

Abstract
Methamphetamine (METH) is a highly addictive compound that induces toxicity of the dopamine (DA) terminals of the neostriatum. Exposure to METH induces long-term deficits in dopamine transporter (DAT) and tyrosine hydroxylase (TH) levels as well as induction of glial fibrillary acidic protein (GFAP) in the caudate putamen (CPu) and the nucleus accumbens (NAc). The primary effect of exposure to METH is elevation of the level of extracellular DA; therefore, we assessed the role of the DA D1 receptor (D1R) and neurokinin-1 receptor (NK-1R) on the expression of toxicity. METH was injected intraperitoneally (10 mg/kg) four times at 2-h intervals (an acute toxic dose), and the mice were sacrificed three days after the treatment. Exposure to METH resulted in marked reduction of DAT sites (reduced to 30 and 21% relative to control in medial and lateral aspects of the CPu) assessed by binding of [125I]RTI-121 by autoradiography or Western blot analysis. Pretreatment with the nonpeptide NK-1R antagonist WIN-51,708 (10 mg/kg) 30 min prior to the first and fourth injections of METH prevented the loss of DAT sites of the CPu. Moreover, pretreatment with WIN-51,708 also prevented the reduction of TH levels induced by METH as well as the induction of GFAP in astrocytes. Pretreatment with the D1R antagonist SCH-23390 (0.25 mg/kg) 30 min before the first and fourth injections of METH conferred partial protection on DAT sites of the CPu. These results demonstrate that receptors postsynaptic to the DA terminals of the CPu are needed in order to express the neurotoxic effects of METH on integral components of the DA terminals of the nigrostriatal projection.
Keywords: Substance P; neurokinin-1 receptor; dopamine D1 receptor; WIN-51,708; SCH-23390; striatum
INTRODUCTION
Methamphetamine (METH) is a psychostimulant that is easy to synthesize and is becoming popular as a drug of abuse in the United States and Europe.1,2 One major social problem associated with METH use is violenc and the law enforcement it entails. Several studies involving METH abusers demonstrated that this drug induces aggressive behavior.3–6 Along with other psychostimulants such as cocaine, METH increases the extracellular concentration of monoamines (noradrenalin, adreanlin, DA, and serotonin) in discrete brain regions associated with motor activity and motivation and reward.7–9 Exposure to high doses of METH produces degeneration of dopaminergic10–12 and serotonergic13,14 terminals in the central nervous system. High doses of METH induce massive depletion of monoamines that can persist for months in rodents. 15 METH-induced depletion of monoamines is associated with reductions in TH, the rate-limiting enzyme of catecholamine synthesis, as well as tryptophan hydroxylase, the enzyme responsible for the biosynthesis of sertonin.16,17 Surprisingly, although METH induces massive and long-lasting depletions of monoamines in rodents, the behavioral impairments produced by high doses of METH are subt1e.15 METH-induced depletions of monoamines are not restricted to rodents. Studies involving nonhuman primates (rhesus monkeys) demonstrate that METH-induced depletions of both DA and serotonin can last up to four years after exposure.18
Because the abuse of METH is rapidly rising, it is desirable to identify agents that prevent METH-induced neurotoxicity without being toxic themselves. Compounds that block glutamate receptors protect from METH but induce toxic effects of their own. 14,19 Pharmacological agents that block neuropeptide receptors in the brain display low toxicity, rendering them potentially useful in the treatment of addiction to psychostimulants. One such class of pharmacological agents is represented by the nonpeptide NK-1R antagonists that readily cross the blood-brain barrier20 and have been demonstrated to prevent METH-induced toxicity in the neostriatum of the mouse brain.21 The neuropeptide Substance P (SP) is the natural ligand for the NK-1R.22 SP is synthesized from the preprotachykinin-A gene along with the related tachykinin peptide neurokinin A by striatonigral projection neurons.23 Antagonists of the NK-1R prevent cocaine-induced overflow of dopamine (DA) in the caudate putamen (CPu) of the rat brain,24 and they attenuate cocaine-induced locomotor activity when given systemically.25 In this paper we demonstrate that the nonpeptide. NK-1R antagonist WIN-51,708 protects striatal dopaminergic terminal markers from exposure to an acute toxic dose of METH. We also report on the finding that pretreatment with the D1R antagonist SCH-23390 protects the striatal dopamine transporter (DAT) from METH. These observations lend support to the hypothesis that events postsynaptic to the DA terminals of the striatum regulate the induction and expression of toxicity to METH.
MATERIALS AND METHODS
Drug Administrations and Temperature Measurements
Ten-week-old male ICR/CD-1 mice weighing 36–44 mg (Taconic, Germantown, NY) were single housed on a 12-h light/dark cycle with food and water available ad libitum. D-METH (Sigma, St. Louis, MO) was dissolved in saline and four injections at 2-h intervals were administered intraperitoneally (i.p.) for one day. SCH-23390 or WIN-51,708 was given 30 min (i.p.) prior to the first and fourth injections of METH or saline. We have found that this paradigm is most effective in preventing the neurotoxic changes induced by multiple administrations of METH.21 Animals were sacrificed by decapitation three days after the last treatment. All animal use procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Hunter College.
Autoradiographic Analysis
Dopamine transporter
The autoradiographic assays were performed as previously described,26 with minor modifications. Briefly, after decapitation the brains were rapidly removed and frozen on dry ice. Twenty-micrometer coronal sections were cut on a cryostat. The sections were incubated with 0.073 nM [125I]RTI-121 (2200 Ci/mmol; New England Nuclear, Boston, MA) in assay buffer (137 mM NaCl, 2.7 mM KCl, 10.14 mM Na2HPO4, 1.76 mM KH2PO4, 10 mM NaI) at room temperature for 60 min. Nonspecific binding was determined with 10 mM GBR-12909. After incubation, the slides were incubated in ice-cold assay buffer twice for 20 min each. The slides were then quickly dipped into ice-cold distilled water for 5 s and air-dried overnight. The slides were apposed to Hyperfilm MP (Amersham Pharmacia, Piscataway, NJ) for 51–53 h together with the [125I]microscale. The binding of [125I]RTI-121 to sections of striatal tissue was quantified by densitometry using a computer-based NIH image analysis system.
Western Blot Analysis of DAT, TH, and GFAP
Protein samples were extracted from the striata as previously described,27 with minor modifications. Briefly, dissected striata were homogenized in a lysis buffer (320 mM sucrose, 5 mM HEPES, 5 nM EDTA, 2 nM aprotinin, 10 nM leupeptin). For DAT Western blotting, the homogenates were centrifuged at 28,000 × g for 5 min and the supernatants were further centrifuged at 28,000 × g for 30 min at 4°C. The pellets were resuspended in lysis buffer and used for analysis. For TH and glial fibrillary acidic protein (GFAP) Western blotting, the homogenates were centrifuged at 10,000 × g for 10 min at 4°C, and the supernatants were used for analysis. Ten micrograms of protein were subjected to 12% SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (BioRad, Hercules, CA). After the blocking of nonspecific binding, membranes were probed with polyclonal anti-DAT (1500, Chemicon, Temecula, CA), monoclonal anti-TH (1:1000, Chemicon, Temecula, CA), or monoclonal anti-GFAP (1:5000, Sigma) antibodies in the presence of 0.5% (for DAT) or 5% (for TH and GFAP) nonfat dry milk in TBS/T buffer at 4°C overnight. The membranes were then incubated with horseradish peroxidase conjugated secondary antibodies for 1 h at room temperature, and the proteins were detected using the ECL Western blotting detection system kit (Amersham Pharmacia Biotech, Piscataway, NJ). Western blotting for α-tubulin (used as an internal standard) was performed with a monoclonal anti-α-tubulin antibody (1:5000, Sigma). Densitometry was performed by an NIH image system, and the relative density of each band obtained for DAT, TH, and GFAP was normalized against that of α-tubulin.
Statistical Analysis
All data were analyzed by ANOVA followed by post hoc analyses using the Fisher’s protected least significant difference test with the significance level set at P < .05.
RESULTS
We assessed the role of the NK-1R and D1R on METH-induced deficits of DA terminal markers using selective antagonists that cross the blood-brain barrier. METH was injected intraperitoneal four times at 2-h intervals, and the animals were sacrificed three days after the treatment. DAT sites of the medial and lateral CPu were reduced to 30 and 21%, respectively, relative to control values at the 10 m g k g dose of METH (Fig. 1). At this dose of METH, DAT sites of the NAc were decreased to 57% relative to control (Fig. 1). The reduction of DAT sites induced by a high dose of METH was more severe in both medial and lateral aspects of the CPu than in the NAc (Fig. 1) as assessed by autoradiography.
FIGURE 1.
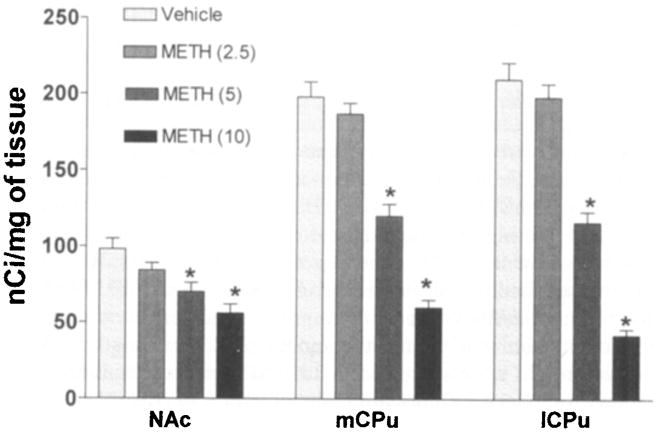
Dopamine transporter sites of the nucleus accumbens (NAc) are more refractory to METH than the lateral and medial aspects of the caudate putamen (1CPu and mCPu, respectively). The mice (6 per group) received four injections of METH at the doses shown in parentheses (mg/kg of body weight) at 2-h intervals and were sacrificed three days after the treatment. Levels of [125I]RTI-121 binding to DAT sites were determined with an image analysis system and are expressed as nCi/mg of tissue. Note that progressive increases in the dose of METH resulted in greater decrements of DAT sites in the lCPu and mCPu than in the NAc. Values over the bars represent standard error of the mean. *P < .05 (Student’s t-test).
The reduction of DAT sites induced by METH was also observed when total protein from the CPu was analyzed by Western blot (Fig. 2A). Moreover, pretreatment of the animals with the nonpeptide NK-1R antagonist WIN-51,708 (10 mg/kg of body weight) 30 min prior to the first and fourth injections of METH effectively prevented the loss of DAT protein from the dopaminergic terminals of the CPu (Fig. 2A). Exposure to METH also resulted in marked reduction of the limiting enzyme of catecholamine biosynthesis TH. The reduction of the level of this enzyme was prevented when the mice received WIN-51,708 30 min prior to the first and fourth injections of METH (Fig. 2B). The induction of GFAP by METH in astrocytes was completely prevented by pretreatment with the NK-1R antagonist WIN-51,708 (Fig. 2C). Exposure to WIN-51,708 alone did not alter the levels of DAT, TH, or GFAP in the CPu. These results demonstrate that blockade of the NK-1R of the CPu by a selective nonpeptide antagonist prevents the initiation and expression of neurotoxicity of the DA terminals that is manifested by long-term deficits in DAT sites and TH levels.
FIGURE 2.
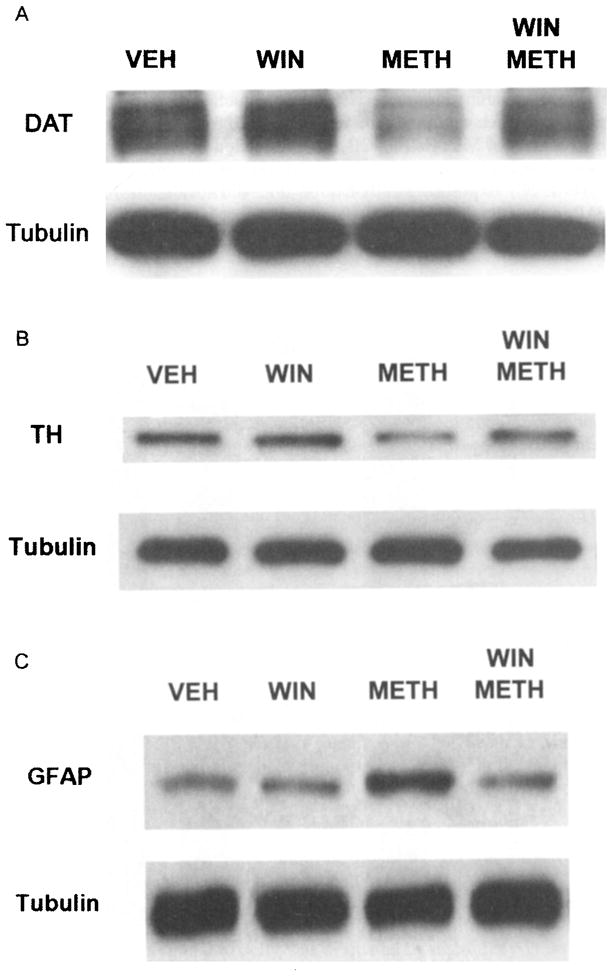
Pretreatment with the nonpeptide NK-1R antagonist WIN-51,708 (WIN) protects dopamine transporters (A, DAT), tyrosine hydroxylase (B, TH), and the induction of glial fibrillary acidic protein (C, GFAP) from METH in the caudate putamen. Mice were sacrificed three days after treatment with METH (10 mg/kg of body weight four times at 2-h intervals). DAT, TH, and GFAP were detected on gels by Western blot analysis. Control mice received vehicle (VEH). A-tubulin was used as an internal standard.
Because the primary consequence of exposure to METH is elevation of extracellular levels of the neurotransmitter DA in the CPu, we assessed the role of the D1R on METH-induced reduction of DAT sites. We injected the D1R antagonist SCH-23390 (0.25 mg/kg of body weight) 30 min before the first and fourth injections of METH. Pretreatment with SCH-23390 resulted in partial protection of the DAT sites of the medial and lateral aspects of the CPu (Fig. 3A and 3B, respectively). SCH-23390 when administered alone was without effect on the levels of DAT sites as assessed by autoradiography (Fig. 3A and 3B).
FIGURE 3.
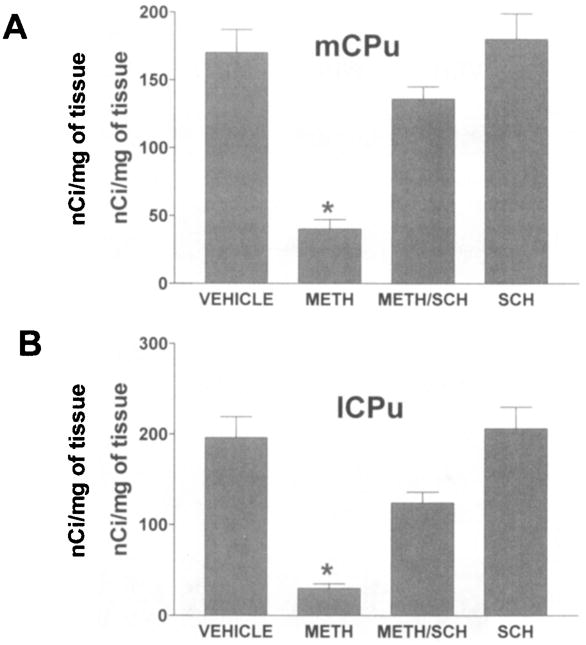
Pretreatment with the dopamine D1 receptor antagonist SCH-23390 (SCH) confers partial protection to DAT sites from METH in both the medial (A) and lateral (B) aspects of the caudate putamen (mCPu and lCPu, respectively). DAT sites were assessed by binding of [125I]RTI-121 to coronal sections through the caudate putamen by autoradiography. The mice (6 per group) were treated with METH (10 mg/kg four times at 2-h intervals) and were sacrificed three days after the treatment. Values over the bars represent standard error of the mean. *P<.05 (Student’s t-test).
DISCUSSION
In the light of the results presented above, it is evident that a functional connection must exist between the striatal NK-1R and the induction of the cascade, culminating in the expression of neurotoxicity to METH. Since the immediate consequence of the accumulation of METH in striatal tissue is augmentation of the extracellular levels of the neurotransmitter DA (DA is the predominant monoamine in this part of the brain), blockade of the DA receptors may confer protection of the presynaptic DA terminals independently of the NK-1R or via a direct mechanism that regulates the release of the neuropeptide SP through the DA receptors, namely the D1R. The role of the NK-1R and the D1R on METH-induced toxicity in the striatum will be discussed below.
Role of the NK-1R in Neurotoxicity to METH
A functional connection exists between the excitatory neurotransmitter glutamate and the neuropeptide SP in a part of the brain that may play a very important role in consolidating the learning aspects of addictive behavior in mammals. The release of SP induces glutamate release in the hippocampus of the rodent brain.28 Several laboratories have demonstrated that the release of glutamate triggers excitatory activities in neurons that ultimately lead to toxicity and cell death in various brain regions.29 The link between glutamate excitotoxicity and SP in the hippocampus is demonstrated by studies utilizing genetic mutants lacking the preprotachykinin-A gene (this gene encodes the neuropeptides SP and neurokinin A). Exposure to the excitotoxin kainate in wild-type mice results in the death of neurons in the hippocampus. However, mice lacking the preprotachykinin-A gene and treated similarly with kainate failed to display hippocampal cell death.30
The involvement of glutamate in METH-induced neurotoxicity and damage in the forebrain has been demonstrated by work from various laboratories.31,32 The impact of glutamate on DA terminal toxicity was evaluated by microdialysis. METH treatment evokes a delayed release of glutamate in the CPu.33 Moreover, agents that attenuate this increase of extracellular glutamate also prevent METH-induced decreases of DA content (a manifestation of toxicity) one week after the treatment with METH.34 The magnitude of toxicity and neural damage induced by METH depends on the amount of glutamate released by this drug. For example, the nucleus accumbens (NAc) displays a much-reduced profile of METH-induced glutamate release relative to the CPu with concomitant abatement of neural damage.34 Neurotoxic regimens of METH induce sustained release of glutamate and are associated with oxidative stress.35 Immunohistochemical studies also provide compelling evidence supporting the glutamate hypothesis of METH toxicity and damage in the neostriatum. Preexposure to the NMDA receptor antagonist MK-801 prevented the depletion of CPu DA terminals by METH as well as the induction of the astrocytic response.14 Konradi, Leveque, and Hyman reported that the induction of immediate early gene expression in CPu neurons by amphetamine requires the NMDA receptor.36
Interaction between the NK-1R and the D1R
The accumulation of METH in striatal tissues triggers the release of massive levels of DA from nigrostriatal terminals.37 The metabolism of excess DA alone can lead to the generation of free radical species. Exposure of rats to a high dose of METH resulted in massive deficits of tissue DA content and its metabolites that was attenuated or prevented by pretreatment with either a D1R or D2R antagonist.38 The same study showed that the damaging effects of METH on serotonin and its metabolites were attenuated by the D1R but not the D2R antagonist.38 A study utilizing in vivo microdialysis to assess the effects of D1R or D2R antagonists on METH-induced DA overflow in the striatum found that either D1R or D2R antagonists, when injected before METH, attenuated DA overflow in response to METH.37 Moreover, these authors correlated the magnitude of striatal METH-induced DA over-flow with the level of depletion of tissue DA content one week after the treatment.37 These studies demonstrated a role for the DA receptors postsynaptic to the nigrostriatal terminals and the development of neurotoxicity to METH in the striatum.
In summary, pretreatment with an NK-1R antagonist prevents the loss of the DA terminal markers such as DAT and TH induced by an acute toxic dose of METH. Pretreatment with the NK-1R antagonist WIN-51,708 abrogates the induction of GFAP in astrocytes of the striatum. The neuroprotective effects of the NK-1R antagonists are independent of METH-induced hyperthermia. In addition, a link is suggested between the D1R and the NK-1R in the presence of METH because a D1R antagonist protects the DAT sites from METH.
Acknowledgments
This work was supported by a Specialized Neuroscience Research Program grant (NS41073) and DA12136 to J.A.A. Support has also come from the Research Centers in Minority Institutions award to Hunter College (NCRR/NIH). We thank T. Austin Brown for expert technical assistance and Gertrude Rivera with the preparation of the manuscript.
References
- 1.Lukas S. DHHS Publication SMA96-8013. 1997. Proceedings of the National Concensus Meeting on the use, abuse and sequelae of abuse of menthaphetamine with implications for prevention, treatment and research. [Google Scholar]
- 2.National Institute on Drug Abuse. Methamphetamine abuse alert. NIDA; Washington, DC: 1999. pp. 15–16. [Google Scholar]
- 3.Carey JT, Mandel J. A San Francisco Bay Area “speed” scene. J Health Soc Behav. 1968;9:164–174. [PubMed] [Google Scholar]
- 4.Ellinwood EH., Jr Assault and homicide associated with amphetamine abuse. Am J Psychiatry. 1971;127:1170–1175. doi: 10.1176/ajp.127.9.1170. [DOI] [PubMed] [Google Scholar]
- 5.Hawks D, et al. Abuse of methylamphetamine. Br Med J. 1969;1:715–721. doi: 10.1136/bmj.2.5659.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szuster RR. Methamphetamine in psychiatric emergencies. Hawaii Med J. 1990;49:389–391. [PubMed] [Google Scholar]
- 7.Fleckenstein AE, Gibb JW, Hanson GR. Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity. Eur J Pharmacol. 2000;406:1–13. doi: 10.1016/s0014-2999(00)00639-7. [DOI] [PubMed] [Google Scholar]
- 8.Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 9.White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–153. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- 10.Lorez H. Fluorescence histochemistry indicates damage of striatal dopamine nerve terminals in rats after multiple doses of methamphetamine. Life Sci. 1981;28:911–916. doi: 10.1016/0024-3205(81)90053-9. [DOI] [PubMed] [Google Scholar]
- 11.Ricaurte GA, et al. Fluoxetine increases long-lasting neostriatal dopamine depletion after administration of d-methamphetamine and d-amphetamine. Neuropharmacology. 1983;22:1165–1169. doi: 10.1016/0028-3908(83)90075-8. [DOI] [PubMed] [Google Scholar]
- 12.Ricaurte GA, et al. Nerve terminal degeneration after a single injection of D-amphetamine in iprindole-treated rats: relation to selective long-lasting dopamine depletion. Brain Res. 1984;291:378–382. doi: 10.1016/0006-8993(84)91273-3. [DOI] [PubMed] [Google Scholar]
- 13.Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- 14.Pu C, Vorhees CV. Protective effects of MK-801 on methamphetamine-induced depletion of dopaminergic and serotonergic terminals and striatal astrocytic response: an immunohistochemical study. Synapse. 1995;19:97–104. doi: 10.1002/syn.890190205. [DOI] [PubMed] [Google Scholar]
- 15.Friedman SD, Castaneda E, Hodge GK. Long-term monoamine depletion, differential recovery, and subtle behavioral impairment following methamphetamine-induced neurotoxicity. Pharmacol Biochem Behav. 1998;61:35–44. doi: 10.1016/s0091-3057(98)00066-5. [DOI] [PubMed] [Google Scholar]
- 16.Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- 17.Hanson GR, et al. Methamphetamine-induced rapid and reversible reduction in the activities of tryptophan hydroxylase and dopamine transporters: oxidative consequences? Ann NY Acad Sci. 1998;844:103–107. [PubMed] [Google Scholar]
- 18.Woolverton WL, et al. Long-term effects of chronic methamphetamine administration in rhesus monkeys. Brain Res. 1989;486:73–78. doi: 10.1016/0006-8993(89)91279-1. [DOI] [PubMed] [Google Scholar]
- 19.Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- 20.Maggi CA. The mammalian tachykinin receptors. Gen Pharmacol. 1995;26:911–944. doi: 10.1016/0306-3623(94)00292-u. [DOI] [PubMed] [Google Scholar]
- 21.Yu J, Cadet JL, Angulo JA. Neurokinin-1 (NK-1) receptor antagonists abrogate methamphetamine-induced striatal dopaminergic neurotoxicity in the murine brain. J Neurochem. 2002;83:613–622. doi: 10.1046/j.1471-4159.2002.01155.x. [DOI] [PubMed] [Google Scholar]
- 22.Mantyh PW, et al. Rapid endocytosis of a G protein-coupled receptor: Substance P evoked internalization of its receptor in the rat striatum in vivo. Proc Natl Acad Sci USA. 1995;92:2622–2626. doi: 10.1073/pnas.92.7.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angulo JA, McEwen BS. Molecular aspects of neuropeptide regulation and function in the corpus striatum and nucleus accumbens. Brain Res Brain Res Rev. 1994;19:1–28. doi: 10.1016/0165-0173(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 24.Noailles PAH, Angulo JA. Neurokinin receptors modulate the neurochemical actions of cocaine. Ann NY Acad Sci. 2002;965:267–273. doi: 10.1111/j.1749-6632.2002.tb04168.x. [DOI] [PubMed] [Google Scholar]
- 25.Kraft M, Noailles P, Angulo JA. Substance P modulates cocaine-evoked dopamine overflow in the striatum of the rat brain. Ann NY Acad Sci. 2001;937:121–131. doi: 10.1111/j.1749-6632.2001.tb03561.x. [DOI] [PubMed] [Google Scholar]
- 26.Tsao LI, et al. Delta opioid peptide [D-Ala2, D-leu5] enkephalin blocks the long-term loss of dopamine transporters induced by multiple administrations of methamphetamine: involvement of opioid receptors and reactive oxygen species. J Pharmacol Exp Ther. 1998;287:322–331. [PubMed] [Google Scholar]
- 27.Deng X, et al. Null mutation of c-fos causes exacerbation of methamphetamine-induced neurotoxicity. J Neurosci. 1999;19:10107–10115. doi: 10.1523/JNEUROSCI.19-22-10107.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, et al. Substance P is expressed in hippocampal principal neurons during status epilepticus and plays a critical role in the maintenance of status epilepticus. Proc Natl Acad Sci USA. 1999;96:5286–5291. doi: 10.1073/pnas.96.9.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snider BJ, Choi DW. Glutamate and neurotoxicity. In: Herman BH, editor. Glutamate and Addiction. Humana Press; Totowa, NJ: 2002. pp. 51–61. [Google Scholar]
- 30.Liu H, et al. Resistance to excitotoxin-induced seizures and neuronal death in mice lacking the preprotachykinin A gene. Proc Natl Acad Sci USA. 1999;96:12096–12101. doi: 10.1073/pnas.96.21.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonsalla PK, Riordan DE, Heikkila RE. Competitive and noncompetitive antagonists at N-methyl-D-aspartate receptors protect against methamphetamine-induced dopaminergic damage in mice. J Pharmacol Exp Ther. 1991;256:506–512. [PubMed] [Google Scholar]
- 32.O’Dell SJ, Weihmuller FB, Marshall JF. MK-801 prevents methamphetamine-induced striatal dopamine damage and reduces extracellular dopamine overflow. Ann NY Acad Sci. 1992;648:317–319. doi: 10.1111/j.1749-6632.1992.tb24567.x. [DOI] [PubMed] [Google Scholar]
- 33.Nash JF, Yamamoto BK. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992;581:237–243. doi: 10.1016/0006-8993(92)90713-j. [DOI] [PubMed] [Google Scholar]
- 34.Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- 36.Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Dell SJ, Weihmuller FB, Marshall JF. Methamphetamine-induced dopamine overflow and injury to striatal dopamine terminals: attenuation by dopamine D1 or D2 antagonists. J Neurochem. 1993;60:1792–1799. doi: 10.1111/j.1471-4159.1993.tb13405.x. [DOI] [PubMed] [Google Scholar]
- 38.Sonsalla PK, Gibb JW, Hanson GR. Roles of D1 and D2 dopamine receptor subtypes in mediating the methamphetamine-induced changes in monoamine systems. J Pharmacol Exp Ther. 1986;238:932–937. [PubMed] [Google Scholar]