

Reprogramming of somatic cells to the pluripotent stem cell state may allow the development of in vitro models of disease and could provide a mechanism for the generation of patient-specific cells of therapeutic interest. Reprogramming of mouse fibroblasts into induced pluripotent stem cells, or iPS cells, has been achieved with overexpression of oct4, sox2, klf4, and c-myc and drug selection for the reactivation of a marker of pluripotency (Maherali et al., 2007; Okita et al., 2007; Takahashi and Yamanaka, 2006; Wernig et al., 2007) (reviewed in Yamanaka, 2007). Here we show that n-myc can substitute for c-myc and that drug selection is dispensable for reprogramming of fibroblasts to pluripotent stem cells. We show that serum-free conditions facilitate reprogramming and that the resulting induced pluripotent stem cells contribute extensively to teratomas and chimeras. Our findings greatly simplify the method for induction of pluripotency and bring it one step closer to clinical applications.
The iPS cell reprogramming method is technically very simple, but suffers from three substantial problems: (1) it requires that the host cell be genetically engineered to express a drug resistance gene driven by a marker of pluripotency, (2) it requires viral-mediated integration of transgenes into the genome, and (3) reactivation of c-myc in differentiated progeny of the induced ES-like cells is common and results in tumor formation (Okita et al., 2007). A major goal in reprogramming is to solve these problems and achieve the generation of normal pluripotent stem cells in the absence of genetic manipulation.
We analyzed the gene expression profiles of pluripotent mouse cells, including the inner cell mass of the blastocyst, ES cells, primordial germ cells, and embryonic germ cells (G. Wei, R.-F. Yeh, M. Hebrok, and M.R.-S., unpublished data). We observed that n-myc, but not c-myc, is highly up-regulated in all pluripotent cells. The study that implicated c-myc in regulation of ES cells relied on the use of dominant-negative and constitutively active c-Myc proteins, which would not functionally discriminate between the various myc family members (Cartwright et al., 2005). n-myc and c-myc are largely interchangeable (Malynn et al., 2000), although n-myc has been reported to be less tumorigenic than c-myc in in vitro assays of transformation (Malynn et al., 2000). For these reasons, we decided to test n-myc in the induction of pluripotency.
We infected rosa26/βgeo;oct4/gfp mouse embryonic fibroblasts (MEFs) with lentiviral vectors that lead to the expression of oct4, sox2, klf4, and n-myc and tested whether any colonies were observed in the absence of drug selection, in standard LIF- and serum-containing ES cell medium. As early as 6 days postinfection, we observed the appearance of colonies, all of which stained strongly for alkaline phosphatase (AP, Figure 1A), a marker of ES cells. The efficiency of formation of AP-positive colonies at 11 days postinfection (82.7 colonies/100,000 cells) was comparable to previous reports (Maherali et al., 2007; Okita et al., 2007; Wernig et al., 2007). However, these colonies (Figure 1B) did not show GFP expression, indicating that reactivation of the oct4 promoter had not yet occurred. We picked 16 colonies, four (25%) of which could be expanded. With further culture, these clones progressively displayed GFP fluorescence (Figure 1C), indicative of more extensive, although still incomplete, reprogramming. Overall, these clones appeared more unstable than regular ES cells, and partially differentiated colonies could be observed in the cultures.
Figure 1. Induction of ES-like Cells by Overexpression of oct4, sox2, klf4, and n-myc in the Absence of Drug Selection.
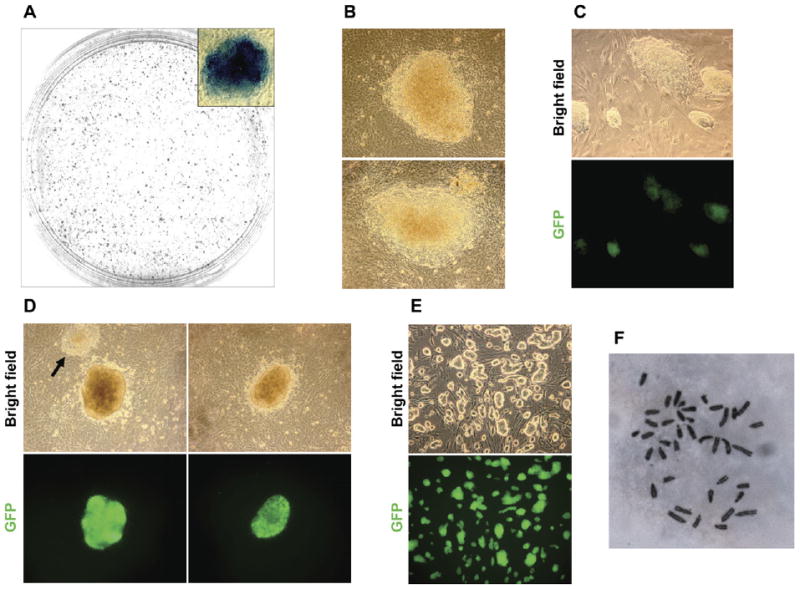
(A) Alkaline phosphatase staining of ES-like colonies induced in the absence of drug selection.
(B) Colonies induced in the presence of serum at 11 days postinfection. No GFP expression was detected.
(C) Clones were picked and expanded in serum-based medium. Patchy GFP expression is seen in some colonies.
(D) Colonies induced in serum-based medium, then replaced with KSR based medium at day 4. Representative colonies from 11 days postinfection are shown. Note that colonies induced in KSR are smaller and more compact than the ones induced in serum (compare to Figure 1B). Furthermore, the majority of ES-like colonies induced in KSR conditions are GFP positive, although some GFP negative colonies are still present (arrow). GFP negative colonies are less compact and have poorly defined edges.
(E) Clones were picked and expanded in KSR-based medium. Clones have excellent ES-cell colony morphology and maintain strong and uniform GFP expression.
(F) Cells of all three clones expanded in KSR-based medium were predominantly diploid. Images in (B), (C), and (D) were taken with a 20× objective, (E) with 10×, and (F) with 100×.
In parallel with the experiments described above, we also tested whether use of serum-free medium would facilitate reprogramming. We chose this approach based on our observation that ES cells, but not MEFs, tolerate knockout serum replacement (KSR) well (data not shown). At 4 days postinfection, FBS-containing medium was replaced with KSR-containing medium. Colonies were first observed at 8 days and counted at 11 days postinfection. The efficiency of AP+ colony formation in these conditions was lower than in the presence of serum (30.7 colonies/100,000 cells). However, the majority of the colonies that did develop were small, round, and compact with well-defined edges and had reactivated the Oct4/GFP reporter (Figure 1D, compare colony morphology with the colonies induced in serum at the same stage, Figure 1B). The colonies induced in KSR could easily be identified and picked based on colony morphology alone (compare the two colonies on Figure 1D, left panels). We picked six colonies 11 days postinfection in serum-free conditions and established three clones (50%) of induced ES-like cells (Figure 1E) that all maintain strong and uniform GFP expression and excellent ES cell-like morphology. All three clones are karyotypically normal diploid cells (Figure 1F).
To test whether the induced cells are pluripotent, we injected two of the clones subcutaneously into immunocompromised mice. Three weeks later, heterogeneous teratomas were observed that were similar in appearance to those resulting from the injection of control ES cells (Figure 2A). Histological analysis of teratomas from both injected clones revealed the presence of ectoderm-, mesoderm-, and endoderm-derived tissues (Figure 2B), indicating that the induced ES-like cells are indeed pluripotent. To further evaluate pluripotency of the induced cells, we injected them into blastocysts to generate chimeras. Staining for β-galactosidase, driven by the rosa26 locus, allowed for the detection of the injected cells and their descendants. Both iPS cell clones generated E13.5 chimeric embryos at a high frequency (six of eight embryos for clone 2K1, and three of four embryos for clone 2K3), with all showing high contribution from donor cells (Figure 2C). Histological analysis of the chimeras revealed contribution to ectoderm, mesoderm, and endoderm-derived organs (Figure 2D), demonstrating that the induced cells are indeed pluripotent stem cells.
Figure 2. Cells Induced in the Absence of Drug Selection Are Pluripotent.
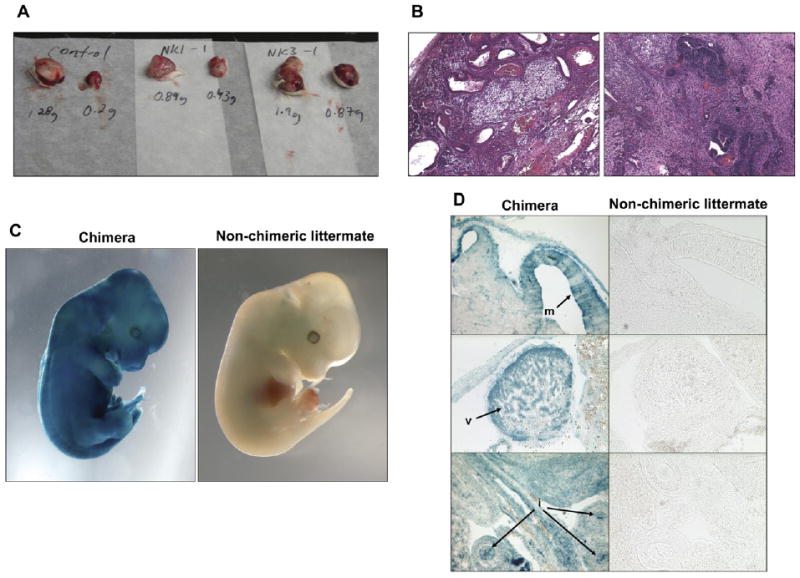
(A) Teratomas generated by clones 2K-1 (middle) and 2K-3 (right) were comparable in weight and appearance to those generated by control ES cells (left).
(B) Teratomas generated by both clones were similar to control ES cells (data not shown) and contained derivatives of all three germ layers, such as cartilage and gut epithelium (left panel) and neural tissue (right panel).
(C) High-contribution chimera resulting from the injection of clone 2K-1 into blastocysts. Similar results were obtained for clone 2K-3.
(D) Sections showing extensive contribution of injected cells to brain (m, midbrain), heart (v, ventricular myocardium), and gut endoderm (i, intestinal epithelium).
Together, these results establish that pluripotency can be induced in fibroblasts in the absence of a drug-selectable ES cell marker. Interestingly, the use of serum-free medium appeared not only to select for reprogrammed cells but also to accelerate reprogramming. That is, there were fewer colonies in serum-free medium but the majority showed oct4/gfp expression, while no GFP positive colonies were observed at the same stage in serum-containing medium. The ability to induce pluripotency in the absence of drug selection will allow reprogramming of a broad range of samples, because no a priori targeting of a drug-selectable cassette is required. This will make the technique broadly accessible to the scientific community. We have also shown that a combination of factors different than the one previously described (Maherali et al., 2007; Okita et al., 2007; Takahashi and Yamanaka, 2006; Wernig et al., 2007), with n-myc substituting for c-myc, is capable of inducing pluripotency. Future studies should address the relative efficacy of n-myc versus c-myc in reprogramming and whether n-myc reactivation, like c-myc, results in tumor formation. The next critical milestones will be to develop approaches that induce pluripotency without viral integration and to translate these methods from mouse to humans.
Supplementary Material
Acknowledgments
We thank Amy Heidersbach and Michael McManus in the Diabetes Center Lentiviral Core Facility for generation of lentiviruses, Alexandre Gaspar-Maia and Connie Wong for assistance with molecular cloning, Xiaoji Chen for karyotyping, and Arnold Kriegstein and Holger Willenbring for discussions. This work was supported by funds to M.V. from NIH/NINDS grant 1F32NS058042-01A1, to R.B. from the University of California, San Francisco Urology Department, Sandler Foundation, and NIH/NINDS grant K08 NS48118, and to M.R.-S. from the UCSF Institute for Regeneration Medicine, Sandler Foundation, NIDDK/DERC, and Juvenile Diabetes Research Foundation. R.B. is a Pew Scholar.
Footnotes
Supplemental Data: Supplemental Data include Experimental Procedures and can be found with this article online at http://www.cellstemcell.com/cgi/content/full/1/3/245/DC1/.
References
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. Development. 2005;132:885–896. doi: 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- Maherali M, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, et al. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Malynn BA, de Alboran IM, O'Hagan RC, Bronson R, Davidson L, DePinho RA, Alt FW. Genes Dev. 2000;14:1390–1399. [PMC free article] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- Yamanaka S. Cell Stem Cell. 2007;1:39–49. doi: 10.1016/j.stem.2007.05.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.