransduction responses and basolateral currents in these cells were then determined by patch clamping. Results indicated very similar electrophysiological profiles for Atoh1-transfected and non-transfected hair cells. These results strongly suggest that expression of Atoh1 is sufficient to induce the formation of a functional hair cell. However, it is important to consider that because of structural instability in transfected hair cells located in non-sensory regions of the cochlea, only hair cells located in or directly adjacent to the sensory epithelium could be analyzed electrophysiologically. The total number of hair cells within the organ of Corti was significantly increased in Atoh1-transfected cochleae, suggesting that at least some of the transfected cells arose from cells that would not have normally developed as hair cells, but since it is impossible to determine which transfected cells would have developed as hair cells even in the absence of exogenous Atoh1 transfection, there remains a small chance that the cells selected for electrophysiological characterization represent cells that were already specified to develop as hair cells. One way to directly address this question would be to transfect Atoh1 into Atoh1 mutant mice. Previous results have demonstrated that Atoh1-transfection is sufficient to induce hair cells in Atoh1 mutants [23], so this approach would ensure that every hair cell arose as a result of transfection.
Pillar cell development is regulated through Fgfr3 and Hey2
The inner and outer hair cell regions of the organ of Corti are separated by the tunnel of Corti, a structure that is only found in mammalian sensory epithelia. The tunnel is formed by single rows of inner and outer pillar cells that extend along the length of the cochlear spiral. While pillar cells clearly fit the broad definition for a supporting cell, their unique morphology and position within the organ of Corti suggests that unique signaling pathways may mediate their development. Previous results have demonstrated that most of the cells within the prosensory population will develop as hair cells if components of the notch signaling pathway are disrupted [27-29]. However, pillar cells are still present in all known notch-pathway mutants, suggesting that other pathways may regulate the formation of this cell type. Recent work has demonstrated that developing pillar cells express the Notch-target Hey2 [30**-32], but that expression of Hey2 is not dependent on notch signaling. If Hey2 is deleted in conjunction with inhibition of notch signaling, then pillar cells will develop as hair cells, suggesting that Hey2 expression is important for maintaining pillar cells but that Hey2 is not regulated through the notch pathway. This study also showed that expression of Hey2 depends instead on activation of the Fgf signaling pathway [30**].
This result is in agreement with independent studies that demonstrated that Fibroblast growth factor receptor 3 (Fgf3) turns on in cells that will develop as pillar cells ([33-36]. Animals with targeted deletions of Fgfr3 have defects in pillar cell development and many pillar cells actually change fates to develop as extra outer hair cells [37-39]. Finally, both animals and humans with FGFR3/Fgfr3 mutations have hearing deficits ([37,38,40-44]. While expression of Hey2 has not been examined in these animals, the results strongly suggest that Hey2 is a direct target of Fgfr3 in the organ of Corti [30]. The results are also consistent with the hypothesis that pillar cells represent a highly specialized type of supporting cells and that unique signaling interactions are required for their development (Fig. 1).
Sine oculis binding protein 1 (Sobp1) regulates cellular pattern in the organ of Corti
One of the most striking aspects of the organ of Corti is the asymmetry in cellular patterning. Moving from its medial to lateral edge, the organ of Corti contains a single row of inner hair cells followed by two rows of pillar cells, then by three or four of outer hair cells and Deiters' cells and finally one or two rows of Hensen's and Claudius' cells. The factors that specify this pattern are unknown, with the exception that disruption of Fgfr1-signaling leads to small patches of loosely organized hair cells, but this is more likely the result of a defect in cell specification rather than patterning. However, a recent study has demonstrated the presence of mirror-image duplications of the organ of Corti in Jackson Circler mice as a result of a spontaneous point mutation in Sobp (also called Jxc1) (Fig. 1)[45]. Sobp is a vertebrate homolog of the Drosophila sine oculis-binding protein and encodes a nuclear zinc-finger protein. While the cochlear phenotype in these animals is variable, in extreme cases the organ of Corti contains single rows of inner hair cells at both its medial and lateral boundaries with a tunnel of Corti located on the inner edge of each inner hair cell row. In the center are as many as six rows of outer hair cells. These results suggest that Sobp regulates cell fate and overall patterning of the organ of Corti. However, it remains to be seen whether Sobp acts as a transcriptional activator to specifically regulate these processes. Sobp is broadly expressed within the cochlear duct, providing limited clues as to its specific role. However, the presence of mirror image duplications of the sensory epithelium is, to date, unique to Sobp mutants, and provides the first clue to the factors that determine asymmetric patterning within the cochlear duct.
Myosin II regulates cochlear outgrowth and the alignment of hair cells
As discussed, the cochlear duct develops as an extension from the ventral region of the otocyst that lengthens and coils to form the mature spiral. At the same time, cells within the duct become aligned into highly ordered rows of hair cells and supporting cells. Previous analyses of mutant mice with shortened cochleae have identified patterning defects in the sensory epithelia of these animals, suggesting a link between outgrowth and cellular patterning [46-49**]. In many cases, the mutated genes leading to these types of patterning defects were members of the planar cell polarity (PCP) pathway, a highly conserved signaling pathway that regulates diverse biological processes, including orientation of hairs on the fly wing, development of the neural tube and the orientation of hair cell stereociliary bundles [50-52]. In addition, the PCP pathway is also known to regulate directed cellular movements, such as cellular migrations related to the simultaneous narrowing and lengthening of an embryo or epithelial sheet, a process referred to as convergence and extension [53-55]. These results suggested that convergence and extension probably also plays a role in cochlear elongation [46,56,57], however the specific motor proteins that regulate the necessary changes in cell shape and position were unknown.
Studies examining the extension of the germ band in Drosophila embryos had identified zipper, the Drosophila homolog of mammalian non-muscle Myosin II heavy chain (MYH/Myh) genes, as a key regulatory factor [58]. In addition, mutations in two of the three mammalian Myosin II genes, MYH9 and MYH14, lead to syndromic and non-syndromic hearing loss in humans [59-62].
Based on these results, Yamamoto and colleagues examined the expression of MYH9, MYH10 and MYH14 in the developing mouse cochlea and used both in vivo and in vitro approaches to determine the effects of inactivation of myosin II [63*]. Results indicated strong expression of MYH10 and MYH14 within the developing organ of Corti and in particular in developing pillar cells. In addition, inhibition of myosin II either in vivo or in vitro resulted in shortened cochleae and patterning defects within the organ of Corti (Fig. 1). In particular, the organ of Corti was shortened and cells failed to become aligned into ordered rows. A definitive link between myosin II and the PCP pathway was not established but the results are consistent with a role for PCP in regulating the orientation or activity of myosin II. The results also demonstrate that outgrowth of the cochlear duct and the generation of cellular pattern within the organ of Corti occur as coordinated processes with a high likelihood of mutual dependence. Future experiments will hopefully elucidate the nature of the interactions between the PCP pathway and myosin II as well as the specific cellular movements that lead to cellular patterning within the organ of Corti.
Conclusions
The organ of Corti is a remarkable example of cellular patterning and morphology. At least two types of hair cells and six types of supporting cells are arranged into a highly ordered pattern of regular rows that extends along the length of the cochlear spiral. The normal formation of this structure is required for auditory function and progressive loss of either hair cells or supporting cells leads to hearing impairment. Although the factors that regulate the diverse signaling interactions that must occur during the development of the organ of Corti remain largely unknown, recent results have provided exciting new insights into at least some of these interactions.
The organ of Corti develops from a limited population of prosensory cells that are restricted to a specific region of the developing cochlear duct. Hedgehog signaling appears to play a role in inhibiting inappropriate prosensory formation, ensuring that sensory cells do not develop in other regions of the cochlea, while Fgf signaling acts to positively induce sensory formation. Once a prosensory region is established, Sox2 and Prox1, in conjunction with notch signaling, regulate the number of cells within the organ of Corti that will develop as hair cells. At the same time, signaling interactions regulated through Fgfr3 and Hey2 act to control the development of the pillar cells,
Acknowledgments
This work was supported by the Intramural Program of the National Institute on Deafness and Other Communication Disorders at the National Institutes of Health. The authors wish to apologize to any of their colleagues whose work was unavoidably excluded from this review due to space constraints.
References
- 1.Cotanche DA. Genetic and pharmacological intervention for treatment/prevention of hearing loss. J Commun Disord. 2008;41:421–443. doi: 10.1016/j.jcomdis.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collado MS, Burns JC, Hu Z, Corwin JT. Recent advances in hair cell regeneration research. Curr Opin Otolaryngol Head Neck Surg. 2008;16:465–471. doi: 10.1097/MOO.0b013e32830f4ab5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kikuchi T, Adams JC, Miyabe Y, So E, Kobayashi T. Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness. Med Electron Microsc. 2000;33:51–56. doi: 10.1007/s007950070001. [DOI] [PubMed] [Google Scholar]
- 4.Barald KF, Kelley MW. From placode to polarization: new tunes in inner ear development. Development. 2004;131:4119–4130. doi: 10.1242/dev.01339. [DOI] [PubMed] [Google Scholar]
- 5.Kelley MW. Regulation of cell fate in the sensory epithelia of the inner ear. Nat Rev Neurosci. 2006;7:837–849. doi: 10.1038/nrn1987. [DOI] [PubMed] [Google Scholar]
- 6.Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–812. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riccomagno MM, Martinu L, Mulheisen M, Wu DK, Epstein DJ. Specification of the mammalian cochlea is dependent on Sonic hedgehog. Genes Dev. 2002;16:2365–2378. doi: 10.1101/gad.1013302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang S, Graham JM, Jr, Olney AH, Biesecker LG. GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat Genet. 1997;15:266–268. doi: 10.1038/ng0397-266. [DOI] [PubMed] [Google Scholar]
- 9.Driver EC, Pryor SP, Hill P, Turner J, Ruther U, Biesecker LG, Griffith AJ, Kelley MW. Hedgehog signaling regulates sensory cell formation and auditory function in mice and humans. J Neurosci. 2008;28:7350–7358. doi: 10.1523/JNEUROSCI.0312-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study identified hearing loss in Pallister-Hall patients, implicating the hedgehog signaling pathway in sensory formation. Additional studies both in vivo and in vitro demonstrated a role for hedgehog signalign in the inhibition of sensory cell formation.
- 10.Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, et al. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116:2442–2455. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiernan AE, Pelling AL, Leung KK, Tang AS, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KS. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- 12.Hume CR, Bratt DL, Oesterle EC. Expression of LHX3 and SOX2 during mouse inner ear development. Gene Expr Patterns. 2007;7:798–807. doi: 10.1016/j.modgep.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mak AC, Szeto IY, Fritzsch B, Cheah KS. Differential and overlapping expression pattern of SOX2 and SOX9 in inner ear development. Gene Expr Patterns. 2009 doi: 10.1016/j.gep.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dabdoub A, Puligilla C, Jones JM, Fritzsch B, Cheah KS, Pevny LH, Kelley MW. Sox2 signaling in prosensory domain specification and subsequent hair cell differentiation in the developing cochlea. Proc Natl Acad Sci U S A. 2008;105:18396–18401. doi: 10.1073/pnas.0808175105. [DOI] [PMC free article] [PubMed] [Google Scholar]; * The effects of Sox2 at the level of specification of cells as hair cells and supporting cells was examined. Analysis of Sox2 hypomorphs identified an over-production of hair cells, suggesting that Sox2 act as a hair cell inhibitor.
- 15.Bermingham-McDonogh O, Oesterle EC, Stone JS, Hume CR, Huynh HM, Hayashi T. Expression of Prox1 during mouse cochlear development. J Comp Neurol. 2006;496:172–186. doi: 10.1002/cne.20944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirjavainen A, Sulg M, Heyd F, Alitalo K, Yla-Herttuala S, Moroy T, Petrova TV, Pirvola U. Prox1 interacts with Atoh1 and Gfi1, and regulates cellular differentiation in the inner ear sensory epithelia. Dev Biol. 2008;322:33–45. doi: 10.1016/j.ydbio.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Pirvola U, Ylikoski J, Trokovic R, Hebert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi T, Ray CA, Bermingham-McDonogh O. Fgf20 is required for sensory epithelial specification in the developing cochlea. J Neurosci. 2008;28:5991–5999. doi: 10.1523/JNEUROSCI.1690-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Fgf20 is shown to be expressed in the developing organ of Corti in a pattern that is consistent with a role in sensory formation and inhibition using a function-clocking antibody leads to a significant decrease in the size of the sensory epithelium.
- 19.Lanford PJ, Shailam R, Norton CR, Gridley T, Kelley MW. Expression of Math1 and HES5 in the cochleae of wildtype and Jag2 mutant mice. J Assoc Res Otolaryngol. 2000;1:161–171. doi: 10.1007/s101620010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- 21.Izumikawa M, Minoda R, Kawamoto K, Abrashkin KA, Swiderski DL, Dolan DF, Brough DE, Raphael Y. Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat Med. 2005;11:271–276. doi: 10.1038/nm1193. [DOI] [PubMed] [Google Scholar]
- 22.Kawamoto K, Ishimoto S, Minoda R, Brough DE, Raphael Y. Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J Neurosci. 2003;23:4395–4400. doi: 10.1523/JNEUROSCI.23-11-04395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods C, Montcouquiol M, Kelley MW. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat Neurosci. 2004;7:1310–1318. doi: 10.1038/nn1349. [DOI] [PubMed] [Google Scholar]
- 24.Zheng JL, Gao WQ. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat Neurosci. 2000;3:580–586. doi: 10.1038/75753. [DOI] [PubMed] [Google Scholar]
- 25.Staecker H, Praetorius M, Baker K, Brough DE. Vestibular hair cell regeneration and restoration of balance function induced by math1 gene transfer. Otol Neurotol. 2007;28:223–231. doi: 10.1097/MAO.0b013e31802b3225. [DOI] [PubMed] [Google Scholar]
- 26.Gubbels SP, Woessner DW, Mitchell JC, Ricci AJ, Brigande JV. Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature. 2008;455:537–541. doi: 10.1038/nature07265. [DOI] [PMC free article] [PubMed] [Google Scholar]; * A new technique for gene transfer through the uterine wall into embryonic mice in vivo was used to force expression of Atoh1 in the otocyst. Resulting transfected hair cells were then identified after birth and electrophysiological techniques were employed to demonstrate functional hair cell characteristics. This is the first demonstration that Atoh1 is sufficient to induce functional hair cells.
- 27.Brooker R, Hozumi K, Lewis J. Notch ligands with contrasting functions: Jagged1 and Delta1 in the mouse inner ear. Development. 2006;133:1277–1286. doi: 10.1242/dev.02284. [DOI] [PubMed] [Google Scholar]
- 28.Kiernan AE, Cordes R, Kopan R, Gossler A, Gridley T. The Notch ligands DLL1 and JAG2 act synergistically to regulate hair cell development in the mammalian inner ear. Development. 2005;132:4353–4362. doi: 10.1242/dev.02002. [DOI] [PubMed] [Google Scholar]
- 29.Lanford PJ, Lan Y, Jiang R, Lindsell C, Weinmaster G, Gridley T, Kelley MW. Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat Genet. 1999;21:289–292. doi: 10.1038/6804. [DOI] [PubMed] [Google Scholar]
- 30.Doetzlhofer A, Basch ML, Ohyama T, Gessler M, Groves AK, Segil N. Hey2 regulation by FGF provides a Notch-independent mechanism for maintaining pillar cell fate in the organ of Corti. Dev Cell. 2009;16:58–69. doi: 10.1016/j.devcel.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Pillar cell progenitors were shown to be prevented from developing as hair cells as a result of expression of Hey2, a notch target gene. However, in the organ of Corti, expression of Hey2 is controlled by Fgfr3 rather than by Notch.
- 31.Li S, Mark S, Radde-Gallwitz K, Schlisner R, Chin MT, Chen P. Hey2 functions in parallel with Hes1 and Hes5 for mammalian auditory sensory organ development. BMC Dev Biol. 2008;8:20. doi: 10.1186/1471-213X-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi T, Kokubo H, Hartman BH, Ray CA, Reh TA, Bermingham-McDonogh O. Hesr1 and Hesr2 may act as early effectors of Notch signaling in the developing cochlea. Dev Biol. 2008;316:87–99. doi: 10.1016/j.ydbio.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peters K, Ornitz D, Werner S, Williams L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol. 1993;155:423–430. doi: 10.1006/dbio.1993.1040. [DOI] [PubMed] [Google Scholar]
- 34.Pirvola U, Cao Y, Oellig C, Suoqiang Z, Pettersson RF, Ylikoski J. The site of action of neuronal acidic fibroblast growth factor is the organ of Corti of the rat cochlea. Proc Natl Acad Sci U S A. 1995;92:9269–9273. doi: 10.1073/pnas.92.20.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacques BE, Montcouquiol ME, Layman EM, Lewandoski M, Kelley MW. Fgf8 induces pillar cell fate and regulates cellular patterning in the mammalian cochlea. Development. 2007;134:3021–3029. doi: 10.1242/dev.02874. [DOI] [PubMed] [Google Scholar]
- 36.Mueller KL, Jacques BE, Kelley MW. Fibroblast growth factor signaling regulates pillar cell development in the organ of corti. J Neurosci. 2002;22:9368–9377. doi: 10.1523/JNEUROSCI.22-21-09368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- 38.Puligilla C, Feng F, Ishikawa K, Bertuzzi S, Dabdoub A, Griffith AJ, Fritzsch B, Kelley MW. Disruption of fibroblast growth factor receptor 3 signaling results in defects in cellular differentiation, neuronal patterning, and hearing impairment. Dev Dyn. 2007;236:1905–1917. doi: 10.1002/dvdy.21192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayashi T, Cunningham D, Bermingham-McDonogh O. Loss of Fgfr3 leads to excess hair cell development in the mouse organ of Corti. Dev Dyn. 2007;236:525–533. doi: 10.1002/dvdy.21026. [DOI] [PubMed] [Google Scholar]
- 40.Hollway GE, Suthers GK, Battese KM, Turner AM, David DJ, Mulley JC. Deafness due to Pro250Arg mutation of FGFR3. Lancet. 1998;351:877–878. doi: 10.1016/S0140-6736(98)24012-8. [DOI] [PubMed] [Google Scholar]
- 41.Mansour SL, Twigg SR, Freeland RM, Wall SA, Li C, Wilkie AO. Hearing loss in a mouse model of Muenke syndrome. Hum Mol Genet. 2009;18:43–50. doi: 10.1093/hmg/ddn311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muenke M, Gripp KW, McDonald-McGinn DM, Gaudenz K, Whitaker LA, Bartlett SP, Markowitz RI, Robin NH, Nwokoro N, Mulvihill JJ, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet. 1997;60:555–564. [PMC free article] [PubMed] [Google Scholar]
- 43.Pannier S, Couloigner V, Messaddeq N, Elmaleh-Berges M, Munnich A, Romand R, Legeai-Mallet L. Activating Fgfr3 Y367C mutation causes hearing loss and inner ear defect in a mouse model of chondrodysplasia. Biochim Biophys Acta. 2009;1792:140–147. doi: 10.1016/j.bbadis.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Toydemir RM, Brassington AE, Bayrak-Toydemir P, Krakowiak PA, Jorde LB, Whitby FG, Longo N, Viskochil DH, Carey JC, Bamshad MJ. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet. 2006;79:935–941. doi: 10.1086/508433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Z, Montcouquiol M, Calderon R, Jenkins NA, Copeland NG, Kelley MW, Noben-Trauth K. Jxc1/Sobp, encoding a nuclear zinc finger protein, is critical for cochlear growth, cell fate, and patterning of the organ of corti. J Neurosci. 2008;28:6633–6641. doi: 10.1523/JNEUROSCI.1280-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature. 2003;423:173–177. doi: 10.1038/nature01618. [DOI] [PubMed] [Google Scholar]
- 47.Etheridge SL, Ray S, Li S, Hamblet NS, Lijam N, Tsang M, Greer J, Kardos N, Wang J, Sussman DJ, et al. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008;4:e1000259. doi: 10.1371/journal.pgen.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, Hamblet NS, Mark S, Dickinson ME, Brinkman BC, Segil N, Fraser SE, Chen P, Wallingford JB, Wynshaw-Boris A. Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development. 2006;133:1767–1778. doi: 10.1242/dev.02347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet. 2008;40:69–77. doi: 10.1038/ng.2007.54. [DOI] [PubMed] [Google Scholar]; ** The first demonstration that formation of the primary cilium, the precursor of the kinocilium, is required for normal planar cell polarity within the inner ear.
- 50.Jones C, Chen P. Primary cilia in planar cell polarity regulation of the inner ear. Curr Top Dev Biol. 2008;85:197–224. doi: 10.1016/S0070-2153(08)00808-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simons M, Mlodzik M. Planar cell polarity signaling: from fly development to human disease. Annu Rev Genet. 2008;42:517–540. doi: 10.1146/annurev.genet.42.110807.091432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodrich LV. The plane facts of PCP in the CNS. Neuron. 2008;60:9–16. doi: 10.1016/j.neuron.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goto T, Davidson L, Asashima M, Keller R. Planar cell polarity genes regulate polarized extracellular matrix deposition during frog gastrulation. Curr Biol. 2005;15:787–793. doi: 10.1016/j.cub.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 54.Carreira-Barbosa F, Concha ML, Takeuchi M, Ueno N, Wilson SW, Tada M. Prickle 1 regulates cell movements during gastrulation and neuronal migration in zebrafish. Development. 2003;130:4037–4046. doi: 10.1242/dev.00567. [DOI] [PubMed] [Google Scholar]
- 55.Carreira-Barbosa F, Kajita M, Morel V, Wada H, Okamoto H, Martinez Arias A, Fujita Y, Wilson SW, Tada M. Flamingo regulates epiboly and convergence/extension movements through cell cohesive and signalling functions during zebrafish gastrulation. Development. 2009;136:383–392. doi: 10.1242/dev.026542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKenzie E, Krupin A, Kelley MW. Cellular growth and rearrangement during the development of the mammalian organ of Corti. Dev Dyn. 2004;229:802–812. doi: 10.1002/dvdy.10500. [DOI] [PubMed] [Google Scholar]
- 57.Chen P, Johnson JE, Zoghbi HY, Segil N. The role of Math1 in inner ear development: Uncoupling the establishment of the sensory primordium from hair cell fate determination. Development. 2002;129:2495–2505. doi: 10.1242/dev.129.10.2495. [DOI] [PubMed] [Google Scholar]
- 58.Bertet C, Sulak L, Lecuit T. Myosin-dependent junction remodelling controls planar cell intercalation and axis elongation. Nature. 2004;429:667–671. doi: 10.1038/nature02590. [DOI] [PubMed] [Google Scholar]
- 59.Donaudy F, Snoeckx R, Pfister M, Zenner HP, Blin N, Di Stazio M, Ferrara A, Lanzara C, Ficarella R, Declau F, et al. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4) Am J Hum Genet. 2004;74:770–776. doi: 10.1086/383285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heath KE, Campos-Barros A, Toren A, Rozenfeld-Granot G, Carlsson LE, Savige J, Denison JC, Gregory MC, White JG, Barker DF, et al. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am J Hum Genet. 2001;69:1033–1045. doi: 10.1086/324267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lalwani AK, Goldstein JA, Kelley MJ, Luxford W, Castelein CM, Mhatre AN. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYH9. Am J Hum Genet. 2000;67:1121–1128. doi: 10.1016/s0002-9297(07)62942-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang T, Pfister M, Blin N, Zenner HP, Pusch CM, Smith RJ. Genetic heterogeneity of deafness phenotypes linked to DFNA4. Am J Med Genet A. 2005;139:9–12. doi: 10.1002/ajmg.a.30989. [DOI] [PubMed] [Google Scholar]
- 63.Yamamoto N, Okano T, Ma X, Adelstein RS, Kelley MW. Myosin II regulates extension, growth and patterning in the mammalian cochlear duct. Development. 2009;136:1977–1986. doi: 10.1242/dev.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Analysis of changes in cochlear patterning in response to genetic or pharmacological inhibition of Myosin II identified a role for developing pillar cells in cochlear patterning and convergence and extension of the cochlear duct.