

Abstract
The early diagnosis of cancer is the critical element in successful treatment and long term favorable patient prognoses. The high rate of mortality is mainly attributed to the tendency for late diagnoses as symptoms may not occur until the disease has metastasized, as well as the lack of effective systemic therapies. Late diagnosis is often associated with the lack of timely sensitive imaging modalities. The promise of nanotechnology is presently limited by the inability to simultaneously seek, treat and image cancerous lesions. This study describes the design and synthesis of fluorescent calcium phosphosilicate nanocomposite particles (CPNPs) that can be systemically targeted to breast and pancreatic cancer lesions. The CPNPs are a ~20nm diameter composite composed of an amorphous calcium phosphate matrix doped with silicate in which a near infra-red imaging agent indocyanine green (ICG) is embedded. In the present studies, we describe and validate CPNP bioconjugation of human holotransferrin, anti-CD71 antibody, and short gastrin peptides via an avidin-biotin- or a novel PEG-maleimide-coupling strategy. The conjugation of biotinylated human holotransferrin (diferric transferrin) and biotinylated anti-CD71 antibody (anti-transferrin receptor antibody) to avidin conjugated CPNPs (Avidin-CPNPs) permits targeting of transferrin receptors, which are highly expressed on breast cancer cells. Similarly, the conjugation of biotinylated pentagastrin to Avidin-CPNPs and decagastrin (gastrin-10) to PEG-CPNPs via PEG-maleimide coupling permits targeting of gastrin receptors, which are over-expressed in pancreatic cancer lesions. These bioconjugated CPNPs have the potential to perform as a theranostic modality, simultaneously enhancing drug delivery, targeting and imaging of breast and pancreatic cancer tumors.
Keywords: bioconjugation, transferrin receptor, gastrin receptor, breast cancer, pancreatic cancer, calcium phosphate, whole animal imaging
Calcium phosphate nanoparticles (CPNPs) have been engineered to be a non-toxic vehicle for the delivery of a diverse range of therapeutic and imaging agents in biological systems.1–4 Previous studies have shown that encapsulation within CPNPs improved the lifetime and quantum properties of fluorescent dyes.1, 4 Initial in vivo imaging trials demonstrated that CPNPs, functionalized with polyethylene glycol (PEG) moieties, accumulated within solid tumors via an enhanced permeation retention (EPR) effect.2 While EPR serves as an effective passive targeting strategy, particular interest lies in the ability to actively target cancerous cells to deliver anti-neoplastic agents, thereby decreasing effective dosage and limiting off-target toxicity.
CPNPs are nontoxic, colloidally stable, nanoscale vehicles that deliver chemotherapeutics and imaging agents. Two exciting aspects of CPNPs as drug delivery vehicles include enterohepatic biliary excretion that minimizes hepatic toxicity and pH-triggered release of active agents. At pH 7.4, the CPNPs are sparingly soluble, but the CPNPs dissolve in the late stage endo-lysosomes at pH 4 to 5.1, 4 The pH response of CPNPs produces two effects. First, it permits a decrease in the effective dose of potentially toxic chemotherapeutic drugs required for optimal therapeutic benefit by increasing the efficiency of drug delivery into cancer cells.3 Second, sequestering the drug in the CPNPs decreases the effective concentration of free drug present in the extracellular fluid where the pH is maintained at approximately pH 7.4 by physiological buffers. This compartmentalization feature for drug delivery produces advantages since acute systemic toxicity to normal cells is limited. Moreover, off-site cytotoxicity may be further ameliorated with target and tissue specific CPNPs.
Scientific investigations have identified cancer cell specific markers with unique phenotypes that can be exploited to target tumors. Of particular interest is the prevalence of transferrin receptors (CD71) on cancerous cells, including breast cancer.5–9 The transferrin receptor is responsible for transporting iron, via interaction with transferrin, into cells as demanded by metabolic need.5, 6 Accordingly, transferrin receptors are found predominately on proliferating cells with elevated metabolic levels, including many cancerous cells, as well as brain capillary endothelial cells, and hematopoietic cells.10, 11 In a manner similar to CD71, gastrin receptors have a predominate prevalence within certain tissues, specifically the gastrointestinal and central nervous systems.12–14 The hormone gastrin binds to a family of G-protein-coupled receptors, also known as the cholecystokinin-2 (CCK2 or CCK-B) receptor family,14, 15 and is typically known as a key mediator of stomach acidity16 and growth of the gastrointestinal tract.17 Intriguingly, CCK2 receptor expression is often increased in many cases of gastrointestinal cancer,13, 18 including pancreatic cancer,19 and, in particular, an increase in the expression of a specific splice variant (CCK2i4sv or CCK-C) of the receptor.20
Despite many new advances in the arsenal of antineoplastic agents, drug resistant, highly metastatic cancers continue to ravage patients.21 As examples, breast cancer is still the second leading cause of death in American women with an estimated 192,370 cases diagnosed in 2009. In this year alone, about 40,610 women will die from breast cancer in the United States. Pancreatic cancer is the fourth leading cause of cancer related deaths in the United States. Approximately 42,470 Americans were diagnosed with pancreatic cancer in the past year and nearly 100% will succumb to this disease.21 It is clear that new modalities are needed that have the capabilities to both improve diagnosis and treatment of cancers. The term theranostic has been coined to describe modalities that can simultaneously diagnose and treat. Our studies describe the in vivo validation of a nano “solution” that can actively target and image breast and pancreatic cancer lesions.
RESULTS AND DISCUSSION
Physical Characterization of CPNPs
Citrate functionalized CPNPs were utilized as a platform for functionalization, which allowed the characterization of bioconjugation via zeta potential analysis (Figure 1). Figure 1 shows the zeta potential distribution of Citrate-CPNPs prior to bioconjugation (blue line), and the zeta potential distributions of the avidin-CPNPs complex (green), maleimide terminated polyethylene glycol (PEG) coated CPNPs (red), and CPNPs conjugated with gastrin 10 via a maleimide PEG coupling (violet). Prior to bioconjugation, the Citrate-CPNPs display a negative mean zeta potential value of −16 ± 1.3 mV, which is consistent with previous reports.1 However, after bioconjugation, the Avidin-CPNPs displayed a relatively high positive mean zeta potential value of +29 ± 8.7 mV. The isoelectric point for avidin is pH 10. Thus, the shift from a negative zeta potential to a positive zeta potential distribution is strong evidence of avidin bioconjugation on the surface of CPNPs. Also, Figure 1 shows the shift in mean zeta potential to +3.0 ± 2.0 mV when coated with the PEG, and then a further shift to +6 ± 3.2 mV when conjugated to gastrin 10.
Figure 1.
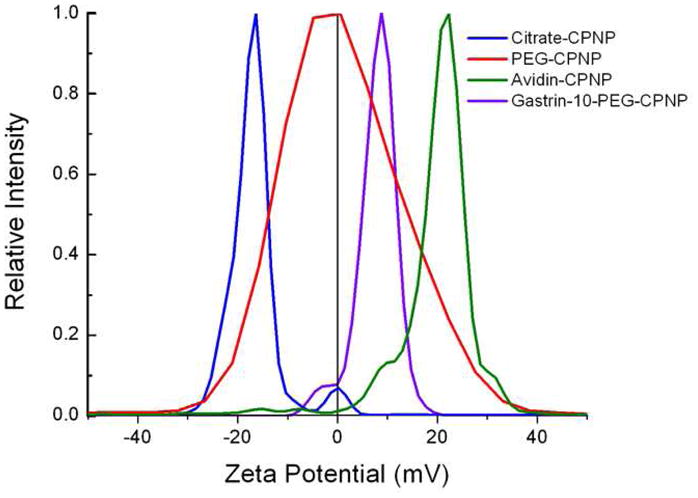
Zeta potential distributions for Citrate-CPNPs, Avidin-CPNPs, PEG-CPNPs, and Gastrin-10-PEG-CPNPs. The Citrate-CPNPs (blue line) displayed a mean zeta potential of −16 ± 1.3 mV, whereas PEGylation shifted the mean zeta potential to +3.0 ± 2.0 mV (red), gastrin 10 conjugation further shifted the mean zeta potential to +6 ± 3.2 mV (violet), and the Avidin-CPNPs (green line) had a mean zeta potential value of +29 ± 8.7 mV. All zeta potential distributions represent the average of three independent experiments.
Further characterization was used to confirm the presence and bioactivity of the bioconjugated Avidin-CPNPs for biotin. A 2,6-ANS titration was used to confirm both the presence of avidin and its associated bioactivity. An analysis of the particle size distributions of the nanoparticles via dynamic light scattering (DLS) revealed that all the various bioconjugated CPNPs had a larger mean hydrodynamic diameter than the non-bioconjugated citrate-CPNPs (Figure 2). Transmission electron microscopy (TEM) analysis indicated that the inorganic particle size was in the range from 10 to 30 nm for all of the CPNPs (inset Figure 2). The smaller size via TEM relative to DLS analyses is consistent with the ability to determine the solid material diameter via the TEM technique in contrast to DLS which gives the hydrodynamic size distribution in colloidal suspension of solid particle, organic layers, and surrounding liquid.
Figure 2.
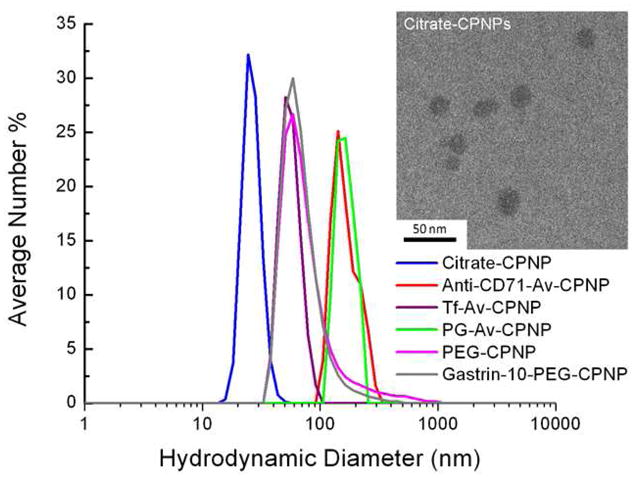
Dynamic light scattering determinations for Citrate-CPNP, Anti-CD71-Avidin-CPNPs (Anti-CD71-Av-CPNP), Human Holotransferrin-Avidin-CPNPs (Tf-Av-CPNP), Pentagastrin-Avidin-CPNPs (PG-Av-CPNP), maleimidePEG-CPNPs (PEG-CPNP), and Gastrin 10-maleimidePEG-CPNPs (Gastrin-10-PEG-CPNP). All dynamic light scattering determinations are the mean of three independent experiments. Inset shows a typical TEM micrograph of Citrate-CPNPs.
Previous studies have demonstrated that a 2,6-ANS assay can be utilized to evaluate the biotin-binding functionality of avidin.22 The 2,6-ANS fluorescent probe binds to avidin, an event that can be measured using fluorescence spectroscopy. Without avidin present, the 2,6-ANS fluorescent probe displays low fluorescence intensity. In the presence of avidin, the binding of 2,6-ANS to the biotin-binding site on avidin produces an increase in fluorescence intensity. The 2,6-ANS was therefore added in increasing concentrations to the avidin-CPNPs, and a concentration-dependent increase in 2,6-ANS fluorescence emission was noted (Figures 3A and 3B). The titration of 2,6-ANS showed an increase in fluorescence up to 1.79 × 106 RFU after the addition of 34 μM 2,6-ANS. Beyond this point of maximum fluorescence intensity the 2,6-ANS self-quenched, at which point biotin was added to displace the 2,6-ANS. Biotin has a greater affinity for the biotin-binding site on avidin than does the 2,6-ANS fluorescent probe. Therefore, biotin additions to the 2,6-ANS-Avidin-CPNP complex displaces the 2,6-ANS from the biotin-binding site on avidin. Since the 2,6-ANS fluorescent probe displays minimal fluorescence when it is not bound to avidin, this displacement produces a decrease in fluorescence (Figures 3C and 3D) to a plateau of 1.08 × 106 RFU after the addition of 1.90 nM biotin. The plateau is present in Figure 3D because of the intrinsic fluorescence of 2,6-ANS. This result demonstrates the successful coupling of biotin to the Avidin-CPNPs. The 2,6-ANS fluorescence emission did not decrease completely as some 2,6-ANS remains bound to the Avidin-CPNPs. While the affinity of avidin for biotin is high, residual reactants and ionic conditions can influence this affinity as it has been established that water participates in displacing biotin from the binding pocket of avidin or similar proteins.23, 24 Nonetheless, this analysis has successfully demonstrated that the Avidin-CPNPs are biofunctional, through binding of 2,6-ANS as well as its displacement by biotin.
Figure 3.
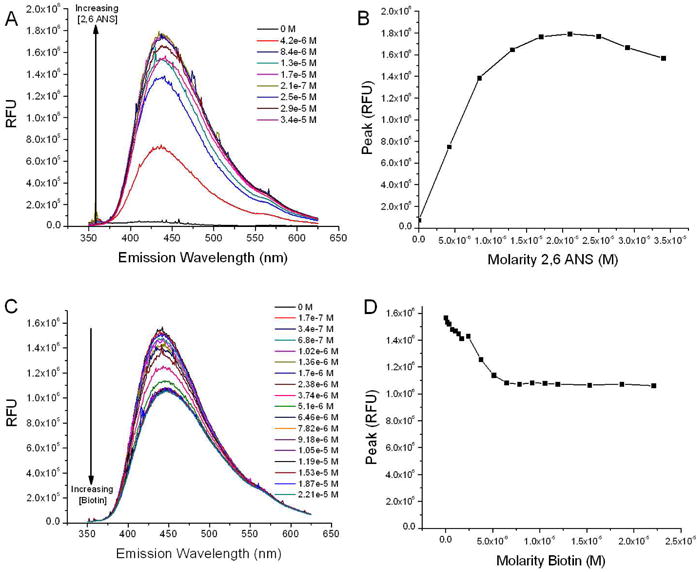
The displacement of 2,6-ANS was utilized to evaluate the coupling of biotin to Avidin-CPNPs. (A) Fluorescence intensities for the first step of the 2,6-ANS assay. The addition of 2,6-ANS to the Avidin-CPNP complex results in a six fold increase in fluorescence as the fluorescent probe binds to the biotin binding site on avidin. The 2,6-ANS was added at increasing concentrations to Avidin-CPNPs and increased fluorescence, indicative of 2,6-ANS bound to avidin, was quantitatively determined. (B) Peak height of fluorescence shown on Figure 3A as a function of 2,6-ANS molarity. (C) Fluorescence intensities for the second step of the 2,6-ANS assay. The addition of biotin to the 2,6-ANS-Avidin-CPNP complex results in a decrease in fluorescence as biotin displaces the fluorescent probe from the biotin binding site on avidin. Biotin was added at increasing concentrations to the 2,6-ANS-Avidin-CPNP complex and a decrease in fluorescence, indicative of biotin displacing 2,6-ANS, was quantitatively determined. (D) Peak height of fluorescence shown on Figure 3C as a function of biotin molarity. All determinations are representative of three independent experiments.
Evaluation of Breast Cancer-Targeted CPNPs In Vivo
Transferrin receptors are expressed on cells with increased metabolic demand, including several cancerous cells. The presence of the transferrin receptor (CD71) on the surface of human MDA-MB-231 cells was determined via flow cytometry and was found to be prevalent on nearly all cells analyzed (Figure 4A). The presence of CD71 on most MDA-MB-231 cells indicated that it would be an ideal surface target, exploited by coupling specific antibodies, or the ligand holotransferrin, to our Avidin-CPNPs. It has been previously shown that the untargeted PEG-CPNPs passively accumulate in breast cancer tumors via the EPR effect.2 This finding was successfully repeated within this trial as a positive control (Figure 4B). Intriguingly, tumors from mice receiving Anti-CD71-Avidin-CPNPs, and not Human Holotransferrin-Avidin-CPNPs or untargeted PEG-CPNPs, were effectively targeted as evidenced by prominent illumination 96 hours following tail vein injection of CPNPs (Figure 4B). It has been reported, and is likely in this circumstance, that the transferrin receptors are saturated with transferrin25 and therefore are unable to bind the Human Holotransferrin-Avidin-CPNPs. This is also supported by the success of the Anti-CD71-Avidin-CPNPs, which recognize an epitope separate from the ligand-binding site on the transferrin receptor. Importantly, the Anti-CD71-Avidin-CPNPs were more effective at targeting the tumors than the passively accumulating PEG-CPNPs based on the relative fluorescence intensity. However, and not surprisingly, the effective targeting was not limited to the tumors, but also to the spleen, which is rich in a diversity of hematopoietic cells (Figure 4C). It was also observed that there was some off-target staining of the stomach (Figure 4C), possibly due to avidin interaction with biotin ingested as part of the mouse’s diet or due to the presence of transferrin receptors on these tissues. Previously, clearance of PEG-CPNPs was reported to occur via hepatobiliary clearance evidenced by predominant staining of the liver within minutes following tail vein injection.2 In the current study, hepatobiliary clearance was validated 24 hours post-tail vein injection of PEG-CPNPs, and showed the progression of signal from the liver and through the intestine as fecal matter (Figure 5). Overall, these findings showed that the transferrin receptor-targeted CPNPs were effective and selective in an in vivo model of breast cancer.
Figure 4.
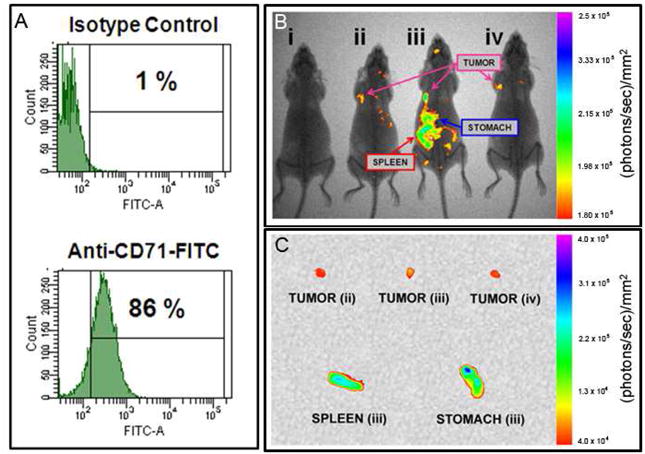
Targeting transferrin receptors in an in vivo subcutaneous-tumor model of breast cancer. (A) Human MDA-MB-231 metastatic breast cancer cells were analyzed via flow cytometry for the presence of the transferrin receptor (CD71). (B) MDA-MB-231 cells were xenografted subcutaneously into athymic nude mice. One week following engraftment, ICG-loaded CPNPs were administered systemically via tail vein injection and near-infrared images were taken 96 hours post-injection. From left to right, mice receiving: (i) free ICG, (ii) ICG-loaded, PEG-CPNPs, (iii) ICG-loaded, Anti-CD71-Avidin-CPNPs, or (iv) ICG-loaded, Human Holotransferrin-Avidin-CPNPs. (C) Excised tumors (mice ii, iii, and iv from panel B), and spleen and stomach (mouse ii). All images are representative of four independent experiments.
Figure 5.
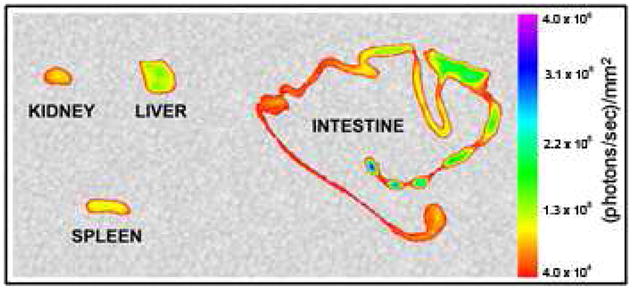
ICG-loaded PEG-CPNP clearance via hepatobiliary secretion. 24 hours post-tail vein injection, the kidney, liver, spleen, and intestine were excised and imaged. Increased signal towards end of intestine as indicated by fecal pellets within intestine. All images are representative of three independent experiments.
Evaluation of BxPC-3 Pancreatic Cell Targeting by Gastrin Receptor-Targeted CPNPs
Increased surface expression of gastrin receptors on pancreatic tumors, and cell lines, was targeted by CPNPs coupled via a PEG linker to a short gastrin peptide (Gastrin-10-PEG-CPNPs). BxPC-3 human pancreatic cancer cells were treated with Gastrin-10-PEG-CPNPs or untargeted PEG-CPNPs for 5 or 60 minutes, followed by a replacement of media for 55 minutes or no change, respectively. Cells were fixed and visualized using a fluorescence microscope set up to analyze a broad range of fluorescence simultaneously. Only BxPC-3 cells exposed for 60 minutes to Gastrin-10-PEG-CPNPs, and no media exchange, displayed fluorescent staining (Figure 6A). Intriguingly, the observed fluorescence was green and blue, indicative of the pH-dependent degradation of CPNPs as they internalize to the endosomal-lysosomal pathway, and release the encapsulated dye (fluorescein). Fluorescein displays a complex pH-dependent equilibrium and emission from its two fluorescent ionic forms, the monoanion and dianion.26, 27 In higher pH environments, such as that in the CPNPs and physiological solutions, the significant emission wavelength is from the dianion (peak excitation 495 nm, green). As pH drops below 6.5, the molecule is deprotonated into its monoanionic form which is excited in the blue (450 nm). Thus, emission signals from the fluorescein-encapsulating CPNPs shift from green toward blue as they experience the pH drop characteristic of the endosomal-lysosomal pathway into the cells, resulting in the dissolution of the particles and release of the fluorophore into the lower pH environment of late stage endosomes.
Figure 6.
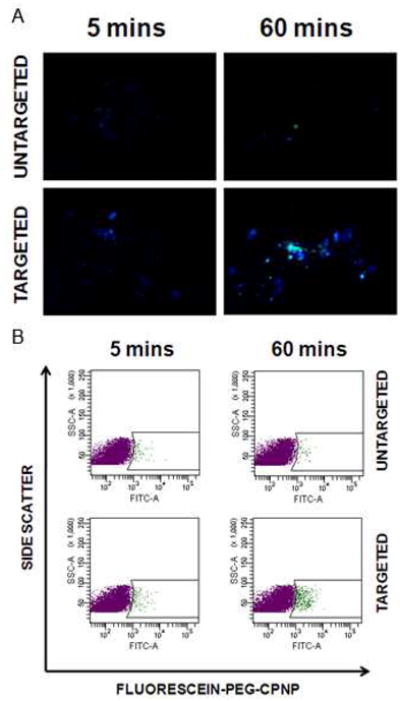
Gastrin receptor-targeted CPNPs effectively targeted human BxPC-3 pancreatic cancer cells. BxPC-3 cells were exposed to fluorescein-loaded untargeted PEG-CPNPs, or Gastrin- 10-PEG-CPNPs, for 5 minutes followed by exchange to fresh media for 55 minutes, or exposure for 60 minutes. (A) Cells were fixed and visualized by microscopy. (B) Cells were fixed and analyzed by flow cytometry with graphs representing 10,000 collected events per sample.
Alternatively, BxPC-3 cells exposed, fixed, and analyzed via flow cytometry (Figure 6B). This further showed that Gastrin- 10-PEG-CPNPs (60 minutes exposure) targeted BxPC-3 cells while untargeted PEG-CPNPs did not.
Evaluation of Pancreatic Cancer-Targeted CPNPs In Vivo
It was found that the untargeted, PEG-CPNPs effectively accumulated 24 hours post-tail vein injection within small BxPC-3 tumors in the pancreas (Figure 7A), and these whole animal images were confirmed by excision of the pancreas (Figure 7B). The Pentagastrin-Avidin-CPNPs, using the avidin-biotin coupling approach, also targeted the pancreatic tumors (Figure 7). However, the Gastrin-10-PEG-CPNPs proved to be much more successful at targeting the pancreatic tumors (Figure 7A), including peritoneal extensions of the primary tumor, as well as the brain which is also rich in gastrin receptors.14 We confirmed targeting of the Gastrin-10-PEG-CPNPs to the brain by excising and imaging the brain during necropsy (Figure 7B). An advantage of the later targeting approach is the covalent attachment, eliminating the possibility of nonspecific avidin interactions in vivo, as well as the PEG, which permits improved systemic retention and decreased immune-reactivity.2 It is also possible that the presence of avidin on the CPNPs does not permit crossing of the blood-brain-barrier, whereas the PEG-maleimide bioconjugation for gastrin-10 may permit penetration of the brain-brain-barrier. It is noteworthy that the untargeted, PEG-CPNPs did not display any significant brain-accumulation in this study. A recent study comparing interleukin-13-targeted nanoliposomes to untargeted nanoliposomes in a cranial model of glioblastoma showed that only targeted nanoliposomes moved significantly across the blood-brain-barrier.28 The current study, although using a different target and different nanoparticles (gastrin receptor-targeted - CPNPs), corroborates the other group’s finding that targeted nanoparticles can cross the blood-brain-barrier. Importantly, the CPNPs are biocompatible and it has been previously shown that they exhibited no specific detrimental effects toward neurons.3 This finding was reiterated in the current study, as no mice receiving any CPNPs showed signs of neurological deficits. Therefore, this portion of the study demonstrated that the CPNPs can be effectively targeted to gastrin receptors in vivo in a model of pancreatic cancer, and further showed the potential for targeting across the blood-brain-barrier.
Figure 7.
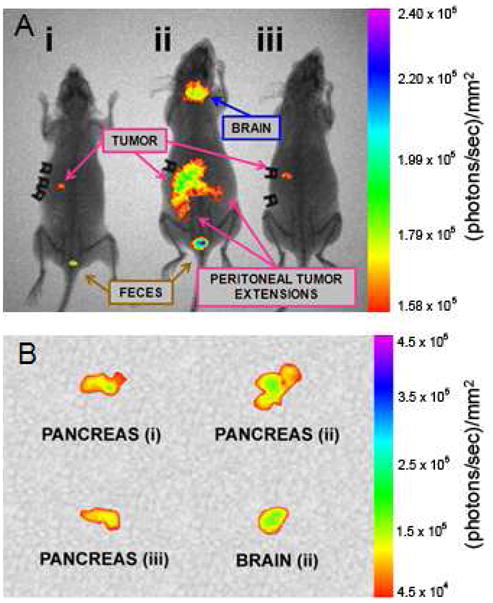
Targeting gastrin receptors in an in vivo orthotopic-tumor model of pancreatic cancer. Human BxPC-3 pancreatic cancer cells were xenografted orthotopically into athymic nude mice. (A) One week following engraftment, ICG-loaded CPNPs were administered systemically via tail vein injection and near-infrared images were taken 24 hours post-injection. From left to right, mice receiving: (i) ICG-loaded, PEG-CPNPs, (ii) ICG-loaded, Gastrin- 10-PEG-CPNPs, or (iii) ICG-loaded, Pentagastrin-Avidin-CPNPs. (B) Excised, tumor-bearing, pancreases from each mouse, and excised brain (mouse ii). All images are representative of at least four independent experiment
CONCLUSIONS
The ability to target nanodelivery systems to specific tissues is important in the development of improved therapeutics for diseases such as cancer. In many studies and clinical circumstances, the efficacies of treatments are limited or the off-target effects are dramatic. Nanoscale therapeutics help to minimize these problems by concentrating smaller doses of therapeutic agents, yet are still limited if not targeted. Recent research has shown that molecular-specific therapeutics, or targeted therapeutic delivery systems are highly efficacious, and may even help to overcome complicating circumstances such as multidrug resistance.11 Often these studies are restricted to in vitro models. However, the true test for active targeting requires in vivo models, in which the delivery modality is administered systemically with the nanodelivery system allowed to freely circulate to localize in the desired tissue to establish efficacy of the targeting strategy and delivery system. The present study demonstrates effective systemic targeting in both a subcutaneous and an orthotopic in vivo model using the CPNP nanodelivery system.
The CPNPs in this study were engineered specifically as non-toxic biocompatible nanoscale delivery vehicles. It has been previously shown that a variety of molecules, including dyes that could be used in tumor detection, or hydrophobic antineoplastic agents such as ceramide could be encapsulated.1–3 Until now, the CPNPs have relied on passive accumulation via the EPR effect for solid tumor detection. This study demonstrates that surface-targeting strategies can be successfully attached to the CPNPs, via two distinct coupling methods, and these bioconjugated CPNPs can effectively target select tissues via surface feature targeting (Figure 8). Specifically, breast cancer tumors were targeted in vivo by targeting transferrin receptors, and orthotopic pancreatic cancer tumors were targeted in vivo by targeting gastrin receptors. Additionally our experiments confirmed that the untargeted, PEG-CPNPs could accumulate moderately, yet could be effectively imaged, within small orthotopic pancreatic tumors, extending the diagnostic imaging capability and therapeutic delivery capabilities to one of the more evasive cancers. This study also showed that gastrin receptor-targeted CPNPs could cross the blood-brain-barrier, which may expand the utility of the CPNPs to therapeutics targeted to glioblastoma or even to neurodegenerative or psychiatric disorders.
Figure 8.
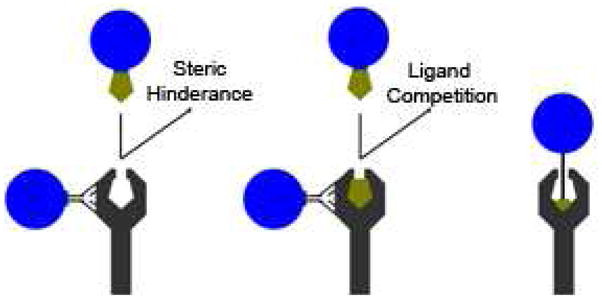
Receptor, or surface feature, targeting strategies and pitfalls. CPNPs were designed to target transferrin receptors (CD71) or gastrin receptors via antibody or ligand coupling via non-covalent (avidin-biotin interactions) and covalent (PEG linker) coupling strategies. Receptor-targeted CPNPs utilizing coupled-antibodies may interact via epitopes separate from ligand-binding sites. Ligand-coupled CPNPs may interact with the receptor, however interference can occur in the form of steric hindrance (avidin or particle interferes with interaction), or ligand competition with the natural ligand.
Even though targeted nanotechnology can be used to target-specific receptors or ligands on cancer cells, there may still be obstacles, as these receptors may be expressed on other tissues. As a case in point to alleviate this short-coming, CPNPs can potentially be ‘loaded’ with selective gene therapies or agents to become cancer-specific. For example, although gastrin receptors are present in both malignant and some normal tissues, only the pancreatic cancer cells produce endogenous gastrin.29 The acid secreting parietal cells of the stomach, imaged in the current investigation with ICG-loaded, Gastrin-10-PEG-CPNPs (covalently coupled), or ICG-loaded, Pentagastrin-Avidin-CPNPs, do not produce endogenous gastrin. The only nonmalignant cells that produce gastrin in adults are the G-cells of the stomach antrum; and, the G-cells do not possess gastrin receptors.30, 31 Previous studies have shown that down regulation of endogenous gastrin expression using RNA interference techniques significantly inhibits growth of pancreatic cancer tumors and metastases.32, 33 One problem with using gene therapy in animals and in humans has been in finding delivery systems that would protect the siRNA from degradation in the peripheral circulation. Since siRNA molecules are readily degraded by nucleases in the peripheral blood and tissues, mechanisms for delivery have been an active area of recent investigation. Viral vectors, especially the adeno-associated viruses (AAVs) and the adenoviruses have been under investigation; however, hepatotoxicity and immunogenicity have been reported.34 The use of a tissue-specific and cancer-selective vehicle such as siRNA-loaded CPNPs coupled for receptor targeting would be ideal as cancer therapeutics.
This study showed the successful bioconjugation of surface targeting strategies to the CPNPs, demonstrating effectiveness, selectivity, and utility in two separate in vivo models. This study will allow the further development of the CPNPs to target a diversity of disorders, including several poor prognosis cancers and possibly even non-solid tumors such as leukemia.
MATERIALS AND METHODS
Preparation of Nanoparticles
CPNPs were prepared by the microemulsion technique and van der Waals-HPLC that have been previously described.1–4 Cyclohexane (C6H12, BHD Chemical Co.), Igepal CO-520 (C13H20O(C2H4O)n=5, Rhodia Chemical Co.), and deionized H2O were used to prepare the microemulsions. Calcium chloride (CaCl2.2H2O, Sigma-Aldrich Co.), disodium hydrogen phosphate (Na2HPO4, Sigma Aldrich Co.), and sodium metasilicate (Na2SiO3, Sigma-Aldrich Co.) were used as particle precursors. Disodium hydrogen citrate dihydrate (HOC(COOH)(CH2COONa)2·2H2O, Sigma-Aldrich Co.) was used as the dispersant. Indocyanine green (ICG) (TCI America Co.) was used as the near infra-red fluorophore in the CPNPs for the animal trials, while fluorescein sodium salt (Sigma-Aldrich Co.) was the visible fluorophore encapsulated for flow cell and in vitro experiments. Neat ethanol was purchased from VWR International. All other chemicals were obtained from Sigma-Aldrich Co., unless otherwise noted.
Two separate microemulsions (1 and 2) were formed with a cyclohexane/Igepal CO-520/water system. The molar ratio of water to surfactant was 4. 650 μL of 1 × 10−2 M CaCl2 in CO2-free deionized H2O was added to 14 mL of a 29 volume percent solution of Igepal CO-520 in cyclohexane to form Microemulsion 1. Similarly, 65 μL of 6 × 10−2 M disodium hydrogen phosphate (Na2HPO4) with 65 μL of 8.2 × 10−3 M sodium metasilicate (Na2SiO3) in CO2-free deionized H2O (pH 7.4) was added to 14 mL of a 29 volume percent solution of Igepal CO-520 in cyclohexane to form Microemulsion 2. A 520 μL aliquot of 0.01 M fluorophore in CO2-free deionized H2O was added into Microemulsion 2 so that the final H2O volume matched that in Microemulsion 1 (650 μL), hence retaining the water to surfactant ratio in each. The individual microemulsions were allowed to equilibrate for 1 hour before 1 and 2 were mixed to form Microemulsion 3. Microemulsion 3 was allowed to undergo micellar exchange for 2 minutes, during which time doped CPNPs precipitated in the micelles. A 225 μL aliquot of 1×10−3 M sodium citrate was added to Microemulsion 3 and allowed to react for 15 minutes. After adding the dispersant, the reverse micelles were dissolved with 50 mL of ethanol adjusted with 1 M KOH before laundering via the van der Waals-HPLC.1–4
The unwashed CPNP suspension was loaded onto a silica HPLC (high performance liquid chromatography) column after the micelles had been dissolved with ethanol; the free organic was laundered with ethanol adjusted with 1 M KOH as the eluent; finally, the particles were eluted using 70:30 ethanol:water by volume. During the washing step, the dye content was monitored at absorption of 785 nm or 495 nm for ICG or fluorescein, respectively. The ethanol washing was continued until the detector reached baseline, indicating removal of the excess dye. The first major peak was collected. The precursor and HPLC solutions were prepared with CO2-free deionized H2O to avoid carbonate contamination in the CPNPs. All solution pH measurements were performed using a Sentron ISFET pH probe calibrated against aqueous standards.
Bioconjugation of CPNPs
To bioconjugate the CPNPs with avidin, a 1 mL aliquot of CPNPs in their 70% ethanol solution was first dried under argon and covered from light until all the solvent evaporated and only the CPNPs remained. This dried sample was then reconstituted back to 1 mL with the addition of 1 mL PBS (0.01 M Phosphate buffer, 0.14 M NaCl, 0.01 M KCl at pH 7.4). This was then followed by the addition of 1 mL of 20 mg/mL 1-ethyl-3-[3-dimethylaminopropyl]-carbodiimide hydrochloride (EDCI) (Sigma-Aldrich Co.) and 1 mL of 6 mg/mL avidin (Rockland Immunochemicals Inc.). Excess avidin was added in order to ensure the surface saturation of CPNPs. This reaction mixture was incubated at 35°C for 24 hours in the dark with continuous stirring at 600 RPM.
After 24 hours, the reaction mixture was centrifuge (Marathon 22K Centrifuge; Fischer Scientific Co.) filtered with a 100 kDa centrifuge filter device (Amicon Ultra-4, PLHK Ultracel-PL Membrane; Millipore Co.) to remove excess unconjugated avidin. Prior to use, the filtration membrane of the centrifuge filter device was washed with deionized H2O in order to minimize non-specific binding. A total of three centrifuge filtrations, each at 1000 × g for 30 minutes, were performed in order to maximize the removal of excess unconjugated avidin. After each centrifuge filtration step, the final volume of the retentate solution was brought back to the starting volume by the addition of PBS.
Multiple types of commercially available centrifuge filter devices, with different filtration membrane materials and chemistry, were evaluated to obtain minimal non-specific adsorption and optimal washing of the CPNPs. The Millipore Amicon centrifuge filter device with 100 kDa nominal molecular weight limit Ultracel YM-100 regenerated cellulose membrane was identified as the product of choice. Centrifuge filter devices with a polyethersulfone filtration membrane were not feasible for this investigation due to significant non-specific binding of the CPNPs to the filtration membrane.
The conjugation of biotinylated human holotransferrin or biotinylated anti-CD71 antibody to avidin-CPNPs permits targeting of the transferrin receptor; the conjugation of biotinylated pentagastrin to avidin-CPNPs permits targeting of the gastrin receptor. A 1 mL aliquot of 3.2 mg/mL biotin conjugated human holotransferrin (Invitrogen Co.), 0.20 mL of 1 mg/mL biotin conjugated anti-CD71 antibody (GeneTex Inc.), or 0.50 mL of 1 mg/mL biotin conjugated pentagastrin (Bachem Co.) (in 0.1 N NH4OH) was added to 1 mL of Avidin-CPNP complex. This reaction mixture was stirred at 600 RPM for 60 minutes at room temperature. The resulting Biomolecule-Avidin-CPNP complex was then filtered to remove excess unconjugated biomolecule (human holotransferrin, anti-CD71 antibody, or pentagastrin). Human holotransferrin-Avidin-CPNPs and anti-CD71-Avidin-CPNPs were filtered by tangential flow diafiltration using a 500 kDa molecular weight cut-off (MWCO) MicroKros hollow fiber tangential flow diafiltration device (Spectrum Laboratories Inc.). Pentagastrin-Avidin-CPNPs were centrifuge filtered with a 100 kDa centrifuge filter device (Amicon Ultra-4, PLHK Ultracel-PL Membrane; Millipore Co.). All the Biomolecule-Avidin-CPNP samples were filtered three times in order to maximize the removal of excess unconjugated biomolecule. After each filtration step, the final volume of the retentate solution was brought back to the starting volume by the addition of PBS.
The conjugation of gastrin-10 to CPNPs via the PEG-maleimide coupling strategy permits targeting of the gastrin receptor as previously described by Sosabowski and colleagues.35 A 9 mL aliquot of citrate-CPNPs was chemically conjugated with maleimide polyethylene glycol amine (PEG-maleimide; JenKem Technology Inc.), through the ethyl-N-(3-dimethylaminopropyl)-N′ hydrochloride carbodiimide reaction (EDCI, Fluka BioChemika ≥99.0% (AT); Sigma-Aldrich Co.). The sample was first stirred at 550 RPM on a combined magnetic stir/hot plate set to 50°C. In a drop wise manner, 1 mL of EDCI (1 mg/mL) followed by 1 mL of PEG-maleimide (10 mg/mL), both in aqueous solutions of CO2-free deionized H2O (pH 7), were added to the sample under continuous stirring, to produce a calculated 6-fold excess for monolayer surface coverage. The particles were allowed to react for 15 hours at 50°C to form amide linkages between the carboxylate surfaces and the PEG-maleimide. The mixture was then centrifuge filtered through a 100 kDa filter (Amicon Ultra-4, PLHK Ultracel-PL Membrane; Millipore Co.) at 5000 × g for 2 minutes to remove any excess EDCI and unreacted PEG-maleimide. The characterization of the resulting Maleimide-PEG-CPNPs in the retentate showed that the CPNPs remained well dispersed after the centrifugation wash. The gastrin-10 peptide has a cysteine residue for covalent attachment. Thus, the gastrin-10 was added at a 5:1 molar excess to the Maleimide-PEG-CPNPs. This solution was incubated overnight at 4 °C, protected from light, to produce Gastrin-10-PEG-CPNPs.
Characterization of Nanoparticles
Particle size distributions for the CPNPs were obtained through dynamic light scattering (DLS) using a Malvern Nano-S Zetasizer. Transmission electron microscopy (TEM) was performed using a JEOL JEM 1200 EXII instrument on dried nanoparticles prepared on a carbon film grid with a copper backing. To verify that avidin was grafted on the CPNPs, zeta potential distributions were obtained using a Brookhaven ZetaPALS zeta potential analyzer. To quantify and test the bioactivity of avidin grafted on CPNPs, a fluorometric assay for avidin-biotin interaction based on the displacement of the fluorescent probe 2-Anilinonaphthalene-6-sulfonic acid (2,6-ANS) was utilized.22
A 4.85 mg/mL solution of 2,6-ANS (Molecular Probes, Invitrogen Co.) was prepared in 1 mL of deionized H2O. A 24 μg/mL solution of biotin was prepared in 10 mL of deionized H2O to produce the biotin solution for the assay. The 2,6-ANS solution, followed by the biotin solution, was titrated in 0.50 μl aliquots into the avidin-CPNP solution to obtain a sufficient number of data points. The reaction mixture was stirred for one minute after the addition of each aliquot to the Avidin-CPNP solution to allow enough time for a homogeneous reaction mixture and to maintain consistency with respect to reaction time throughout the experiment. The fluorescence spectra were recorded with a PTI fluorometer in which emitted radiation is collected at 90 degrees with a photomultiplier tube (PMT) detector. The sample is excited by a xenon arc lamp whose illumination passes through a 5 nm bandwidth slit and a monochrometer to select the excitation wavelength. For the emission scan, the excitation wavelength was set to 328 nm and the emission wavelength range was set to 350–625 nm.
Cell Culture
Human MDA-MB-231 breast cancer cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic solution (Invitrogen, Carlsbad, CA). Human BxPC-3 pancreatic cancer cells were cultured in RPMI-1640 supplemented with 10% FBS and 1% antibiotic/antimycotic solution. All cell cultures were maintained at 37°C and 5% CO2. Cells were harvested by trypsin/EDTA detachment for subculture or tumor engraftment.
Fluorescence Microscopy
Cells were grown on cover slips under normal media conditions, and then incubated with targeted or untargeted CPNPs followed by a media exchange. Cells were fixed in 2% (w/v) paraformaldehyde, and mounted onto glass slides. Cells were visualized using a Nikon Eclipse E400 microscope through a 40X objective using a combination Nikon DAPI/FIT-C filter cube and recorded on a Nikon Coolpix 995 digital camera.
Flow Cytometry
Cells were detached from tissue culture-ware, surface Fc receptors blocked with appropriate IgG, incubated with specific antibodies (anti-human CD71-FITC or-PE, eBiosciences, San Diego, CA), and fixed in 2% (w/v) paraformaldehyde. Samples were analyzed on a BD Biosciences (San Jose, CA) LSR II Special Order flow cytometry in the Penn State College of Medicine Flow Cytometry Core, utilizing appropriate compensation controls. Data analysis was performed using BD Biosciences FACS Diva software.
In Vivo Tumor Xenograft
All animal procedures were approved by the Pennsylvania State University Institutional Animal Care and Use Committee. Four to six week old female athymic nude mice were purchased from Harlan (Indianapolis, IN). Subcutaneous breast cancer xenografts were prepared as previously described.2 Briefly, 1×107 MDA-MB-231 cells, prepared in 100 μL of growth media, were engrafted by subcutaneous injection. Orthotopic pancreatic cancer xenografts were prepared as previously described.32 Briefly, mice were fully anesthetized with a mixture of ketamine-HCl (129 mg/kg) and xylazine (4 mg/kg) injected intramuscularly. A small incision was made in the left flank, the peritoneum was dissected and the pancreas exposed. Using a 27-gauge needle, 1×106 BxPC-3 cells, prepared in 100 μL of Hank’s balanced salt solution, were injected into the pancreas. All xenografted or orthotopic tumors were allowed to establish for one week prior to experimentation.
In Vivo Imaging
CPNPs, or controls, were diluted 1:10 into sterile isotonic NaCl and 100 μL was injected via tail vein into tumor-bearing mice. Equivalent ICG concentrations were determined prior to injection via absorption spectroscopy (2×10−6 M prior to dilution). Whole animal imaging was performed as previously described.2 Briefly, anesthesia was induced and maintained by inhalation of 5% IsolSol (Vedco, St. Joseph, MO) in 100% oxygen. Near-infrared transillumination images (755 nm excitation, 830 nm emission, 3 minute exposure) and corresponding X-ray images were obtained with an In Vivo FX whole animal imaging station (Carestream Health, Rochester, NY). Signal distribution relative to anatomy was illustrated by merging near-infrared and X-ray images.
Acknowledgments
This work was funded, in part, by a grant from the Children’s Miracle Network to BMB to evaluate targeted nanotherapies and NIH grant NIH R01 CA117926 to JPS. This work was also partially supported by Keystone Nano, Inc. James Adair and Mark Kester are CSO and CMO, respectively.
References
- 1.Morgan TT, Muddana HS, Altınoğlu Eİ, Rouse SM, Tabaković A, Tabouillot T, Russin TJ, Shanmugavelandy SS, Butler PJ, Eklund PC, et al. Encapsulation of Organic Molecules in Calcium Phosphate Nanocomposite Particles for Intracellular Imaging and Drug Delivery. Nano Letters. 2008;8(12):4108–4115. doi: 10.1021/nl8019888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altınoğlu Eİ, Russin TJ, Kaiser JM, Barth BM, Eklund PC, Kester M, Adair JH. Near-Infrared Emitting Fluorophore-Doped Calcium Phosphate Nanoparticles for in Vivo Imaging of Human Breast Cancer. ACS Nano. 2008;2(10):2075–2084. doi: 10.1021/nn800448r. [DOI] [PubMed] [Google Scholar]
- 3.Kester M, Heakal Y, Fox T, Sharma A, Robertson GP, Morgan TT, Altınoğlu Eİ, Tabaković A, Parette MR, Rouse SM, et al. Calcium Phosphate Nanocomposite Particles for in Vitro Imaging and Encapsulated Chemotherapeutic Drug Delivery to Cancer Cells. Nano Letters. 2008;12:4116–4121. doi: 10.1021/nl802098g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muddana HS, Morgan TT, Adair JH, Butler PJ. Photophysics of Cy3-Encapsulated Calcium Phosphate Nanoparticles. Nano Letters. 2009;4:1559–1566. doi: 10.1021/nl803658w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daniels TR, Delgado T, Helguera G, Penichet ML. The Transferrin Receptor Part Ii: Targeted Delivery of Therapeutic Agents into Cancer Cells. Clin Immunol. 2006;121(2):159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The Transferrin Receptor Part I: Biology and Targeting with Cytotoxic Antibodies for the Treatment of Cancer. Clin Immunol. 2006;121(2):144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Omary MB, Trowbridge IS, Minowada J. Human Cell-Surface Glycoprotein with Unusual Properties. Nature. 1980;286:888–891. doi: 10.1038/286888a0. [DOI] [PubMed] [Google Scholar]
- 8.Shindelman JE, Ortmeyer AE, Sussman HH. Demonstration of the Transferrin Receptor in Human Breast Cancer Tissue. Potential Marker for Identifying Dividing Cells. Int J Cancer. 1981;27:329–334. doi: 10.1002/ijc.2910270311. [DOI] [PubMed] [Google Scholar]
- 9.Sutherland R, Delia D, Schneider C, Newman R, Kemshead J, Greaves M. Ubiquitous Cell-Surface Glycoprotein on Tumor Cells Is Proliferation-Associated Receptor for Transferrin. Proc Natl Acad Sci US A. 1981;78:4515–4519. doi: 10.1073/pnas.78.7.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gosk S, Vermehren C, Storm G, Moos T. Targeting Anti-Transferrin Receptor Antibody (Ox26) and Ox26-Conjugated Liposomes to Brain Capillary Endothelial Cells Using in Situ Perfusion. J Cereb Blood Flow Metab. 2004;11:1193–1204. doi: 10.1097/01.WCB.0000135592.28823.47. [DOI] [PubMed] [Google Scholar]
- 11.Huwyler J, Drewe J, Krähenbuhl S. Tumor Targeting Using Liposomal Antineoplastic Drugs. Int J Nanomedicine. 20083(1):21–29. [PMC free article] [PubMed] [Google Scholar]
- 12.Smith JP, Fantaskey A, Liu G, Zagon IS. Identification of Gastrin as a Growth Peptide in Human Pancreatic Cancer. Am J Physiol. 1995;268:R135–R141. doi: 10.1152/ajpregu.1995.268.1.R135. [DOI] [PubMed] [Google Scholar]
- 13.Smith JP, Stock EA, Wotring MG, McLaughlin PJ, Zagon IS. Characterization of the CCK-B/Gastrin-Like Receptor in Human Colon Cancer. Am J Physiol. 1996;271:R796–R805. doi: 10.1152/ajpregu.1996.271.3.R797. [DOI] [PubMed] [Google Scholar]
- 14.Wank SA, Pisegna JR, de Weerth A. Brain and Gastrointestinal Cholecystokinin Receptor Family: Structure and Functional Expression. Proc Natl Acad Sci USA. 1992;89(18):8691–5. doi: 10.1073/pnas.89.18.8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kopin AS, Lee YM, McBride EW, Miller LJ, Lu M, Lin HY, Kolakowski LF, Jr, Beinborn M. Expression Cloning and Characterization of the Canine Parietal Cell Gastrin Receptor. Proc Natl Acad Sci USA. 1992;89:3605–3609. doi: 10.1073/pnas.89.8.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soll AHTB. Receptors That Regulate Gastric Acid-Secretory Function. 3. Vol. 1. Raven Press; New York: 1994. pp. 1139–1170. [Google Scholar]
- 17.Johnson LR, McCormack SA. Regulation of Gastrointestinal Mucosal Growth. 3. Vol. 1. Raven Press; New York: 1994. pp. 611–642. [Google Scholar]
- 18.Smith JP, Shih AH, Wotring MG, McLaughlin PJ, Zagon IS. Characterization of CCK-B/Gastrin-Like Receptors in Human Gastric Carcinoma. Int J Oncol. 1998;12:411–419. [PubMed] [Google Scholar]
- 19.Smith JP, Liu G, Soundararajan V, McLaughlin PJ, Zagon IS. Identification and Characterization of CCK-B/Gastrin Receptors in Human Pancreatic Cancer Cell Lines. Am J Physiol. 1994;266:R277–R283. doi: 10.1152/ajpregu.1994.266.1.R277. [DOI] [PubMed] [Google Scholar]
- 20.Smith JP, Verderame MF, McLaughlin P, Martenis M, Ballard E, Zagon IS. Characterization of the CCK-C (Cancer) Receptor in Human Pancreatic Cancer. Int J Mol Med. 2002;10:689–694. [PubMed] [Google Scholar]
- 21.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer Statistics. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 22.Mock DM, Langford G, Dubois D, Criscimagna N, Horowitz P. A Fluorometric Assay for the Biotin-Avidin Interaction Based on Displacement of the Fluorescent Probe 2-Anilinonaphthalene-6-Sulfonic Acid. Anal Biochem. 1985;151(1):178–81. doi: 10.1016/0003-2697(85)90068-5. [DOI] [PubMed] [Google Scholar]
- 23.Livnah O, Bayer EA, Wilchek M, Sussman JL. Three-Dimensional Structures of Avidin and the Avidin-Biotin Complex. Proc Natl Acad Sci USA. 1993;90(11):5076–5080. doi: 10.1073/pnas.90.11.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morag E, Bayer EA, Wilchek M. Reversibility of Biotin-Binding by Selective Modification of Tyrosine in Avidin. Biochem J. 1996;316(Pt 1):193–199. doi: 10.1042/bj3160193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pardridge WM. Brain Drug Delivery and Blood-Brain Barrier Transport. Drug Delivery. 1993;1:83–101. [Google Scholar]
- 26.Sjoback R, Nygren J, Kubista M. Absorption and Fluorescence Properties of Fluorescein. Spectrochim Acta A. 1995;6(51):L7–L21. [Google Scholar]
- 27.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer; Baltimore, MD: 2006. [Google Scholar]
- 28.Madhankumar AB, Slagle-Webb B, Wang X, Yang QX, Antonetti DA, Miller PA, Sheehan JM, Connor JR. Efficacy of Interleukin-13 Receptor-Targeted Liposomal Doxorubicin in the Intracranial Brain Tumor Model. Mol Cancer Ther. 2009;8(3):648–54. doi: 10.1158/1535-7163.MCT-08-0853. [DOI] [PubMed] [Google Scholar]
- 29.Smith JP, Shih A, Wu Y, McLaughlin PJ, Zagon IS. Gastrin Regulates Growth of Human Pancreatic Cancer in a Tonic and Autocrine Fashion. Am J Physiol. 1996;270:R1078–R1084. doi: 10.1152/ajpregu.1996.270.5.R1078. [DOI] [PubMed] [Google Scholar]
- 30.Brand SJ, Fuller PJ. Differential Gastrin Gene Expression in Rat Gastrointestinal Tract and Pancreas During Neonatal Development. J Biol Chem. 1988;263:5341–5347. [PubMed] [Google Scholar]
- 31.Rozengurt E, Walsh JH. Gastrin, Cck, Signaling, and Cancer. Annu Rev Physiol. 2001;63:49–76. doi: 10.1146/annurev.physiol.63.1.49. [DOI] [PubMed] [Google Scholar]
- 32.Matters GL, Harms JF, McGovern CO, Jayakumar C, Crepin K, Smith ZP, Nelson MC, Stock H, Fenn CW, Kaiser J, et al. Growth of Human Pancreatic Cancer Is Inhibited by Down-Regulation of Gastrin Gene Expression. Pancreas. 2009;38:e151–e161. doi: 10.1097/MPA.0b013e3181a66fdc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith JP, Verderame MF, Ballard EN, Zagon IS. Functional Significance of Gastrin Gene Expression in Human Cancer Cells. Regul Pept. 2004;117:167–173. doi: 10.1016/j.regpep.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 34.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in Mice Due to Oversaturation of Cellular MicroRNA/Short Hairpin RNA Pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 35.Sosabowski J, Lee M, Dekker B, Simmons B, Singh S, Bereford H, Hagan S, McKenzie A, Mather S, Watson S. Formulation Development and Manufacturing of a Gastrin/CCK-2 Targeting Peptide as an Intermediate Drug Product for Clinical Imaging Study. Eur J Pharm Sci. 2007;31:102–111. doi: 10.1016/j.ejps.2007.02.007. [DOI] [PubMed] [Google Scholar]