

Abstract
Cartwheel cells are glycinergic auditory interneurons which fire Na+- and Ca2+-dependent spike bursts, termed complex spikes, and which synapse on both principal cells and one another. The reversal potential for glycine (Egly) can be hyperpolarizing or depolarizing in cartwheel cells, and many cells are even excited by glycine. We explored the role of spike activity in determining Egly in mouse cartwheel cells using gramicidin perforated-patch recording. Egly was found to shift toward more negative potentials after a period of complex spiking or Ca2+ spiking induced by depolarization, thus enhancing glycine's inhibitory effect for ∼30 s following cessation of spiking. Combined perforated patch electrophysiology and imaging studies showed that the negative Egly shift was triggered by a Ca2+-dependent intracellular acidification. The effect on Egly was likely caused by bicarbonate-Cl− exchanger-mediated reduction in intracellular Cl−, as H2DIDS and removal of HCO3−/CO2 inhibited the negative Egly shift. The outward Cl− flux underlying the negative shift in Egly opposed a positive shift triggered by passive Cl− redistribution during the depolarization. Thus, a Ca2+-dependent mechanism serves to maintain or enhance the strength of inhibition in the face of increased excitatory activity.
Introduction
Glycinergic and GABAergic synapses typically mediate neural inhibition. Nevertheless, while many produce hyperpolarizing IPSPs, it is well known that glycinergic or GABAergic inputs can be depolarizing, exerting shunting inhibition or excitation in developing or mature neurons (Marty and Llano, 2005; Ben-Ari et al., 2007). This range of effects arises from two characteristics. First, reversal potentials for glycine or GABA-mediated IPSPs (Egly/GABA) vary widely [e.g., −85 mV in Purkinje neurons (Chavas and Marty, 2003), −37 mV in some adult hypothalamic neurons (DeFazio et al., 2002)]. Second, Egly/GABA is generally close to the resting potential, so that variation in resting potential can switch the polarity of glycine/GABA effects (Marty and Llano, 2005). Glycine/GABAA receptor channels are permeable to Cl− and, to a lesser extent, HCO3− (Bormann et al., 1987). The heterogeneity of Egly/GABA among neurons has thus been attributed to the difference in intracellular Cl−, assuming that intracellular [HCO3−], determined by intracellular pH, is constant. Cl− imaging has revealed that intracellular Cl− concentration ([Cl−]i) is correspondingly high in cells having depolarized EGABA (Duebel et al., 2006; Rocha-González et al., 2008), and that decrease in [Cl−]i may occur during developmental periods when EGABA shifts negative (Kuner and Augustine, 2000; Berglund et al., 2006).
Neuronal Cl− levels may be regulated by cation-Cl− cotransporters, KCC2 and NKCC1, and Na+-independent and Na+-driven Cl−-HCO3− exchangers, as well as by Cl− channels (Farrant and Kaila, 2007). Therefore, steady-state Egly/GABA is dependent on the balance of Cl− extrusion and accumulation mechanisms. For example, KCC2 expression increases during the period of developmental negative EGABA shift in pyramidal neurons (Rivera et al., 1999; Yamada et al., 2004). NKCC1, but not KCC2, is expressed in some neurons having depolarized EGABA (DeFazio et al., 2002; Kim and Chung, 2007). Egly/GABA may be modulated by passive redistribution of Cl− (Kaila and Voipio, 1987; Staley et al., 1995; Ehrlich et al., 1999; Billups and Attwell, 2002) or by KCC2 or NKCC1 (Fiumelli et al., 2005; Brumback and Staley, 2008). Interestingly, although Cl−-HCO3− exchangers have been identified in neurons (Kopito et al., 1989; Schwiening and Boron, 1994; Grichtchenko et al., 2001; Brett et al., 2002), their role in regulating [Cl−]i or Egly/GABA has received little attention (Gulácsi et al., 2003), perhaps due to the assumption that intracellular pH, which determines the driving force for HCO3−, is stable.
The cartwheel cell (CWC) is a glycinergic interneuron in the dorsal cochlear nucleus (DCN). CWCs form synapses among themselves and with the principal cells of DCN (Wouterlood and Mugnaini, 1984; Berrebi and Mugnaini, 1991), mediating strong feedforward inhibition of principal cells upon somatosensory stimulation (Davis and Young, 1997). Their electrical signature is the complex spike, a burst of fast spikes atop a Ca2+-dependent slow depolarization (Zhang and Oertel, 1993; Manis et al., 1994; Golding and Oertel, 1996; Kim and Trussell, 2007). Here, we investigated glycinergic responses in CWCs with the gramicidin perforated-patch method and found that complex-spike activity triggered a negative shift in Egly. This shift occurred as a result of a Ca2+-dependent acidification and a consequent decrease in [Cl−]i or [HCO3−]i, most likely involving the activity of Na+-driven Cl−-HCO3− exchanger. To our knowledge, this is the first demonstration that the anion exchanger, working against an activity-dependent intracellular acid load, can control the glycinergic/GABAergic reversal potential.
Materials and Methods
Slice preparation and recording.
Brainstem slices containing the DCN were prepared from ICR mice aged 17–27 d (Harlan). Mice were anesthetized with isoflurane and then decapitated in accord with the regulations of the Institutional Animal Care and Use Committee of Oregon Health & Science University. Subsequently, a block of brainstem was isolated and horizontal slices of 200 μm thickness were cut with a vibrating slicer (VT1200S, Leica). Dissection and slicing were done in a warm (∼30°C) solution saturated with 95% O2/5% CO2 and composed of either of the following (in mm): (1) 129 NaCl, 3 KCl, 1.2 KH2PO4, 2.4 CaCl2, 1.3 MgSO4, 20 NaHCO3, 3 HEPES, 10 glucose (ACSF), or (2) 73 sucrose, 81 NaCl, 3 KCl, 1.2 NaH2PO4, 1 CaCl2, 0.7 MgCl2, 1.3 MgSO4, 0.5 ascorbic acid, 25 NaHCO3, 3 HEPES, 10 glucose. The latter solution was used in approximately half of the dissections. The slices were kept in ACSF at 34.5°C for the first hour and then left at room temperature.
ACSF was the standard bathing medium for recordings, except for the following. For inducing intracellular acidification, modified ACSFs were used, in which NaCl was reduced by 20 mm and an equiosmolar amount (∼22 mm, referred to as “20 mm” in Results) of either Na-methanesulfonate (NaCH3O3S) or Na-propionate was introduced. For inducing intracellular alkalinization, 10 mm trimethylamine hydrochloride (TMA) replaced equimolar NaCl in the ACSF. A 3 m KCl/3% agarose salt bridge reference electrode was used in experiments using these three modified ACSFs. For bicarbonate- and CO2-free condition, the ACSF was buffered only with HEPES and equilibrated with 100% O2 (“HEPES/O2”): 20 mm NaHCO3 was replaced with 20 mm HEPES (in addition to the original 3 mm), and glucose was increased to 20 mm to achieve a similar osmolarity to that of ACSF (∼303 mOsmol/L). The pH of HEPES/O2 was adjusted with NaOH at 34°C to 7.32–7.34, which is the pH range of ACSF equilibrated with 5% CO2/95% O2, measured at 34°C.
DCN cells in the slice were visualized by infrared differential interference contrast videomicroscopy on an Olympus BX51WI microscope with a 60× water-immersion objective (0.9 NA, LUMPlanFL, Olympus). The ACSF or other saline solution was perfused at 2–3 ml/min through the recording chamber by a peristaltic pump (Minipulse 3, Gilson), and the temperature of the solution at the recording chamber was maintained at 34 ± 0.5°C by an in-line heater (SH27B, Warner Instruments). Medium-sized cells in the molecular and fusiform cell layers of DCN were identified as CWCs if they showed complex spikes spontaneously or upon injection of depolarizing current. The data presented in this study were obtained with gramicidin perforated-patch recording (Rhee et al., 1994). The standard pipette solution for perforated patch recording contained (in mm) 140 KCl, 10 NaCl, and 10 HEPES (pH adjusted to 7.25 with KOH, 290 mOsmol/L). Gramicidin was then added to this solution on the day of experiment at a final concentration of 40–100 μg/ml from a stock solution of 30–50 mg/ml DMSO. For simultaneous imaging, one or two AM fluorescent dyes (see below) were added along with gramicidin. The maximum % v/v of DMSO reached in the recording solution was 0.48% when two AM dyes were included. The tip of the recording pipette was filled with the standard solution (without gramicidin or indicator dyes). The recording pipettes had a resistance of 3–6 MΩ when filled with the standard solution, and they were prepared by pulling thick-walled filamented borosilicate glass capillaries (1B120F-4, World Precision Instruments) and wrapped with Parafilm along one-third of the pipette's length from the tip to reduce capacitance. The detection of patch rupture was by a sudden offset in membrane potential. The liquid junction potential was not corrected, as discussed previously (Kim and Trussell, 2007).
Recordings were made with a BVC-700A (Dagan) or MultiClamp 700B amplifier (Molecular Devices) in conjunction with Digidata 1322A digitizer (Molecular Devices) and ClampEx software (pClamp 9.2, Molecular Devices). After the electrode had formed a seal (>1 GΩ) on the cell membrane in voltage clamp (v-clamp), the progression of perforation (reduction in series resistance, Rs) was monitored in current clamp (i-clamp) by periodic bridge balancing and by observing the growth in amplitudes of spontaneous fast spikes. The effect of glycine on spontaneous activity or on membrane potential (Vm) was monitored early on during perforation because it could be distinguished even with a relatively high Rs. Glycine or GABA was pressure-ejected (Picospritzer II, General Valve) from a patch pipette pointed toward the cell body from 25 to 30 μm away. The duration and pressure of a puff were adjusted for each cell to elicit a 500 ms response: the ranges were 5–20 ms and 0.5–2 psi with 2 mm glycine, 100–300 ms and 2–4 psi with 0.5 mm glycine or 0.5 mm GABA. Care was taken to minimize glycine/GABA applications, to avoid causing a significant change in intracellular [Cl−] or [HCO3−] due to the flux through the glycine/GABA receptors. Glycine and GABA solutions were prepared in ACSF except during experiments involving HCO3−/CO2 removal, for which glycine was dissolved in HEPES-buffered saline. Glycinergic/GABAergic postsynaptic potential (PSP) was evoked by 200 μs 30–60 V pulses given through a glass stimulating electrode filled with ACSF. The recorded cell was kept in i-clamp mode except when Egly was measured. After the glycine response of spontaneous activity/Vm had been examined, the Na+ channel blocker tetrodotoxin (TTX) (0.4 μm) and glutamate receptor blockers DNQX (6,7-dinitroquinoxaline-2,3-dione, 10 μm) and APV (2-amino-5-phosphonovaleric acid, 100 μm) were added, and a bias current was given to keep Vm at −75 to −80 mV. I-clamp recording was sampled at 20 kHz and low-pass filtered at 10 kHz. The pipette capacitance was compensated in both i-clamp and v-clamp mode.
Egly measurements.
Voltage ramps (v-ramp) in v-clamp were used for measurement of Egly and were sampled at 40 kHz and low-pass filtered at 10 kHz. The v-ramp protocol consisted of four consecutive runs of a 1-s-long unit stimulus that included a short −5 mV step followed by a 130 ms depolarizing ramp spanning 18–20 mV around the expected Egly of the recorded cell (Vhold = −75 mV) (supplemental Fig. S1Bi, available at www.jneurosci.org as supplemental material). Glycine was puffed after the −5 mV step during the first and third runs. The average ramp voltage where the glycine responses and control responses crossed was taken as the raw Egly. The Rs during an Egly measurement was estimated from dividing 5 mV by the average amplitude of the capacitative transient current (in nanoamperes) of the −5 mV step. The raw Egly was corrected for the voltage error from the Rs with the following formula: Egly = raw Egly − [(average clamp current at raw Egly) × Rs]. The inclusion criteria for Egly data was Rs <60 MΩ. The Rs dropped to 20–40 MΩ within 40 min of forming a seal in most cells used in Egly measurements.
The resting Egly (measured >150 s after any depolarization-induced spiking) was measured after an initial ∼10 s clamp to −75 mV. As shown in supplemental Fig. S1Di, available at www.jneurosci.org as supplemental material, a slow, negative drift in the resting Egly was found in most cells. As the perforated-patch recording pipette contained 150 mm Cl−, the negative drift was opposite to that expected if the pipette Cl− leaked into the cell. The negative drift in Egly was also recognized in i-clamp mode from the change in Vm response to glycine over time. The negative drift occurred both with the dye-free and dye-containing recording solution and was still observed when different pipette salts (145 K-gluconate, 4 NaCl, 4 NaOH, 10 HEPES or 140 KCl, 6 MgCl2, 4 KOH, 10 HEPES) were used. Occasionally, a positive drift in resting Egly was seen under control conditions and was followed within a few minutes by the rupture of perforated patch; data from these recordings were not included.
We did not determine resting Egly by plotting the amplitude of glycine-induced currents versus holding potential in v-clamp because the Egly seemed to drift toward the holding potentials, particularly when the new potential was below −80 mV or above −60 mV, suggestive of a passive Cl− conductance. The v-ramp protocol was adopted to minimize the drift of Egly during measurements and to allow the activity-induced change in Egly to be followed (see Results).
Fluorescent imaging.
Imaging experiments with the pH-sensitive dye SNARF-5F (SNARF) were generally performed with a monochromator-based imaging system (Polychrome V, Till Photonics) attached to the Olympus BX51WI microscope. The excitation wavelength was 547 nm, and emission was long-pass filtered above 600 nm (Chroma E590LPv2). Fluorescent images were acquired with an IMAGO QE cooled charge-coupled device camera (Till Photonics) controlled by the TILLvisION 4.0 software. SNARF-5F was loaded into a cell by including the AM ester form (43 μm) in the recording pipette from which it diffused through the perforated patch and was de-esterified inside the cell (Filosa et al., 2002). The fluorescent intensity of the recorded cell increased with time as the dye accumulated (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). To minimize phototoxicity and to keep the average raw intensity of cell body in different image series within a narrow range, the images were obtained (at 1 Hz) with 4 × 4 binning, and the exposure time was adjusted as the baseline intensity increased (e.g., 150 ms for first imaging series collected ∼30 min after the seal, gradually shortened to 50 ms over the next hour). For simultaneous pH-Ca or pH-Cl imaging, two-photon scanning microscopy was used. Images of 256 × 256 pixels were acquired with the Ultima system by Prairie Technologies using a Chameleon Ultra II Ti:sapphire pulsed laser (Coherent) (Roberts et al. 2008). For pH and Ca2+ imaging, the AM forms of SNARF-5F and either Fura-2 (100 μm) or Fluo-4 (76 μm) were included in the recording pipette for loading. Ca2+ dyes did not load as well as SNARF-5F through the patch membrane, but Fluo-4 loading was improved by adding Pluronic F-127 (final concentration, 0.017%). The excitation wavelength was 800 nm for simultaneous pH-Ca imaging with Fluo-4, and 780 nm with Fura-2. For concurrent pH and Cl− imaging, Cl−-sensitive dye MQAE [N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide] was loaded into the DCN slice by incubating the slice in 3 mm dye in ACSF for 8 min at 34°C, and SNARF-5F was loaded from the recording pipette. MQAE and SNARF-5F were both excited by 750 nm laser light. For pH and Ca2+ imaging, fluorescent emission was split into two photomultiplier tubes using a dichroic mirror and bandpass filters for red (SNARF-5F) and green (Fluo-4 and Fura-2) light. For pH and Cl− imaging, the green bandpass filter was removed to collect all emission below 560 nm for the weak MQAE signal. As shown in an example of simultaneously recorded MQAE and SNARF-5F images (see Fig. 6Bi below), some of MQAE's fluorescence was caught in the red channel for SNARF-5F, but its impact on the SNARF signal was probably negligible, considering the weak MQAE signal. The baseline MQAE fluorescence did not noticeably decrease, as expected from dye leakage or bleaching, either during a 150 s run of experiment or over the ∼1 h recording period. However, as experiments evoking a change in MQAE signal were repeated over time, the response magnitude became attenuated. With time, the typical inhomogeneous MQAE staining of the cell body (Marandi et al., 2002) also became more homogeneous. We suspect that the time-dependent loss of response is due to MQAE's conversion to a hydrolyzed (de-esterified) form having a reduced Cl− sensitivity (Verkman, 1990; Koncz and Daugirdas, 1994). No glycine puff was given during two-photon imaging experiments.
Figure 6.

Ca2+-dependent intracellular acidification. A, Simultaneous monitoring of pHi and Egly with respect to 8 s of simple spiking (red) and complex spiking (black) in one cell. Arrowheads indicate glycine (2 mm) responses. The pH-sensitive dye SNARF-5F's signal is the average fluorescence intensity of a region of interest drawn inside the cell body. An intracellular acidification, manifested as a decrease in SNARF's signal, occurred during both simple spiking and complex spiking. The black bar above SNARF traces indicates the duration of 8 s depolarizing current injection. B, The intracellular acidification was also seen with Ca2+ spiking (in TTX) and inhibited by zero-Ca2+ (replaced with Co2+ or Mg2+) or 300 μm Cd2+. Bi–ii, Simultaneously recorded SNARF signal and Egly from two cells. The 8 s depolarizing current injections evoking Ca2+ spikes or just depolarization after Ca2+ channel block are marked with thick bars above SNARF traces. The amount of injected current is shown beside each symbol along with the number of evoked Ca2+ spikes in parenthesis. An example of complete block (Bi) and incomplete block (Bii) of the depolarization-induced acidification by zero-Ca2+ is shown. Biii, Relation between the change in SNARF signal to the peak negative Egly shift in control condition and the peak positive Egly shift in Ca2+ block condition for eight cells. C, The changes in pHi and [Ca2+]i induced by 8 s complex spiking were detected by simultaneous two-photon imaging of SNARF-5F and Fluo-4 or Fura-2. Examples from two different cells are shown. The duration of 8 s depolarizing current injection evoking complex spikes is indicated by the shaded rectangle. The excitation wavelengths were 800 nm (Ci) and 780 nm (Cii).
For both single-photon and two-photon image series obtained, the “signal” was extracted from the average intensity (in arbitrary units) of a region of interest drawn along the periphery of the cell body (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). The average intensity of a background region was subtracted from the SNARF signal but not from other dyes' signals because the signal-to-noise ratio was worsened by background subtraction. The time plot of fluorescence signal over a 150–200 s period was corrected for the up-sloping baseline by fitting a straight line along the control period and subtracting the line from the signal. The initial intensity value before the line subtraction (F0) was then added back to all points, and the plot of (F − F0)/F0 (“ΔF/F”) was produced. For the 800 s image series involving weak acid or weak base challenge, the fitted straight line was not subtracted but used as F0 in calculating (F − F0)/F0 to prevent the overestimation of ΔF in the latter part of the 800 s period. Although a linear increase in the baseline fluorescence was assumed for convenience, not infrequently the baseline-corrected SNARF-5F signals were found to deviate from 0 at the end of a 160 s series when an intensity-attenuating response (acidification) was expected to have terminated. In such cases it was unclear whether it was a true signal or due to the failure of linear baseline correction. We mostly focused on the peak change in signal occurring within 30 s from the control period, during which the linear extension of baseline is less likely to fail. Occasionally, a small, abrupt increase in SNARF signal occurred that was not associated with a stimulus or a change in Vm (see Fig. 5A, black, and 6A and 8Aii, gray, below); the origin of this change is unknown. The peak signal in a time plot was selected by eye for single-photon SNARF-5F data and by curve fitting or from the intersection of two fitted straight lines for the two-photon MQAE or SNARF-5F data, respectively.
Figure 5.
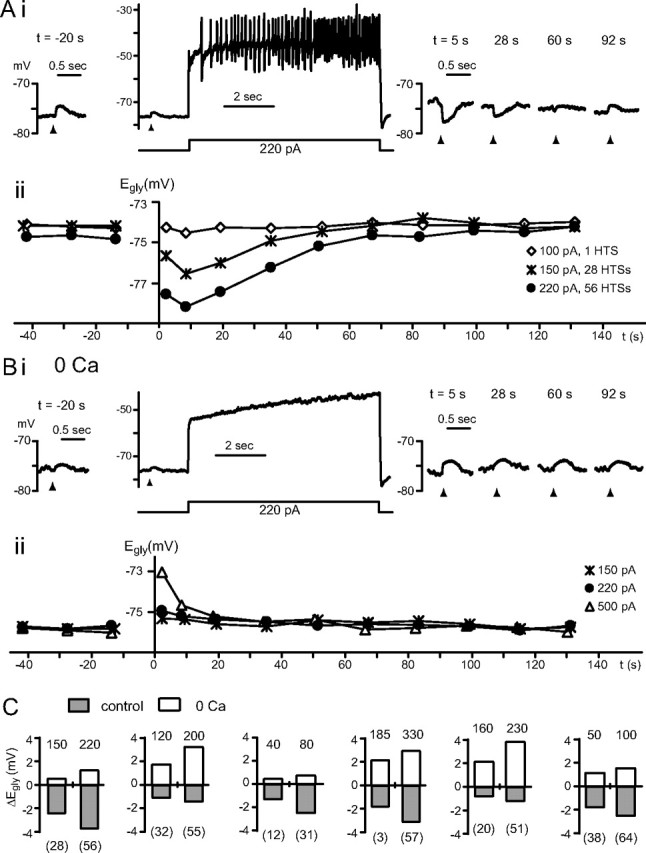
Ca2+-dependent and -independent change in Egly. A, B, Data from one cell comparing glycine responses and Egly measurements in control condition (A) with those in zero-Ca2+ (B). Time 0 is the moment the 8 s depolarizing current injection terminated. Ai, After 56 high-threshold Ca2+ spikes induced with a 220 pA injection, the glycine response shifted negative. Aii, The Egly series measured along with the Vm recording in Ai is shown (●) with two other series obtained with different amounts of current injections. The negative Egly shifts peaked at 8.5 s. HTSs, High-threshold spikes. Bi, ii, The Vm and Egly series in zero-Ca2+ (replaced with Mg2+) are displayed in the same way as in A. After a depolarization in zero-Ca2+, no negative shift but a positive shift in Egly occurred. Bias current was −40 pA in Ai and −55 pA in Bi. C, Bar graphs showing the peak change in Egly after an 8 s current injection (amount in pA indicated above each bar) in control condition and in zero-Ca2+ from six cells. The number of Ca2+ spikes evoked is shown in parentheses under each bar belonging to control conditions. All data were obtained in TTX.
Drug application.
All the pharmacological agents except glycine and GABA were applied by bath perfusion. DNQX (10 μm) and APV (100 μm) were coapplied with TTX. H2DIDS (4,4′-diisothiocyano-1,2-diphenylethane-2,2′-disulphonic acid) was directly dissolved in ACSF on the day of each experiment, but other drugs were diluted from a stock solution in water (TTX, APV, glycine, GABA) or DMSO (all others). Drugs were obtained from Sigma-Aldrich with the exception of TTX (Alomone Labs), APV, DNQX (Ascent Scientific), and H2DIDS (Invitrogen). Carboxyeosin diacetate used was from Sigma-Aldrich (n = 3) or Invitrogen (n = 2). All fluorescent dyes and Pluronic F-127 were from Invitrogen. When CdCl2 or CoCl2 was used, KH2PO4 in the bathing solution was replaced with KCl to prevent precipitation.
Data analysis.
Data were analyzed with Clampfit (Molecular Devices), Microsoft Excel, and KyPlot (KyensLab). Numerical values were given as mean ± SD where available. Two-tailed t test (paired or unpaired) or Kolmogorov–Smirnov test was used to compare two groups of data, and one-way ANOVA and the multiple-comparisons test (Tukey–Kramer test) were used for three or more groups of data. The level of significance was at 0.05 for all statistical tests.
Immunohistochemistry.
Mice, of ages from postnatal day 23 (P23) to P27, were anesthetized with isoflurane and then perfused transcardially with PBS followed by 4% paraformaldehyde (in PBS). The brain was removed and postfixed in 4% paraformaldehyde at 4°C for 2.5 h. Thirty-micrometer-thick coronal brainstem sections containing the DCN were cut using Leica VT1000S and then boiled in 10 mm sodium citrate, pH 6.0, for 20 min using a microwave oven. After cooling, sections were blocked for 1 h in 2% normal goat serum/0.2% Triton X-100 (in PBS) and then incubated overnight at 4°C with one of two clones of mouse monoclonal antibodies against the SLC4A8 gene product, which is human NDCBE (clones 1G10 and 6E11; Abnova). Clone 1G10 was diluted at 1:100 and 6E11 at 1:75 in the block solution. Clone 1G10 has been tested by the manufacturer to be cross-reactive to mouse protein in Western blot analysis. The next day, sections were washed in PBS and incubated with goat anti-mouse IgG conjugated to Alexa Fluor 488 (Invitrogen; 1:500 in block solution). After 2 h, sections were washed in PBS, mounted on gelatin-coated slides, and dried. Section were then delipidized by going through ascending series of alcohols and xylene and descending series of alcohols and water, in sequence. Slides were coverslipped using Fluoromount G medium (Southern Biotech). Confocal laser-scanned images of sections were obtained with an Olympus FV1000 microscope with a 60× oil-immersion objective (NA, 1.42) under the control of Olympus Fluoview-1000 software.
Results
Glycine response of cartwheel cells
Depolarizing, excitatory glycine responses have been reported in CWCs using microelectrode or extracellular cell-attached recording [Golding and Oertel, 1996 (mice, P18–26); Tzounopoulos et al., 2004 (mice, P18–22)]. We reevaluated the prevalence of depolarizing glycine responses using gramicidin perforated-patch recording and found that cells varied in their response from depolarization to hyperpolarization, suggestive of variable glycine reversal potential (Egly). For spontaneously spiking cells, the effect of glycine was categorized as excitatory, inhibitory, or mixed. Excitation (Fig. 1A) was recognized by an increase in spike frequency with an obvious depolarization. Mixed responses (Fig. 1B) consisted of a depolarized pause in spiking followed by higher-frequency firing at the decay of the response. Reducing the puff pressure or duration for this group did not reveal an increase but rather a decrease or no change in spiking at the onset of response. This is expected if Egly is a few millivolts below the spike threshold, such that shunting depolarization provided a platform for higher-frequency spiking after glycine receptors close (Gulledge and Stuart, 2003). An inhibitory glycine response (Fig. 1C), a decrease or pause in spiking, was observed, with the Vm driven to levels from ∼5 mV below the fast-spike threshold (∼−65 mV) to −84 mV. The glycine responses in CWCs that did not spike spontaneously (Vm −81.4 ± 2.8, n = 127) consisted of depolarizing or hyperpolarizing deflections of Vm; in some of these cases (11 of 127), spikes were evoked at the peak of a depolarizing response. The proportions of cells showing the three types of glycine response are shown in Table 1; for the standard pipette solution (“no dye”), the excitatory, mixed, and inhibitory proportions were 31, 12, and 57%, respectively (n = 113).
Figure 1.

Three types of glycine response in spontaneously spiking CWCs. A–C, Examples of excitatory (A), mixed (B), and inhibitory (C) responses to a glycine puff during spontaneous activity. The duration of the glycine puff is indicated with a bar, and the glycine concentrations were 0.5, 0.5, 0.5, 0.5, 2, and 2 mm for the six cells in left-to-right, top-to-bottom order.
Table 1.
Numbers and proportions of glycine response types in spontaneously spiking CWCs
| n (%) | exc. (%) | mix. (%) | inh. (%) | |
|---|---|---|---|---|
| No dye | 113 | 35 (31.0) | 14 (12.4) | 64 (56.6) |
| Complex spiking | 30 (26.5) | 1 (3.3) | 3 (10.0) | 26 (86.7) |
| Simple spiking | 83 (73.5) | 34 (41.0) | 11 (13.3) | 38 (45.8) |
| AM dye | 129 | 22 (17.1) | 23 (17.8) | 84 (65.1) |
| Complex spiking | 17 (13.2) | 0 (0) | 3 (17.6) | 14 (82.4) |
| Simple spiking | 112 (86.8) | 22 (19.6) | 20 (17.9) | 70 (62.5) |
| Total | 242 | 57 (22.3) | 37 (15.3) | 148 (61.2) |
| Complex spiking | 47 (19.4) | 1 (2.1) | 6 (12.8) | 40 (85.1) |
| Simple spiking | 195 (80.6) | 56 (28.7) | 31 (15.9) | 108 (55.4) |
Percentage values in parentheses in the n column are the proportion of complex-spiking or simple-spiking cells. exc., Excitatory; mix., mixed; inh., inhibitory.
Several potential factors, either biological or experimental, could influence the distribution of response polarity. For example, higher proportion of inhibitory responses was observed in cells recorded with a pipette solution containing an AM-ester dye (SNARF-5F or Fluo-4, Table 1, see data below on imaging). Another potential factor that could impact the distribution is postnatal age, as intracellular [Cl−] is higher during the first 1–2 weeks after birth, resulting in transient excitatory response to GABA or glycine (Ben-Ari et al., 2007). However, no significant difference was found in the age distribution of all cells showing excitatory responses vs. inhibitory responses (Kolmogorov–Smirnov test, p = 0.99; 22.4 ± 2.4 d, n = 57, excitation vs 22.4 ± 2.6 d, n = 148, inhibition). No difference was present in the ages of mice recorded with or without AM dyes (Kolmogorov–Smirnov test, p = 0.65; 22.3 ± 2.5 d, n = 113, no dye vs 22.5 ± 2.5 d, n = 129, with dye). Thus, age is not a factor affecting glycine responses in our dataset. More interestingly, we observed an association between the type of glycine response and whether the cell's spontaneous activity included complex spikes or was all simple spiking. In the current study, 19% of spontaneously spiking cells (47 of 242) were complex spiking, and among these the excitatory response was observed in only one, while the inhibitory response was seen in 85.1% of them (Table 1). This suggests a possible relation between Egly and complex spike activity, as described below.
Activity-dependent shift in the glycine response
A series of experiments were designed to obtain a qualitative description of the effects of spike activity on glycine responses, and the results suggested the cellular mechanisms tested later in this study. Four cells showing depolarizing glycine responses, two simple-spiking cells and two silent cells, were induced to fire complex spikes for a long period by sustained depolarizing current injection (50–150 pA for 57–137 s in different cells), and their glycine responses were monitored in 10–15 s intervals. As shown in Figure 2A, the glycine responses shifted negative, i.e., becoming less excitatory, or more inhibitory, as complex spiking continued, and when the spiking was terminated, a hyperpolarizing glycine response was observed, which shifted back to depolarizing over the next 100–200 s. These results suggest a negative shift in Egly.
Figure 2.
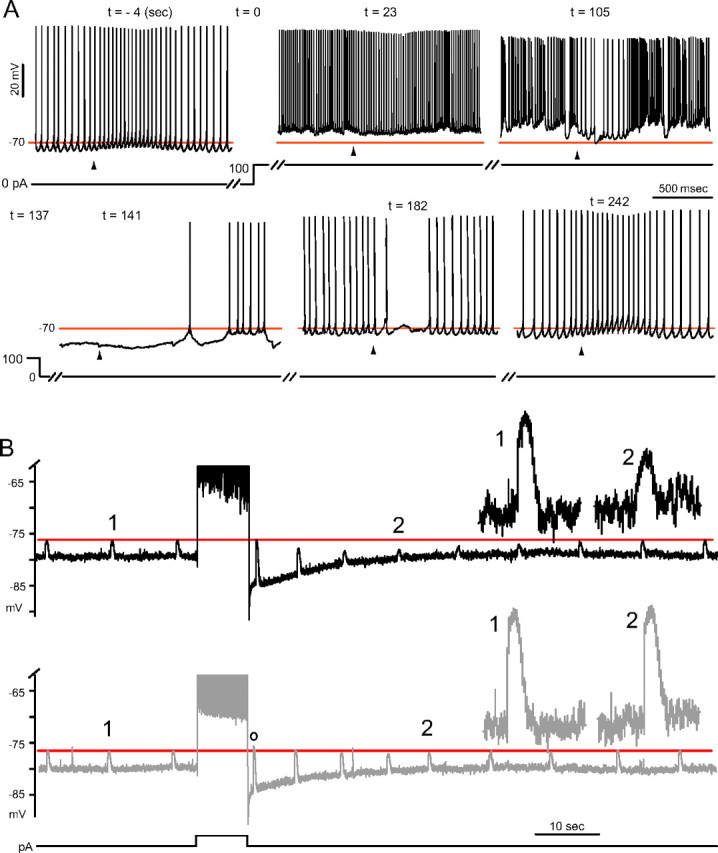
Activity-dependent shifts in the glycine response. A, An example of negative glycine response shift occurring with prolonged complex spiking. An all-simple-spiking cell with an excitatory glycine response was injected 100 pA at t = 0. Complex spikes began appearing at t = 27 s. The weakly inhibitory effect of glycine at t = 23 s became more inhibitory (t = 105 s) as complex spiking continued. After termination of depolarization, the glycine responses were hyperpolarizing until the fourth response (t = 182), and the return of the excitatory response took ∼100 s (t = 242 s). Arrowheads indicate time of a 500 μm glycine puff, and the horizontal line is drawn at −70 mV for reference. B, Comparison of the change in glycine responses following simple and complex spiking. An all-simple-spiking cell with an inhibitory glycine (2 mm) response was silenced with −110 pA bias current and induced to fire late complex spikes (top) or trains of simple spikes (bottom), with an 8 s step of 250 pA and 170 pA, respectively. The horizontal line is aligned to the peaks of glycine responses before the evoked activity. Insets show magnified responses at time points marked “1” and “2.” A positive shift in the glycine response occurred (o) immediately after simple spiking.
To contrast the relative effects of simple and complex spikes, the type of spikes in the train were controlled by the magnitude of a given duration of the stimulus. To facilitate the comparison of glycine response amplitudes, cells were silenced to −80 mV, and six cells showing depolarizing glycine response at −80 mV were chosen for further analysis. As shown in Figure 2B (top), although a reversal to a hyperpolarizing glycine response did not occur, complex spiking resulted in a reduction in the peaks of glycine responses (contrast responses 1 and 2; mean shift −1.7 ± 0.7 mV, n = 6) 20–30 s after the stimulus ended. In contrast, after simple spiking (including cases where the single onset complex spike was present) (Fig. 2B, bottom) such a reduction was barely noticeable (−0.1 ± 0.2 mV, n = 6, p = 0.005, paired t test). Interestingly, however, the peak of the first glycine response after simple spiking (o) was 0.1–3.3 mV more positive than that before the stimulus, despite riding on a prominent afterhyperpolarization (four of six cells). A positive shift in Egly is expected to develop during a long depolarization and to decay on repolarization in the presence of a passive Cl− conductance, as [Cl−]i would change along with the Vm. A similar negative shift was seen in GABAA component of the responses to puffing 500 μm GABA after complex spiking (8–14 s duration, n = 7 cells, data not shown).
CWCs receive mixed glycinergic/GABAergic synapses from other CWCs (Roberts et al., 2008). We evoked glycinergic/GABAergic PSPs by stimulating (0.5–0.7 Hz) in the deep layer of DCN in the presence of glutamate receptor blockers (100 μm APV, 10 μm DNQX). Complex spiking induced a reversal in the polarity of evoked PSPs from depolarizing to hyperpolarizing, indicating a negative shift in Egly (n = 8) (Fig. 3A,B). For three cells in which the evoked PSPs were hyperpolarizing even when Vm was brought near −80 mV, 10 s of complex spiking made the PSPs still more hyperpolarizing (Fig. 3B, cell 3). The difference in the average amplitude of PSPs between the preactivity period and the 15th to 20th second postactivity period was from −0.4 to −2.2 mV in 10 cells (average, −1.3 mV, complex spiking induced for 10∼16 s by 100∼250 pA).
Figure 3.

Change in the reversal potential of glycinergic PSPs. A, Glycinergic/GABAergic PSPs were evoked at 0.5 Hz in a cell exhibiting spontaneous bursts of simple spikes. Four segments from a 150-s-long Vm recording are shown in sequence. Complex spiking was induced after 20 s of control period by 75 pA injection (depol) for 30 s. The PSPs were depolarizing at a Vm of −79 mV during the pre-depolarization period. Just after the depolarization and complex spiking, PSPs were hyperpolarizing even at −81 mV but became depolarizing at −79 mV again over the next 100 s. PSPs were recorded in the presence of 100 μm APV and 10 μm DNQX. B, Evoked glycinergic/GABAergic PSPs shifting negatively after complex spiking. The bias currents during the pre-depolarization period for cells 1, 2, and 3 were −45, −60, and 0 pA, respectively, and the current was adjusted after depolarization to hold the Vm close to the pre-depolarization level.
The CWC's complex spike requires Ca2+ channels (Kim and Trussell, 2007), and thus a complex spike generates a larger rise in intracellular Ca2+ than does a simple spike (Molitor and Manis, 2003; Roberts et al., 2008). To investigate whether the Ca2+ influx is sufficient for the negative shift in the glycine response to occur, high-threshold Ca2+ spikes were evoked in the presence of Na+ channel blocker TTX, 0.4 μm. Ca2+ spikes were evoked in seven cells that had a depolarizing glycine response at Vm = −80 mV, and glycine was puffed every 8 s (Fig. 4A). In all seven cells, the glycine response increased by the third puff during Ca2+ spiking. After Ca2+ spike trains, the Vm was depolarized compared with the pre-Ca2+ spiking level, lacking the afterhyperpolarization seen after complex spiking. This depolarization actually helped reveal the negative shift in Egly as the peaks of hyperpolarizing glycine responses after Ca2+ spiking (at −78 to −93 mV in five cells) were clearly more negative than the depolarizing peaks of glycine responses before Ca2+ spiking (at −79 to −74 mV).
Figure 4.

Ca2+ spiking leads to Egly shifts. A, Glycine responses (2 mm, marked by arrowheads) shifted negative during a prolonged period of Ca2+ spiking. Spikes were evoked for 42 s by +70 pA from −30 pA bias. A negative shift and recovery in Egly was evident as the polarity of glycine responses changed from depolarizing to hyperpolarizing after Ca2+ spiking and back to depolarizing. The hyperpolarization to −80 mV at 2 s into the stimulus was an afterhyperpolarization of the first Ca2+ spike, and the fluctuation of Vm during 10 s after Ca2+ spiking is thought to be a manifestation of the intrinsic bistability of CWCs. B, Plot of the peak negative (neg) shift in Egly versus the number of high-threshold Ca2+ spikes evoked during an 8 s challenge protocol. Dots represent 148 Egly series from 83 CWCs. 104 series are from 55 cells recorded with AM dye solution and 44 series from 28 cells without dye. C, The shifts in Egly at 2 s after an 8 s Ca2+ spiking (i), complex spiking (ii), or simple spiking (iii) are plotted against the peak negative shift of the Egly series. Dots in each plot represent single Egly series from different cells, with n = 44, 29, and 8 for i, ii, and iii, respectively. All data were obtained in TTX except for that shown in Cii, iii.
Measurement of Egly
We used a voltage-ramp protocol to measure Egly and document the time course of its change after spike-train stimuli (supplemental Fig. S1Bi, available at www.jneurosci.org as supplemental material; also see Materials and Methods). To avoid the slow negative drift in Egly (supplemental Fig. S1Di, available at www.jneurosci.org as supplemental material; also see Materials and Methods), representative resting Egly was assessed as early as possible during patch perforation, as long as Rs was <60 MΩ (in TTX). With this criterion, the mean resting Egly from 164 cells was −74.3 ± 5.8 mV. Table 2 lists Egly values subgrouped with respect to spontaneous activity, response to glycine, and presence of AM dye in the recording pipette. The difference between spiking cells' Egly and that of silent cells was insignificant (t test, p = 0.60). Egly was most negative in cells with inhibitory responses and most positive for those with excitatory responses. The difference among the three response groups was significant (one-way ANOVA, p < 0.001). While the Egly difference between the excitatory group and mixed group was not significant in pairwise comparison (Tukey–Kramer test, p = 0.56), the differences in the other pairwise group comparisons were significant (p < 0.001). The mean Egly of complex spiking cells, which showed a higher proportion of inhibitory glycine responses, was not significantly more negative than that of simple spiking cells (−76.2 ± 5.6, n = 17, vs −73.7 ± 5.8, n = 84; p = 0.12, t test). For comparison with CWCs, we examined glycine responses in fusiform cells which are the principal neuron of the DCN and are postsynaptic to CWCs. All showed hyperpolarizing, inhibitory responses to glycine (n = 28) and the Egly measured in four cells was −83.9 ± 0.7 mV (in TTX). That the fusiform cell showed more negative Egly than the CWC and only inhibitory responses to glycine is in agreement with Golding and Oertel (1996).
Table 2.
Resting Egly of cartwheel cells
| n | Egly(mV) | SD | Range | |
|---|---|---|---|---|
| Total | 164 | −74.3 | 5.8 | −87.4, −58.0 |
| Spiking | 101 | −74.0 | 5.9 | −87.0, −58.0 |
| exc. | 24 | −68.5 | 4.6 | −75.9, −58.0 |
| mix. | 16 | −69.9 | 3.0 | −77.1, −65.5 |
| inh. | 61 | −77.3 | 4.5 | −87.0, −68.3 |
| Silent (Vm −81.4 ± 2.8 mV) | 63 | −74.5 | 5.8 | −87.4, −60.8 |
| No dye | 92 | −73.1 | 5.3 | −85.0, −58.0 |
| SNARF–AM | 72 | −75.6 | 6.3 | −87.4, −63.2 |
See supplemental Discussion, available at www.jneurosci.org as supplemental material, for the possible cause of more negative Egly in AM dye-loaded cells. exc., Excitatory; mix., mixed; inh., inhibitory.
A protocol using mixed voltage- and current-clamp recording modes was used to monitor the time course of Egly shifts in relation to a period of activity. Vm and glycine responses were recorded in current clamp for 150–180 s interrupted every 15 s or less by the ramp protocol; an ∼8 s burst of complex or Ca2+ spiking was induced at t = 35 s (supplemental Fig. S1A–C, available at www.jneurosci.org as supplemental material). Shown in Figure 4B is the distribution of peak negative Egly shifts (difference between the most negative Egly after Ca2+ spiking and the mean of pre-Ca2+ spiking values) versus the number of Ca2+ spikes evoked during the 8 s depolarization (n = 148 measurements; Pearson's r = −0.66, p < 0.001). Initially, we evoked enough Ca2+ spiking in each cell to recognize a clear negative shift in glycine responses during current clamp after the stimulus (supplemental Fig. S1A–C, available at www.jneurosci.org as supplemental material) (Fig. 5A) and confirmed that the Egly shifted negative. However, for cells with the largest shifts (>5 mV), Egly would often fail to recover fully within 120 s after the Ca2+ spiking or would settle to a more negative level than the control level. Additionally in some cases Ca2+ spiking would continue beyond the 8 s current injection. Therefore, we adjusted the current injection in each cell to prevent runaway spiking and to achieve ≤5 mV of negative Egly shift. As shown in Figure 5A, the most negative shift in Egly occurred at one of the first three measurements after Ca2+ spiking (at 2, 8.5, or 19.5 s, mean 9.3 ±.5.9 s, n = 61 series with ≥1.5 mV peak negative shifts from 61 cells). Restoration of Egly occurred over 100–130 s, and single exponential fits for recovery gave a mean time constant of 35 ± 11 s (n = 34 cells).
The 8 s challenge protocol was also run in cells in the absence of TTX to examine the Egly shift with complex or simple spiking. Egly shifted negative by up to 4 mV after an 8 s stimulus evoked with a maximum of 250 pA injection (Fig. 4Cii). In 32 Egly series (from 32 cells, 29 with AM dye and 3 without dye) where >1 mV peak negative shift occurred with complex spiking, the time of peak was most often (18 of 32 cells) at the 19.5 s point, and two had the peak at 34 s. Thus, the average peak time (16.3 ± 7.8 s, n = 32) was later than with Ca2+ spiking. The first data point (2 s) after complex spiking was not as negatively shifted as that after Ca2+ spiking, and showed a small positive shift in some cases. Figure 4C shows the maximal negative shift in Egly plotted against the shift at the 2 s time point for individual experiments obtained with Ca2+ spiking (limited to 1–4 mV shifts for comparison with complex spiking-induced shifts), complex spiking and simple spiking. These data show that with only Ca2+ spiking, the magnitude of peak negative shifts was greater, and an initial positive shift was rarely observed. Such initial positive shifts were more common with complex spiking, and especially with simple spiking; as discussed below, these positive shifts appear to reflect passive elevation in intracellular Cl− that are opposed by a Ca2+-dependent mechanism.
Ca2+-dependent and passive Egly shifts
To test whether Ca2+ influx triggers the negative shift of Egly, Ca2+ was removed from the ACSF (“zero-Ca2+”, MgCl2 substitution of CaCl2, in TTX, n = 8). Removal of Ca2+ depolarized Vm (restored to between −75 and −80 mV with bias current), and shifted resting Egly from +2 to −4 mV in different cells. As illustrated in Figure 5B, 8 s current injection in TTX and zero-Ca2+ depolarized the cells strongly but without spiking activity. Following the stimulus, there was no negative shift in Egly, even with currents as large as 500 pA (n = 8 cells). Rather, the Egly immediately shifted positive after the strongest depolarization, and this positive shift decayed within 60 s (Fig. 5Bii). While the negative shift in Egly when spikes were present peaked at 8.5 s or 19.5 s after stimulus in six of the eight cells, the positive shift in zero-Ca2+ peaked at 2 s (the first measured time point) in seven of eight cells. The simplest explanation for the transient positive shift in Egly is a rise in intracellular Cl− through a passive conductance during the depolarization. Such a Cl− conductance is expected also to mediate the influx of Cl− during the control condition with Ca2+ spiking. The fact that the Egly was often more negative than the control level soon after (at 2 s) Ca2+ spiking and that it could drop further during the next 10 or 20 s indicates that Cl− was actively removed or that a constitutive Cl− accumulation was inhibited by a Ca2+-triggered mechanism. Figure 5C illustrates in six cells the contrasting direction of Egly shifts induced by the same amount of current injection in control and zero-Ca2+, as well as the tendency for both directions of shifts to increase with the amount of current injected. The average of the largest negative shift from each cell was −2.4 ± 2.2 mV and that for the positive shift was 2.2 ± 1.2 mV. Thus, the Ca2+-dependent process opposed a passive process, shifting Egly negative by nearly 5 mV in this protocol.
A hypothesis for the mechanism of Ca2+-dependent negative Egly shift
A negative shift in Egly is expected to result from a reduction in intracellular Cl− and/or HCO3−. HCO3− is less permeable than Cl− through glycine/GABA receptors (Bormann et al., 1987), and at constant PCO2, the intracellular concentration, [HCO3−]i, is expected to be set by the intracellular pH (pHi) (Roos and Boron, 1981). On the other hand, [Cl−]i in neurons may be regulated by KCC2, NKCC1, and the anion (Cl−-HCO3−) exchangers, either Na+-independent (AE) or Na+-driven (NDCBE) forms (Farrant and Kaila, 2007) (see Fig. 9Di). To date, the specific expression of each transporter species in CWCs is not known, but we assumed that all four kinds are functioning to develop a hypothesis for the mechanism of Ca2+-dependent negative Egly shift. For KCCs or NKCCs to cause a decrease in [Cl−]i with Ca2+ spiking, their activity needs to be increased or decreased, respectively, with a rise in intracellular Ca2+. However, given the depolarized Vm during Ca2+ spiking, extracellular K+ could elevate, and the electrochemical driving force thus may not be in favor of KCC transporting Cl− out of the cell. Moreover, blockade of NKCC with bumetanide did not prevent the effect of Ca2+ spiking on Egly (supplemental Results, available at www.jneurosci.org as supplemental material). The Cl−-HCO3− exchangers, AE and NDCBE, have been studied mostly in the context of pHi regulation: AE mediates influx of Cl− while exporting HCO3− activated by intracellular alkalinization, and NDCBE, known as an acid extruder, moves Cl− out in exchange for HCO3− driven by the Na+ gradient (Chesler, 2003; Romero et al., 2004). Intracellular acidification has been shown to occur with spiking activity or depolarization in various types of neurons often in a Ca2+-dependent way (Trapp et al., 1996a), and the proposed mechanisms are replacement of H+ by Ca2+ in intracellular binding sites, mitochondrial Ca2+ uptake leading to H+ release and cytosolic Ca2+ removal by Ca2+-H+ ATPases of plasma membrane (PMCA) or endoplasmic reticulum (SERCA) (for review, see Ballanyi and Kaila, 1998; Chesler, 2003).
Figure 9.
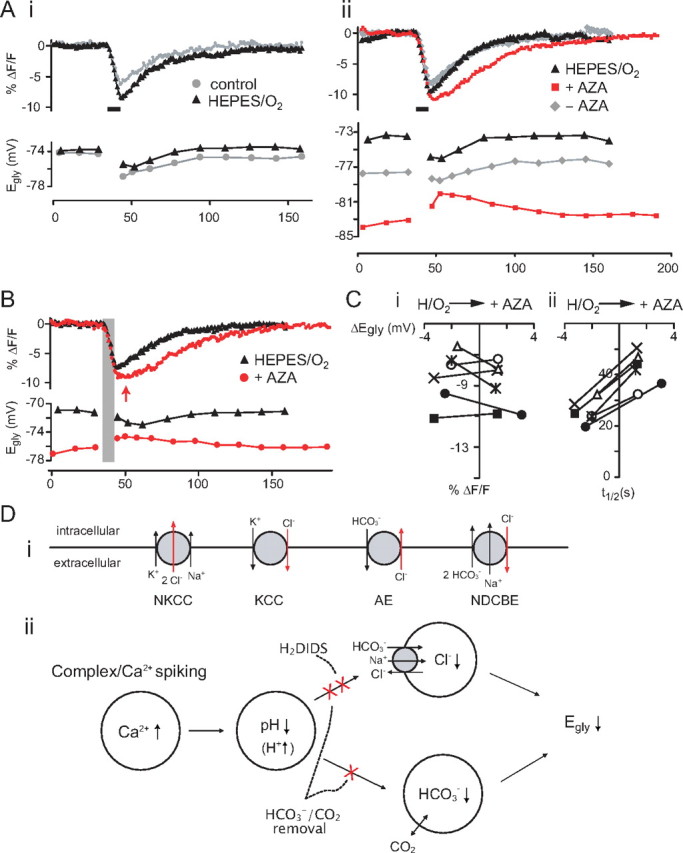
Block of the negative Egly shift by HCO3−/CO2 removal. A, The effect on Egly and pHi of removing HCO3−/CO2 from perfusion (HEPES/O2) (i) and then adding AZA (50 μm) (ii) in one cell. Superimposed control–treatment pairs of Egly/pHi series were chosen on the basis of similar numbers of evoked Ca2+ spikes during the 8 s depolarization. The periods of 8 s Ca2+ spiking are marked with thick bars. Ai, The injected current and number of evoked Ca2+ spikes were 180 pA/16 and 120 pA/18 Ca2+ spikes for the control and HEPES/O2, respectively. Aii, The injected current and number of evoked Ca2+ spikes were 130 pA/30, 110 pA/31, and 110 pA/15 Ca2+ spikes for HEPES/O2, after AZA addition (+AZA) and 17 min after removing AZA (−AZA), respectively. The effects of AZA were fully reversible if it had been applied for less than ∼15 min. B, Another example showing the slowed pHi recovery and positive Egly shift after addition of AZA in HEPES/O2 after Ca2+ spiking. Time of peak acidification in AZA is indicated with an arrow, and the period of Ca2+ spiking is shaded. The injected current and number of evoked Ca2+ spikes were 170 pA/23 and 160 pA/22 Ca2+ spikes for HEPES/O2 and +AZA, respectively. C, Plot of peak acidification (i) and half-recovery time (ii) against the peak negative Egly shift and the peak positive Egly shift for HEPES/O2 and +AZA for six cases (from different cells) in which the numbers of evoked Ca2+ spikes were similar between the two conditions. The number of evoked Ca2+ spikes in +AZA condition ranged from 20 to 47 in the six cases. The difference in peak acidification was not significant, but the half-recovery time was significantly lengthened in AZA added to HEPES/O2. D, Chloride transporters potentially affecting neuronal [Cl−]i (i), and the proposed mechanism of activity-dependent negative shift in Egly in CWCs (ii). KCC, K+-Cl− cotransporters; NKCC, Na+-K+-Cl− cotransporters; AE, Na+-independent anion (Cl−-HCO3−) exchanger; NDCBE, Na+-driven Cl−-HCO3− exchanger, also known as NDAE.
Therefore, we propose that complex or Ca2+ spiking leads to a Ca2+-dependent intracellular acidification, which leads NDCBE to extrude Cl− and cause a negative shift in Egly (see Fig. 9Dii). Also contributing to the negative shift in Egly could be the lowering of [HCO3−]i during the intracellular acidification (Kaila et al., 1993), provided that PCO2 is constant and carbonic anhydrase is present, thus promoting fast equilibration of the reaction, CO2 + H2O ↔ H2CO3 ↔ H+ + HCO3− (Roos and Boron, 1981). We tested key elements of this hypothesis, examining whether (1) the pHi decreased with Ca2+/complex spiking and the decrease was Ca2+ dependent, (2) Egly could shift negative with an intracellular acidification not associated with electrical activity, and (3) blocking NDCBE could eliminate the negative Egly shift.
Activity-dependent and Ca2+-dependent intracellular acidification
Changes in pHi were monitored with the fluorescent indicator SNARF-5F (SNARF). To maintain the perforated patch condition, the dye was introduced into the cell by including the AM ester form in the recording pipette, which we found would diffuse through the perforated patch and become de-esterified. This loading method led to a steady increase in the baseline intracellular fluorescence over the course of the recording due to accumulation of the de-esterified indicator (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). A change in pHi was detected by measuring SNARF emission at wavelengths >600 nm, where the fluorescence intensity decreased with a decrease in pH. SNARF images were taken at 1 Hz during a run of the 8 s challenge protocol to follow simultaneously the change in pHi and Egly after complex/Ca2+ spiking. Shown in Figure 6A are the concurrent changes in SNARF signal and Egly induced by simple (red) and complex (black) spiking in one cell. Similar profiles were observed in nine other cells. In all cells, the pHi fell during both complex and simple spiking, and the recovery to near control level occurred within the next 120 s beginning immediately after the spikes terminated. The degree of acidification, however, was greater with complex spiking than with simple spiking in each cell, with the peak SNARF signal 119–240% (mean, 171%) larger with complex spiking (p < 0.001, paired t test, n = 10). Complex spiking was evoked with 150–250 pA in different cells, and the resulting acidification and peak negative Egly shift were −9.5 ± 1.6 in %ΔF/F (n = 10) and −1.9 ± 0.2 mV (n = 6). The corresponding values for simple spiking, evoked with 70–200 pA in the same group of cells, were −5.7 ± 1.1 in %ΔF/F (n = 10) and −0.8 ± 0.2 mV (n = 6). The fact that complex spiking led to a larger pHi decrease than simple spiking suggests that the intracellular acidification may be proportional to the increase in [Ca2+]i. However, that an obvious negative Egly shift was only observed with complex spiking suggests that the intracellular acidification may need to be larger than a threshold level to be associated with a negative Egly shift.
Ca2+ spiking (in TTX) was found to cause the same response pattern, a drop in pHi during the activity, with recovery beginning upon termination of the activity (Fig. 6Bi,ii, controls). As illustrated in Figure 6Bi,ii, the acidification elicited by Ca2+ spiking under control conditions became unnoticeable (Fig. 6Bi) (n = 5 cells) or largely reduced (Fig. 6Bii) (n = 3 cells) when the same amount of current injection was given in zero-Ca2+ (replaced with Co2+ or Mg2+) or in 300 μm Cd2+. Switching to solutions containing 2.4 mm Co2+ (zero-Ca2+) or 300 μm Cd2+ caused the resting Egly to shift positive by 4–7 mV with an apparent shrinkage of the cell body. Nevertheless, there was an additional positive shift in Egly immediately after depolarization (n = 8 cells), just as in the previous experiments without SNARF. The %ΔF/F values at the end of the 8 s current injection are plotted against the peak negative Egly shift (in control condition) and the peak positive Egly shift (in Ca2+ block) for the eight cells in Figure 6Biii. The average reduction in acidification by Ca2+ block was 6.6 ± 0.9 in %ΔF/F for the 8 data series in the plot. In Ca2+ block conditions, another current injection 100–250 pA larger than that used for evoking Ca2+ spikes was given in seven of the eight cells. A larger current injection, i.e., larger depolarization, caused a more positive Egly shift in all cells, but no change in the pHi response (five of the seven cells) (Fig. 6Bi,ii).
Change in [Ca2+]i and pHi were monitored simultaneously with respect to an 8 s complex spiking taking advantage of the overlapping two-photon excitation spectra of the fluorescent indicators. The Ca2+ indicator Fluo-4 (n = 3) or Fura-2 (n = 5) was loaded into the cell along with SNARF by including their AM forms in the recording pipette. As shown in Figure 6C, the rise and fall in [Ca2+]i was rapid, and thus restricted to the period of spiking, in sharp contrast to the prolonged decrease in pHi. The [Ca2+]i peaked at the end of the depolarization and then within 5 s fell to ∼10–15% (three cells with Fluo-4) or 20–25% (five cells with Fura-2) of the peak. The relatively fast clearance of the Ca2+ rise compared with that of H+ (pHi recovery) after complex spiking suggests that the slow recovery of pHi is not secondary to a gradual recovery of Ca2+ but rather attributable to the rate of H+ removal and the intracellular H+ buffering capacity. These data confirm that both intracellular acidification and negative Egly shift are Ca2+ dependent.
Activity-independent changes in pHi and Egly
We next explored whether the pHi decrease is a necessary intermediate in the process of Ca2+-induced negative Egly shift. Preventing or reducing the pHi decrease during Ca2+ spiking by blocking the PMCA and SERCA was attempted using carboxyeosin, an inhibitor of Ca2+-H+ ATPases (Gatto et al., 1995). Unfortunately, this compound (diacetate form bath-applied at 40 or 80 μm) was not useful, due both to its intrinsic fluorescence, which precluded monitoring of pHi with SNARF, and to its suppression of Ca2+ spikes, with only 1–3 spikes evoked at the onset of an 8 s depolarization (see Choi and Eisner, 1999).
We asked instead whether Egly shifts negative with an activity-independent intracellular acidification induced by a weak acid (Roos and Boron, 1981) in CWCs. Kaila et al. (1993) had shown that the IPSP reversal potential shifted negative or positive during application of weak acid or weak base, respectively, in neocortical pyramidal cells. Sodium propionate, 20 mm, was perfused for 100–120 s while cells were held at −75 mV in voltage clamp (in TTX), with periodic Egly measurements. To maintain the same [Cl−]o throughout the period of Egly measurements, cells were bathed in a solution containing 20 mm Na-methanesulfonate instead of Na-propionate before and after the propionate challenge (Kaila et al., 1993). Simultaneous records of pHi change and Egly during propionate wash-in and washout were obtained from five cells as shown in Figure 7Ai. In all cases, pHi fell during propionate perfusion (peak acidification of −8.4 ± 3.1 in %ΔF/F) and this change was correlated with a negative shift in Egly (peak shift of −4.9 ± 1.6 mV compared with the mean Egly during 100 s before propionate). Upon propionate washout, a small overshoot in pH above baseline level was observed (3.7 ± 2.6 in %ΔF/F), accompanied in four of five cases by a concurrent overshoot in Egly, 1.6 ± 0.6 mV.
Figure 7.

Activity-independent pHi and Egly change and simultaneous Cl and pH imaging. A, Examples showing the concurrent changes in Egly (left ordinate) and pHi (right ordinate) induced during and after 20 mm sodium propionate (Ai) and 10 mm TMA (Aii) perfusion for inducing intracellular acidification and alkalinization, respectively. SNARF images were obtained at 0.5 Hz while cells were held at −75 mV in v-clamp in TTX. B, Simultaneous two-photon imaging of intracellular pH and Cl− with SNARF and MQAE. Excitation wavelength, 750 nm. Bi, Two images of one area taken at the same time through the red emission channel (SNARF) and green channel (MQAE). MQAE was bulk-loaded into DCN slice, and SNARF was loaded from the recording pipette on the left of the cell at the lower left. Inhomogeneous staining with MQAE is typical. Bii, Simultaneous records of pHi (SNARF) and Cl−i (MQAE) obtained from two different cells. An 8 s complex spiking was evoked during the period of gray rectangle with the indicated amount of current. The peak of each signal is indicated with an arrow.
We then applied the weak base TMA (10 mm) to see if an alkalinization would shift Egly in the depolarizing direction in CWCs. A parallel positive shift in Egly was observed along with the TMA-induced intracellular alkalinization in all four cells examined (Fig. 7Aii) (peak shifts were 1.9 ± 0.3 mV in Egly and 4.0 ± 0.9 in %ΔF/F). Moreover, an undershoot occurred upon TMA washout in pHi and Egly, of −5.1 ± 0.9 in %ΔF/F and −2.6 ± 0.9 mV, respectively. The overshoot and undershoot in pHi during washout of weak acid and weak base may reflect the activation of pHi regulation mechanisms during intracellular acidification and alkalinization, respectively (Roos and Boron, 1981). With both weak acid and weak base challenges, the Egly was found to shift negative with a pHi decrease and to shift positive with a pHi increase. The Spearman's correlation coefficient (ρ) between Egly and decimated SNARF signal (in %ΔF/F) was calculated for each run of propionate or TMA challenge. With propionate, the average correlation coefficient for the full duration (800 s) of the trial was 0.61 ± 0.09 (p < 0.05 for all five cases) and that for the 135 s period of wash-in to washout was 0.88 ± 0.10 (p < 0.05 for all five cases). For TMA runs, the coefficient for whole duration was 0.73 ± 0.11 (p < 0.05 for all four cases) and that for the 160 s period of wash-in and out was 0.78 ± 0.23 (p < 0.05 in three cases, p > 0.05 in one). Thus, the activity-independent pHi decrease or increase caused negative or positive shifts in Egly in CWCs (see supplemental Results, available at www.jneurosci.org as supplemental material, for the issue of possible changes in [Ca2+]i by weak acid/base).
Simultaneous monitoring of Cl−i and pHi
pHi may affect Egly through the NDCBE/AE-mediated change in [Cl−]i, and/or through the passive change in [HCO3−]i. To examine whether [Cl−]i actually falls during the activity-dependent negative Egly shift, we used MQAE, a fluorescent dye quenched by Cl− and relatively insensitive to pH changes (Verkman, 1990; Marandi et al., 2002). MQAE loading of CWCs was done by incubating slices in the indicator. MQAE (3 mm) was loaded for 8 min at 34°C; longer incubations or higher concentrations led to excessive depolarization and inability to maintain firing. With SNARF loaded from the recording pipette, simultaneous monitoring of pHi and Cl−i by two-photon imaging (Fig. 7Bi) was done in CWCs held silent in current clamp. The MQAE fluorescence was found to increase (a decrease in Cl−i) after sufficient complex spiking in each cell examined, and the return to baseline took place over similar time scale as that of pHi (Fig. 7Bii). Eight seconds of depolarization-induced simple spiking examined in four cells did not induce a change in MQAE fluorescence, while complex spiking did so in the same cells (data not shown). The peak increase in MQAE signal was 3.3–7.3 in %ΔF/F (average, 4.7; n = 15) after an 8 s complex spiking evoked by 100–250 pA in different cells, and the corresponding peak decrease in SNARF signal ranged from −6.4 to −13.5 in %ΔF/F (average, −8.8). Interestingly, the peak in MQAE fluorescence occurred at 15.4 ± 5.4 s from the end of complex spiking, while the peak acidification was at 1.9 ± 1.5 s (average difference, 13.5 ± 5.3 s, paired t test, p < 0.001, n = 15). The large difference in time course for MQAE and SNARF signals argues against the possibility that MQAE fluorescence might have originated from a decrease in non-Cl− quencher anion, such as HPO42− and HCO3−, whose concentration is dependent on pHi (Koncz and Daugirdas, 1994). The time-to-peak of MQAE fluorescence (15 s) was similar to that of the Egly shift (16.3 s), indicating that a change in Egly reflects a change in [Cl−]i.
Block of the negative Egly shift by H2DIDS
Anion exchangers NDCBE and AE are known to be sensitive to block by disulfonic stilbene derivatives such as DIDS and SITS (Romero et al., 2004). Using H2DIDS, an analog of DIDS, to block NDCBE, we asked whether the change in Egly can be blocked despite activity-dependent intracellular acidification. H2DIDS (100 μm) attenuated the glycine responses but did not prohibit determination of Egly. Several effects of H2DIDS were observed before testing its effects on the activity-dependent shift. In H2DIDS, the resting Egly (in TTX) changed by −3.6 ± 2.0 mV (from control resting Egly ranging from −72 to −85 mV, p = 0.007, paired t test, n = 6) and Vm hyperpolarized (not measured but evident from the positive shift in holding current at −75 mV). H2DIDS increased the Ca2+ spikes number in response to current injection, and often the spiking did not cease immediately after termination of the stimulus. Therefore, cells were kept in voltage clamp at −75 mV throughout a run of simultaneous pHi and Egly monitoring except for 10 s when a glycine puff and an 8 s depolarization were given in current clamp. The greater number of Ca2+ spikes evoked in H2DIDS led to a larger pHi decrease than under control condition in each cell examined (219 ± 58% increase in number of spikes evoked with the same current, 70–300 pA in different cells, and 168 ± 34% larger acidification peak in SNARF signal, n = 6) (Fig. 8A,B). Importantly, however, the negative Egly shift was eliminated by H2DIDS after the Ca2+ spiking ended [+0.4 to +1.5 mV (mean, +0.9) shift compared with −1.4 to −4.1 mV (mean, −2.6) in control conditions, n = 5, p = 0.001, paired t test] (Fig. 8C). The small positive Egly shift peaked at 2 s point after Ca2+ spiking in H2DIDS and is probably due to accumulation of Cl− through a passive H2DIDS-insensitive mechanism. The elimination of negative Egly shift in H2DIDS excludes the possibility that the negative Egly shift in control conditions was primarily caused by the reduction in [HCO3−]i during the pHi decrease rather than by the reduction in [Cl−]i, consistent with our conclusions with MQAE.
Figure 8.

Block of the negative Egly shift by H2DIDS. A, Simultaneously obtained SNARF signal and Egly from one cell are shown for control condition (Ai) and in100 μm H2DIDS (Aii) along with the Vm trace of Ca2+ spiking. Thick bars indicate periods of 8 s Ca2+ spiking. Arrowheads indicate the time of 2 mm glycine puff. The number of evoked Ca2+ spikes is shown in parenthesis. The bias currents during the 8 s current injection were −80 pA and −55 pA for i and ii, respectively. B, Another example showing the difference in magnitude and time course of pHi change between the control condition and in H2DIDS. The period of 8 s Ca2+ spiking evoked with 70 pA is bound by a rectangle. Numbers of evoked Ca2+ spikes are shown in parenthesis. The arrow indicates the time of peak acidification in H2DIDS. C, Plot of peak acidification (in SNARF's ΔF/F) against the peak negative Egly shift in control condition and the peak positive Egly shift in H2DIDS. Five cases (pairs) from different cells are shown. The same depolarizing current injection was used for control and H2DIDS conditions in each of the five cases, but the amount ranged from 70 to 300 pA in different cells. The average of peak negative Egly shifts in controls is −2.6 ± 1.1 mV, and that of peak positive shifts in H2DIDS is 0.9 ± 0.4 mV for the five cases in the plot.
The recovery time course of pHi appeared slower in H2DIDS in all cases examined, and this was evident in the cell of Figure 8A, in which an acidification of similar magnitude to that in control experiment was obtained with a smaller current injection in H2DIDS. In addition, the peak acidification in H2DIDS did not occur at or immediately after the end of 8 s depolarization (at 1.3 ± 0.6 s in controls, n = 6) but was delayed to a later time than in control conditions (difference in time to peak, 15.2 ± 8.2 s, n = 6) (Fig. 8A,B). This suggests that the production of acid continues beyond the duration of depolarizing current injection, and the mechanism inhibited by H2DIDS normally works to remove the acid fast enough for the pHi to begin rising immediately after the depolarization. Though the H2DIDS-sensitive acid removal mechanism may be the Na+-HCO3− cotransporter (Romero et al., 2004) as well as the NDCBE, that the slowed pHi recovery occurred together with the block of negative Egly shift in H2DIDS supports the involvement of NDCBE.
Block of the negative Egly shift by HCO3−/CO2 removal
Although we interpreted the effects of H2DIDS in terms of the block of NDCBE, the drug may also have inhibited KCC. For example, H2DIDS and DIDS block KCC in red blood cells (Delpire and Lauf, 1992; Culliford et al., 2003). To confirm that the Cl−-HCO3− exchangers, rather than the KCC, is involved in the negative Egly shift after Ca2+ spiking, we investigated whether removal of HCO3− and CO2 could block the negative Egly shift. In addition to disabling NDCBE, HCO3− removal is expected to eliminate the contribution of lowered [HCO3−]i to the negative Egly shift during a pHi decrease. For HCO3−/CO2-free condition, NaHCO3 in ACSF was replaced with equimolar HEPES, and the solution was gassed with 100% O2 (HEPES/O2). After >10 min of perfusion with HEPES/O2, the resting Egly of CWCs shifted by +0.4 ± 1.8 mV (range, −3.4 ∼ +2.7 mV, n = 12, p = 0.50 with paired t test), and the excitability slightly increased in 9 of 12 cells. Although reduced, the negative Egly shift with 8 s Ca2+ spiking persisted in HEPES/O2 in all cells examined (Fig. 9Ai), contrary to our expectation (supplemental Results, available at www.jneurosci.org as supplemental material). However, it is possible that CO2 may still be produced by oxidative metabolism in cells despite perfusion of a nominally HCO3−/CO2-free solution (Voipio and Ballanyi, 1997). Hydration of CO2 could then generate sufficient HCO3− in the slice to drive NDCBE. Therefore, we minimized endogenous HCO3− production by blocking carbonic anhydrase which catalyzes the hydration of CO2. Acetazolamide (AZA), a membrane-permeable inhibitor of carbonic anhydrase, added in HEPES/O2 at 50 μm caused the resting Egly of CWCs to shift negative by 4.6 ± 2.6 mV (from −64 to ∼ −81 mV, n = 10, p < 0.001, paired t test). The excitability increased in AZA/HEPES/O2 to such a degree that cells would not stop Ca2+ spiking after an 8 s depolarization; thus, Egly/pHi series were obtained in voltage clamp as described above for H2DIDS experiments. In AZA/HEPES/O2, the negative Egly shift was absent after an 8 s Ca2+ spiking in all 10 cells examined (Fig. 9Aii). Instead, Egly was positively shifted, which peaked at 8–55 s after spiking rather than immediately (∼2 s) (9 of 10 cells) (Fig. 9Aii,B). The maximal positive Egly shift over several runs of Egly/pHi series with different current injections in single cells ranged from 1.6 to 3.1 mV (average, 2.3 mV, after 22–96 Ca2+ spikes evoked by 27–190 pA in different cells, n = 10).
AZA/HEPES/O2 also had effects indicative of reduced control of pHi. During experiments with AZA/HEPES/O2, attempts were made to evoke similar numbers of Ca2+ spikes in AZA to that before AZA addition by reducing the amount of injected current during Egly/pHi series. Figure 9C shows the peak acidification (i) and half-time of pHi recovery (ii) plotted against the peak negative and positive Egly shift for HEPES/O2 and AZA/HEPES/O2, respectively, for six cells in which the numbers of evoked Ca2+ spikes were similar between the two conditions (≤3 spike difference). The difference between the HEPES/O2 and AZA/HEPES/O2 conditions in peak acidification was not significant (p = 0.24, paired t test), but the half-recovery time was longer by 166 ± 21% in AZA (p < 0.001, paired t test) as was the time of peak acidification (delayed by 3.2 ± 1.7 s in AZA, p = 0.006, paired t test). Thus, like H2DIDS in HCO3−/CO2-buffered conditions, the addition of AZA in HEPES/O2 slowed the pHi recovery. We also tested AZA in HCO3−/CO2-buffered condition. Resting Egly again shifted negative by 1.8 ± 1.0 mV in AZA (p = 0.002, paired t test, n = 8), and the excitability increased. After Ca2+ spiking under AZA, the Egly shifted negative, and the recovery from acidification began without delay in all cells examined. Comparing control–AZA pairs of Egly/pHi series with similar numbers of evoked Ca2+ spikes (one to seven more spikes in AZA, n = 5 cells), however, the negative Egly shift was smaller by 1.2 ± 0.5 mV in AZA (p = 0.005, paired t test) while the peak acidification was 139 ± 20% larger in AZA (p = 0.002, paired t test). This effect, reduced coupling between pHi decrease and Egly shift, was similar to that observed in HEPES/O2 (supplemental Results, available at www.jneurosci.org as supplemental material). Taken together, these results suggest that the mechanism responsible for the negative Egly shift is H2DIDS sensitive and HCO3− dependent, but may be able to function at low bicarbonate level (see Discussion).
Immunohistochemical detection of NDCBE in the DCN
To confirm that NDCBE protein is indeed expressed in CWCs, we probed the DCN with two different clones of monoclonal antibodies against human NDCBE followed by fluorescent labeling with a secondary antibody. As expected from NDCBE's homeostatic function and as reported by Chen et al. (2008), widespread staining was found in the DCN, the adjoining brainstem and in the cerebellum. The two clones of NDCBE antibody resulted in identical staining pattern. In the DCN, the molecular layer was diffusely stained, and many cells, from small packed cells to large cells, were labeled over the entire DCN (Fig. 10A). No labeling was observed when the primary antibody was omitted (Fig. 10B). The labeling in DCN included numerous round, medium-sized cell bodies in the molecular and outer fusiform cell layer, which corresponds to the size and distribution of CWCs (Wouterlood and Mugnaini, 1984; Berrebi and Mugnaini, 1991). Unexpectedly, the strongest labeling was found in the unipolar brush cell (UBC), identified by its single tufted dendrite (Diño and Mugnaini, 2008), both in the DCN and in the cerebellum (Fig. 10A,C). In the cerebellum, Purkinje cells were stained brightly in the soma and the primary dendrite as in previous reports (Damkier et al., 2007; Chen et al., 2008), and granule cells were also stained, as seen by a thin ring of labeled cytoplasm around the nuclei (Fig. 10C) (Chen et al. 2008).
Figure 10.

Detection of NDCBE in the DCN by indirect immunofluorescence. A, A confocal laser-scanned image showing labeling of NDCBE in the DCN. Putative CWCs are concentrated in the middle row of the image, and two of them are labeled with asterisks. Arrows, UBCs. B, Another DCN section processed at the same time with the one in A, but without the primary antibody. Orientation of the DCN section in both A and B is the ependymal surface toward the bottom. C, A piece of cerebellum attached to the brainstem section containing the DCN of A shows labeling. Purkinje cells are marked with asterisks and one UBC is marked with an arrow. Images of cerebellar sections not treated with the primary antibody were as dark as the one in B (data not shown). Scale bar, 20 μm. NDCBE antibody was the 1G10 clone (1:100). Laser and confocal settings were identical in all three micrographs.
Discussion
We have identified a novel postsynaptic mechanism for enhancement of the effectiveness of inhibition during prolonged spiking activity or transiently following a period of increased activity. A shift in EGABA/IPSP dependent on postsynaptic activity has been reported in hippocampal neurons (Fiumelli et al., 2005; Brumback and Staley, 2008) and subthalamic neurons (Wang et al., 2006). However, while the EGABA/IPSP shift in both of these regions is Ca2+ dependent, it differs from the situation in CWC in several major respects. These reported shifts were long-lasting (>30 min) and often in the direction opposite to that seen in CWC. EGABA shifts in hippocampal cells were depolarizing and were induced with several minutes of 10–25 Hz firing. In subthalamic neurons, negative or positive shift in EIPSP appeared after evoking rebound bursts at 0.1 Hz for 100 s depending on intraburst frequency. Unlike the positive Egly shift in CWCs, the positive shift in this study was eliminated by application of Ca2+ channel blockers. The proposed mechanism for the EGABA shift in hippocampal neurons was Ca2+-mediated downregulation of KCC2 (Fiumelli et al., 2005) or change in the set-point of NKCC following lowering of [Na+]i (Brumback and Staley, 2008). It may be that the mechanism we have identified is of general significance but precedes the longer-lasting Cl− shifts described above. Given its dependence on dynamic control of intracellular Cl− and pH, the drop in Egly we describe may have been missed in studies that do not employ perforated patch recording.
Negative Egly shift following intracellular acidification
Glycine responses ranged from depolarizing excitation to hyperpolarizing inhibition, reflecting the range of resting Egly (−58 to −87 mV). These differences may be due to variation in the resting pHi and [Cl−]i among CWCs (supplemental Discussion, available at www.jneurosci.org as supplemental material). The negative Egly shift following complex spiking likely arises from NDCBE-mediated decrease in [Cl−]i, given that the negative Egly shift was sensitive to H2DIDS and removal of HCO3−. The contribution of lowered [HCO3−]i to the activity-dependent negative Egly shift does not appear as important as the decrease in [Cl−]i based on the following observations. (1) When Cl−-HCO3− exchangers were blocked by H2DIDS, the decrease in pHi was greater than control, but Egly did not shift negative. (2) In nominally HCO3−/CO2-free Ringer (HEPES/O2), where the impact of change in [HCO3−]i on Egly is expected to be greatly reduced even if a few-millimolar level of intracellular and extracellular HCO3− is considered (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material), the Egly could shift negative similar to control conditions. It appears that the Na+-driven Cl−-HCO3− exchange could run at a reduced rate with a few-millimolar extracellular HCO3− in HEPES/O2, such that the Egly shifted negative by the fall in [Cl−]i. This is consistent with the reported Km or Ki for [HCO3−]o of Na+-driven or Na+-independent Cl−-HCO3− exchange of 1–10 mm (Boron et al., 1981; Boron and Russell, 1983; Olsnes et al., 1986; Vaughan-Jones, 1986; Cassel et al., 1988). However, the amount of decrease in [Cl−]i associated with the activity-dependent negative Egly that we observed, in normal HCO3−/CO2-buffered condition, may be small (<2 mm). The relation between Egly, [Cl−]i, and [HCO3−]i according to the Goldman–Hodgkin–Katz equation predicts that a drop in Egly occurring with a pHi decrease involves less decrease in [Cl−]i than that occurring without a pHi change, and even an increase in [Cl−]i can be associated with the negative Egly shift, depending on the magnitude of pHi decrease (supplemental Discussion and Fig. S3, available at www.jneurosci.org as supplemental material).
The different contributors to Cl− flux and their pH and voltage dependence likely underlie the time course of change in Egly. During spiking, passive influx of Cl− is expected to oppose efflux mediated by NDCBE. In the case of prolonged complex/Ca2+ spiking and large intracellular acidification, NDCBE-mediated Cl− efflux may dominate the passive influx leading to a fall in [Cl−]i. If the spiking-induced acidification did not drive NDCBE sufficiently, [Cl−]i may rise above baseline, and the outcome may be the small depolarizing shift in Egly observed immediately after simple spiking and in some cases of complex spiking. After the voltage is restored and pHi begins to normalize, remaining activity of NDCBE may become more prominent, which could account for the decrease in [Cl−]i during the initial 10 s or so, i.e., the delayed peak in negative Egly shift. It is possible that AE, KCC, and NKCC, which are inhibited at acidic pHi (Olsnes et al., 1986; Boyarsky et al., 1988; Leem et al., 1999; Russell, 2000; Bergeron et al., 2003), may also be involved and contribute to the time course of Egly change.
Activity-induced intracellular acidification
Decrease in intracellular pH occurring with depolarization, spike firing, or Ca2+ rise, has been observed in a wide variety of neurons (for review, see Ballanyi and Kaila, 1998; Chesler, 2003). The pHi decrease during evoked spiking in CWCs required Ca2+ entry, as depolarization given in zero-Ca2+ or after Ca2+ channel blockade induced little or no acidification. We did not investigate the mechanism of Ca2+-dependent pHi decrease in CWCs, but it could occur via multiple pathways, consistent with previous studies: displacement of H+ by Ca2+ in intracellular binding sites, mitochondrial Ca2+/H+ exchange, PMCA or SERCA-mediated Ca2+extrusion, and stimulation of metabolic acid production (Ballanyi and Kaila, 1998; Chesler, 2003). Consideration of the magnitude of change in Cl− and the proposed coupling between Ca2+, H+, bicarbonate and Cl− leads to the conclusion that total Ca2+ flux during spike trains must have been quite large. Direct measurements of ion concentration and buffer capacity will be needed to confirm these relations. A novel aspect of present study was that the spiking-induced pHi decrease was demonstrated with the gramicidin perforated-patch recording in mammalian neurons, while many previous studies used whole-cell recording (Trapp et al., 1996a, b; Meyer et al., 2000; Willoughby and Schwiening, 2002; Ritucci et al., 2005). We found that complex spiking-induced acidification was smaller and decayed faster in cells recorded whole-cell than in those recorded in perforated-patch condition (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). Willoughby and Schwiening (2002) showed in whole-cell recorded cerebellar Purkinje cells that acidic pHi transients induced by depolarization or spiking were greater in dendrites than in soma. Given the large dendritic Ca2+ signals characteristic of CWCs (Roberts et al. 2008), complex/Ca2+ spiking could induce a larger negative Egly shift in dendrites than we recorded at the soma, provided that NDCBE is expressed in dendrites.
Functional relevance of shifts in Egly
Golding and Oertel (1996) proposed that weakly excitatory glycinergic and GABAergic input in CWCs at rest could become inhibitory when the cell fires at high rates (i.e., during strong depolarization); this model assumed that during intense activity the voltage would be positive to a constant value of Egly of approximately −55 mV. A similar view in relation to change in EGABA was offered by Chavas and Marty (2003) regarding the polarity of GABAergic input to cerebellar molecular layer interneurons, which also was both excitatory and inhibitory. Unlike our study, Golding and Oertel (1996) did not observe cells with hyperpolarizing Egly at rest. However, hyperpolarizing PSPs were seen at resting Vm of CWCs by Manis et al. (1994), and CWC spiking increased after strychnine or bicuculline in vivo (Davis and Young, 2000). Using perforated patch, Mancilla and Manis (2009) also observed hyperpolarizing IPSPs in CWCs of young (P10–12) rats; however the resting potential in the cells they reported was relatively depolarized (mean, −53 mV). In contrast, their mean reversal potential was −67 mV, within the range of those we report here. These differences among studies may also be related to variation in the resting pattern of spontaneous activity of cells. We found that few spontaneously complex spiking cells showed excitatory responses, and many of these showed inhibitory responses to glycine. Indeed, in the work by Davis and Young (2000), only complex spiking units were selected for recording, and most CWCs in the work by Manis et al. (1994) were predominantly complex spiking. It could be that the presumably more negative resting Egly in these cells is associated with a lower resting pHi or the dominance in activity of KCC over NKCC.
While the prolonged high-frequency complex spiking used in the present study may only rarely occur in physiological settings, it is likely that pHi will fall during briefer periods of activity. Because such periods of activity will be associated with passive influx of Cl−, we suggest that the Ca2+-dependent mechanism will serve to minimize passive changes in Egly. However, when such protracted spike activity does occur, the increase in glycinergic/GABAergic inhibition through the negative Egly shift is expected to reduce the frequency of complex spikes in a CWC. For cells postsynaptic to a CWC, the DCN principal cells or other CWCs, this mechanism would limit the frequency of the large PSP bursts originating from presynaptic complex spikes (Tzounopoulos et al., 2004; Roberts et al., 2008) and thus limit the maximal glycinergic inhibition or excitation coming from a CWC. From the perspective of an inhibitory interneuron network, the use-dependent variation in resting Egly which enables both excitatory and inhibitory connections may be more effective in stabilizing inhibition to principal neurons (Chavas and Marty, 2003).
Footnotes
This work was supported by National Institutes of Health Grant R37NS28901. We thank Sidney Kuo for help with immunohistochemistry.
References
- Ballanyi K, Kaila K. Activity-evoked changes in intracellular pH. In: Kaila K, Ransom BR, editors. pH and brain function. New York: Wiley; 1998. pp. 291–308. [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Bergeron MJ, Gagnon E, Wallendorff B, Lapointe JY, Isenring P. Ammonium transport and pH regulation by K+-Cl-cotransporters. Am J Physiol Renal Physiol. 2003;285:F68–F78. doi: 10.1152/ajprenal.00032.2003. [DOI] [PubMed] [Google Scholar]
- Berglund K, Schleich W, Krieger P, Loo LS, Wang D, Cant NB, Feng G, Augustine GJ, Kuner T. Imaging synaptic inhibition in transgenic mice expressing the chloride indicator, Clomeleon. Brain Cell Biol. 2006;35:207–228. doi: 10.1007/s11068-008-9019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrebi AS, Mugnaini E. Distribution and targets of the cartwheel cell axon in the dorsal cochlear nucleus of the guinea pig. Anat Embryol (Berl) 1991;183:427–454. doi: 10.1007/BF00186433. [DOI] [PubMed] [Google Scholar]
- Billups D, Attwell D. Control of intracellular chloride concentration and GABA response polarity in rat retinal ON bipolar cells. J Physiol. 2002;545:183–198. doi: 10.1113/jphysiol.2002.024877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron WF, Russell JM. Stoichiometry and ion dependencies of the intracellular-pH-regulating mechanism in squid giant axons. J Gen Physiol. 1983;81:373–399. doi: 10.1085/jgp.81.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron WF, McCormick WC, Roos A. pH regulation in barnacle muscle fibers: dependence on extracellular sodium and bicarbonate. Am J Physiol. 1981;240:C80–C89. doi: 10.1152/ajpcell.1981.240.1.C80. [DOI] [PubMed] [Google Scholar]
- Boyarsky G, Ganz MB, Sterzel RB, Boron WF. pH regulation in single glomerular mesangial cells. II. Na+-dependent and -independent Cl(-)-HCO3-exchangers. Am J Physiol. 1988;255:C857–C869. doi: 10.1152/ajpcell.1988.255.6.C857. [DOI] [PubMed] [Google Scholar]
- Brett CL, Kelly T, Sheldon C, Church J. Regulation of Cl–HCO3- exchangers by cAMP-dependent protein kinase in adult rat hippocampal CA1 neurons. J Physiol. 2002;545:837–853. doi: 10.1113/jphysiol.2002.027235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumback AC, Staley KJ. Thermodynamic regulation of NKCC1-mediated Cl− cotransport underlies plasticity of GABAA signaling in neonatal neurons. J Neurosci. 2008;28:1301–1312. doi: 10.1523/JNEUROSCI.3378-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel D, Scharf O, Rotman M, Cragoe EJ, Jr, Katz M. Characterization of Na+-linked and Na+-independent Cl-/HCO3- exchange systems in Chinese hamster lung fibroblasts. J Biol Chem. 1988;263:6122–6127. [PubMed] [Google Scholar]
- Chavas J, Marty A. Coexistence of excitatory and inhibitory GABA synapses in the cerebellar interneuron network. J Neurosci. 2003;23:2019–2031. doi: 10.1523/JNEUROSCI.23-06-02019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LM, Kelly ML, Parker MD, Bouyer P, Gill HS, Felie JM, Davis BA, Boron WF. Expression and localization of Na-driven Cl-HCO(3)(-) exchanger (SLC4A8) in rodent CNS. Neuroscience. 2008;153:162–174. doi: 10.1016/j.neuroscience.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Choi HS, Eisner DA. The effects of inhibition of the sarcolemmal Ca-ATPase on systolic calcium fluxes and intracellular calcium concentration in rat ventricular myocytes. Pflugers Arch Eur J Physiol. 1999;437:966–971. doi: 10.1007/s004240050868. [DOI] [PubMed] [Google Scholar]
- Culliford S, Ellory C, Lang HJ, Englert H, Staines H, Wilkins R. Specificity of classical and putative Cl-transport inhibitors on membrane transport pathways in human erythrocytes. Cell Physiol Biochem. 2003;13:181–188. doi: 10.1159/000072420. [DOI] [PubMed] [Google Scholar]
- Damkier HH, Nielsen S, Praetorius J. Molecular expression of SLC4-derived Na+-dependent anion transporters in selected human tissues. Am J Physiol Regul Integr Comp Physiol. 2007;293:R2136–R2146. doi: 10.1152/ajpregu.00356.2007. [DOI] [PubMed] [Google Scholar]
- Davis KA, Young ED. Granule cell activation of complex-spiking neurons in dorsal cochlear nucleus. J Neurosci. 1997;17:6798–6806. doi: 10.1523/JNEUROSCI.17-17-06798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KA, Young ED. Pharmacological evidence of inhibitory and disinhibitory neuronal circuits in dorsal cochlear nucleus. J Neurophysiol. 2000;83:926–940. doi: 10.1152/jn.2000.83.2.926. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Heger S, Ojeda SR, Moenter SM. Activation of A-type gamma-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16:2872–2891. doi: 10.1210/me.2002-0163. [DOI] [PubMed] [Google Scholar]
- Delpire E, Lauf PK. Kinetics of DIDS inhibition of swelling-activated K-Cl cotransport in low K sheep erythrocytes. J Membr Biol. 1992;126:89–96. doi: 10.1007/BF00233463. [DOI] [PubMed] [Google Scholar]
- Diño MR, Mugnaini E. Distribution and phenotypes of unipolar brush cells in relation to the granule cell system of the rat cochlear nucleus. Neuroscience. 2008;154:29–50. doi: 10.1016/j.neuroscience.2008.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duebel J, Haverkamp S, Schleich W, Feng G, Augustine GJ, Kuner T, Euler T. Two-photon imaging reveals somatodendritic chloride gradient in retinal ON-type bipolar cells expressing the biosensor Clomeleon. Neuron. 2006;49:81–94. doi: 10.1016/j.neuron.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Lohrke S, Friauf E. Shift from depolarizing to hyperpolarizing glycine action in rat auditory neurones is due to age-dependent Cl- regulation. J Physiol. 1999;520:121–137. doi: 10.1111/j.1469-7793.1999.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Filosa JA, Dean JB, Putnam RW. Role of intracellular and extracellular pH in the chemosensitive response of rat locus coeruleus neurones. J Physiol. 2002;541:493–509. doi: 10.1113/jphysiol.2001.014142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumelli H, Cancedda L, Poo MM. Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron. 2005;48:773–786. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Gatto C, Hale CC, Xu W, Milanick MA. Eosin, a potent inhibitor of the plasma membrane Ca pump, does not inhibit the cardiac Na-Ca exchanger. Biochemistry. 1995;34:965–972. doi: 10.1021/bi00003a031. [DOI] [PubMed] [Google Scholar]
- Golding NL, Oertel D. Context-dependent synaptic action of glycinergic and GABAergic inputs in the dorsal cochlear nucleus. J Neurosci. 1996;16:2208–2219. doi: 10.1523/JNEUROSCI.16-07-02208.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grichtchenko II, Choi I, Zhong X, Bray-Ward P, Russell JM, Boron WF. Cloning, characterization, and chromosomal mapping of a human electroneutral Na+-driven Cl-HCO3 exchanger. J Biol Chem. 2001;276:8358–8363. doi: 10.1074/jbc.C000716200. [DOI] [PubMed] [Google Scholar]
- Gulácsi A, Lee CR, Sík A, Viitanen T, Kaila K, Tepper JM, Freund TF. Cell type-specific differences in chloride-regulatory mechanisms and GABAA receptor-mediated inhibition in rat substantia nigra. J Neurosci. 2003;23:8237–8246. doi: 10.1523/JNEUROSCI.23-23-08237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Excitatory actions of GABA in the cortex. Neuron. 2003;37:299–309. doi: 10.1016/s0896-6273(02)01146-7. [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature. 1987;330:163–165. doi: 10.1038/330163a0. [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J Physiol. 1993;464:273–289. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, Chung LY. Dual GABAergic synaptic response of fast excitation and slow inhibition in the medial habenula of rat epithalamus. J Neurophysiol. 2007;98:1323–1332. doi: 10.1152/jn.00575.2007. [DOI] [PubMed] [Google Scholar]
- Kim Y, Trussell LO. Ion channels generating complex spikes in cartwheel cells of the dorsal cochlear nucleus. J Neurophysiol. 2007;97:1705–1725. doi: 10.1152/jn.00536.2006. [DOI] [PubMed] [Google Scholar]
- Koncz C, Daugirdas JT. Use of MQAE for measurement of intracellular [Cl-] in cultured aortic smooth muscle cells. Am J Physiol. 1994;267:H2114–H2123. doi: 10.1152/ajpheart.1994.267.6.H2114. [DOI] [PubMed] [Google Scholar]
- Kopito RR, Lee BS, Simmons DM, Lindsey AE, Morgans CW, Schneider K. Regulation of intracellular pH by a neuronal homolog of the erythrocyte anion exchanger. Cell. 1989;59:927–937. doi: 10.1016/0092-8674(89)90615-6. [DOI] [PubMed] [Google Scholar]
- Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- Leem CH, Lagadic-Gossmann D, Vaughan-Jones RD. Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J Physiol. 1999;517:159–180. doi: 10.1111/j.1469-7793.1999.0159z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancilla JG, Manis PB. Two distinct types of inhibition mediated by cartwheel cells in the dorsal cochlear nucleus. J Neurophysiol. 2009;102:1287–1295. doi: 10.1152/jn.91272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis PB, Spirou GA, Wright DD, Paydar S, Ryugo DK. Physiology and morphology of complex spiking neurons in the guinea pig dorsal cochlear nucleus. J Comp Neurol. 1994;348:261–276. doi: 10.1002/cne.903480208. [DOI] [PubMed] [Google Scholar]
- Marandi N, Konnerth A, Garaschuk O. Two-photon chloride imaging in neurons of brain slices. Pflugers Arch. 2002;445:357–365. doi: 10.1007/s00424-002-0933-7. [DOI] [PubMed] [Google Scholar]
- Marty A, Llano I. Excitatory effects of GABA in established brain networks. Trends Neurosci. 2005;28:284–289. doi: 10.1016/j.tins.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Meyer TM, Munsch T, Pape HC. Activity-related changes in intracellular pH in rat thalamic relay neurons. Neuroreport. 2000;11:33–37. doi: 10.1097/00001756-200001170-00007. [DOI] [PubMed] [Google Scholar]
- Molitor SC, Manis PB. Dendritic Ca2+ transients evoked by action potentials in rat dorsal cochlear nucleus pyramidal and cartwheel neurons. J Neurophysiol. 2003;89:2225–2237. doi: 10.1152/jn.00709.2002. [DOI] [PubMed] [Google Scholar]
- Olsnes S, Tønnessen TI, Sandvig K. pH-regulated anion antiport in nucleated mammalian cells. J Cell Biol. 1986;102:967–971. doi: 10.1083/jcb.102.3.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Ebihara S, Akaike N. Gramicidin perforated patch-clamp technique reveals glycine-gated outward chloride current in dissociated nucleus solitarii neurons of the rat. J Neurophysiol. 1994;72:1103–1108. doi: 10.1152/jn.1994.72.3.1103. [DOI] [PubMed] [Google Scholar]
- Ritucci NA, Dean JB, Putnam RW. Somatic vs. dendritic responses to hypercapnia in chemosensitive locus coeruleus neurons from neonatal rats. Am J Physiol Cell Physiol. 2005;289:C1094–C1104. doi: 10.1152/ajpcell.00329.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl-co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Roberts MT, Bender KJ, Trussell LO. Fidelity of complex spike-mediated synaptic transmission between inhibitory interneurons. J Neurosci. 2008;28:9440–9450. doi: 10.1523/JNEUROSCI.2226-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha-González HI, Mao S, Alvarez-Leefmans FJ. Na+,K+,2Cl-cotransport and intracellular chloride regulation in rat primary sensory neurons: thermodynamic and kinetic aspects. J Neurophysiol. 2008;100:169–184. doi: 10.1152/jn.01007.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero MF, Fulton CM, Boron WF. The SLC4 family of HCO 3- transporters. Pflugers Arch. 2004;447:495–509. doi: 10.1007/s00424-003-1180-2. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Schwiening CJ, Boron WF. Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na(+)-dependent Cl(-)-HCO3-exchange. J Physiol. 1994;475:59–67. doi: 10.1113/jphysiol.1994.sp020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Trapp S, Lückermann M, Brooks PA, Ballanyi K. Acidosis of rat dorsal vagal neurons in situ during spontaneous and evoked activity. J Physiol. 1996a;496:695–710. doi: 10.1113/jphysiol.1996.sp021720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Lückermann M, Kaila K, Ballanyi K. Acidosis of hippocampal neurones mediated by a plasmalemmal Ca2+/H+ pump. Neuroreport. 1996b;7:2000–2004. doi: 10.1097/00001756-199608120-00029. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Vaughan-Jones RD. An investigation of chloride-bicarbonate exchange in the sheep cardiac Purkinje fibre. J Physiol. 1986;379:377–406. doi: 10.1113/jphysiol.1986.sp016259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS. Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol Cell Physiol. 1990;259:C375–C388. doi: 10.1152/ajpcell.1990.259.3.C375. [DOI] [PubMed] [Google Scholar]
- Voipio J, Ballanyi K. Interstitial PCO2 and pH, and their role as chemostimulants in the isolated respiratory network of neonatal rats. J Physiol. 1997;499:527–542. doi: 10.1113/jphysiol.1997.sp021946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kitai ST, Xiang Z. Activity-dependent bidirectional modification of inhibitory synaptic transmission in rat subthalamic neurons. J Neurosci. 2006;26:7321–7327. doi: 10.1523/JNEUROSCI.4656-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D, Schwiening CJ. Electrically evoked dendritic pH transients in rat cerebellar Purkinje cells. J Physiol. 2002;544:487–499. doi: 10.1113/jphysiol.2002.027508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouterlood FG, Mugnaini E. Cartwheel neurons of the dorsal cochlear nucleus: a Golgi-electron microscopic study in rat. J Comp Neurol. 1984;227:136–157. doi: 10.1002/cne.902270114. [DOI] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Oertel D. Cartwheel and superficial stellate cells of the dorsal cochlear nucleus of mice: intracellular recordings in slices. J Neurophysiol. 1993;69:1384–1397. doi: 10.1152/jn.1993.69.5.1384. [DOI] [PubMed] [Google Scholar]