

Abstract
Chemotactic factors known as chemokines play an important role in the pathogenesis of Multiple Sclerosis (MS). Transgenic expression of TNFα in the central nervous system (CNS) leads to the development of a demyelinating phenotype (TNFα-induced demyelination; TID) that is highly reminiscent of MS. Little is known about the role of chemokines in TID but insights derived from studying this model might extend our current understanding of MS pathogenesis and complement data derived from the classic autoimmune encephalomyelitis (EAE) model system. Here we show that in TID, chemokines and their receptors were significantly increased during the acute phases of disease. Notably, the CCL2 (MCP-1)-CCR2 axis and the closely related ligand-receptor pair CCR1-CCL3 (MIP-1α) were among the most up-regulated during disease. On the other hand, receptors like CCR3 and CCR4 were not elevated. This significant increase in the levels of chemokines/receptors correlated with robust immune infiltration of the CNS by inflammatory cells, i.e., macrophages, and immune cells particularly T and B cells. Immunostaining and confocal microscopy, along with in vitro studies revealed that astrocytes were a major source of locally produced chemokines and expressed functional chemokine receptors such as CCR2. Using an in vitro system we demonstrate that expression of CCR2 was functional in astrocytes and that signaling via this receptor lead to activation of NF-kB and Akt and was associated with increased astrocyte survival. Collectively, our data suggests that transgenic murine models of MS are useful to dissect mechanisms of disease and that in these models, up-regulation of chemokines and their receptors may be key determinants in TID.
Keywords: Multiple sclerosis, chemokines, TNFα, biomarker, inflammation, transgenic mice
Introduction
Multiple Sclerosis (MS) affects approximately 350,000 people in the United States and 1.1 million people worldwide (Noseworthy et al., 2000). After trauma, MS is the most common central nervous system disease that strikes young adults (Noseworthy et al., 2000). Despite the existence of some therapeutic alternatives, MS is currently a progressive disease entity, with 90 percent of cases entering a progressive phase (Dyment and Ebers, 2002; Hafler, 2004; Noseworthy et al., 2000). MS is characterized by patchy complex inflammatory demyelinating lesions, some degree of neuronal degeneration, and astroglial proliferation (Dyment and Ebers, 2002; Hafler, 2004; Noseworthy et al., 2000). It is thought that in MS pathogenesis several cellular processes such as recruitment and activation of monocytes/macrophages and T cells, as well as activation of resident glial populations, i.e., microglia and astrocytes, contribute to the demyelinating process (Dyment and Ebers, 2002; Hafler, 2004; Noseworthy et al., 2000). In this context, the family of chemoattractants known as chemokines may play a key role by regulating process such as leukocyte infiltration into the central nervous system (CNS) (Huang et al., 2000; Trebst and Ransohoff, 2001).
Chemokines, an expanding family of small proteins (8–12 kDa) that signal through G protein-coupled receptors (GPCRs), are involved in cell attraction, maturation and activation (Charo and Ransohoff, 2006). The receptors for these molecules have been shown to trigger numerous downstream signaling cascades, and are thought to contribute to numerous disease processes involving the CNS (Biber et al., 2002). A powerful approach for dissecting the role of the chemokine system in MS pathogenesis has been, to genetically inactivate mice for a given chemokine or chemokine receptor such as the chemokine CCL2 and Chemokine Receptor (CCR)-2 respectively; and to use the murine experimental autoimmune encephalomyelitis (EAE) model of MS (Fife et al., 2000; Huang et al., 2001; Izikson et al., 2000; Mahad and Ransohoff, 2003). The EAE model is based on the immunization of mice with peptides or myelin extracts emulsified in Complete Freunds Adjuvant (CFA). Mice lacking CCL2 or CCR2 are significantly protected against the development of EAE (Fife et al., 2000; Huang et al., 2001; Izikson et al., 2000). The information obtained so far has been extremely useful and has raised the possibility of the implementation of therapy for MS based on chemokine/receptor blockade. Nevertheless, the EAE model has several limitations. For instance, it relies on the use of CFA as part of the immunization protocol and this adjuvant can commonly lead to confounding effects (Steinman and Zamvil, 2005). In this light, it is not surprising that the protective role of CCR2 in EAE was recently shown to be dependent on the immunization protocol and the strain of the mice used (Gaupp and Berrios, 2002).
Thus, we surmised that the analysis of the role of the chemokine system in MS pathogenesis in a model system that was not dependent on immunization with CFA for disease induction would be highly informative. To address this possibility, we adopted two unique murine models of MS in our laboratory. These well established models are based on loss of regulation of TNFα production and were developed by engineering the transgenic expression of TNFα in astrocytes or progenitor cells in the nervous tissue (Akassoglou et al., 1998; Akassoglou et al., 1997; Probert et al., 1997; Probert et al., 1995). Here we refer to the development of MS-like pathology in these models TNFα-induced demyelination or TID. In these models, TID depends on the activation of resident cells and leukocyte recruitment into the nervous tissue without the need for immunization with CFA.
A pivotal role for TNFα in the pathogenesis of inflammatory demyelinating disease of the CNS has been suggested in several studies of MS in humans and animal models (Akassoglou et al., 1999; Akassoglou et al., 1998; Kassiotis et al., 1999b; Probert et al., 1997). Induction of EAE with Myelin Basic Protein (MBP) in TNFα deficient mice or p55TNF-R deficient mice is considerably delayed (Akassoglou et al., 1999; Akassoglou et al., 1998; Kassiotis et al., 1999b; Probert et al., 1997). The contribution of TNFα to the pathogenic processes underlying the development of MS could be both direct, i.e., by inducing demyelination, as well as indirect, i.e., by promoting cell infiltration (Probert et al., 2000). With respect to the potential direct effects of TNFα, it has been shown that this cytokine is overexpressed in the serum and cerebrospinal fluid of MS patients and by resident and infiltrating cells at sites of CNS injury (Hofman et al., 1989; Sharief and Hentges, 1991). TNFα can induce selective cytotoxicity of oligodendrocytes in vitro and myelin damage in brain slices (Akassoglou et al., 1999; Akassoglou et al., 1998; Kassiotis et al., 1999b; Probert et al., 1997), and in vivo in TNFα transgenic mice (Akassoglou et al., 1998), and has therefore been directly implicated in the demyelinating process. With respect to the indirect contribution of TNFα to MS pathogenesis; it is clear that this cytokine plays a unique role in the initiation and maintenance of local cell recruitment, which is mediated in part, by the coordinated expression of chemokines by resident cells and their receptors in infiltrating leukocytes
Here, using genetic murine models of MS, we show that the chemokine system is up-regulated during the process leading to leukocyte infiltration and tissue damage. Moreover, our data suggest that non-leukocyte derived chemokines, i.e., astrocyte derived, can play a major role in MS pathogenesis. Finally, we demonstrate that astrocytes express functional chemokine receptors such as CCR2 that can affect their activation or survival.
Materials and Methods
Mice, reagents and clinical scoring
Tg6074 and TgK21, along with other lines of Tg mice were generated as previously described (Akassoglou et al., 2004; Akassoglou et al., 1998; Probert and Akassoglou, 2001; Probert et al., 1996; Probert et al., 1997; Probert et al., 1995). Colonies of TgK21 and Tg6074 mice were established at the University of Texas Health Science Center at San Antonio (UTHSCSA). PCR based genotyping was used to identify Tg mice. The TgK21 expresses an uncleavable human TNFα protein under the control of Glial Fibrillary Acidic Protein (GFAP) promoter, on the surface of astrocytes. For maintenance purposes, TgK21 and Tg6074 mice were crossed with mice deficient for TNF receptor I (TNFRI−/−) (The Jackson Laboratory) to obtain TgK21/TNFRI+/− mice in which the development of neurological disease is prevented. For experiments, TNFα transgenic mice were crossed again onto a wild-type TNFRI+/+ background. In Tg6074 mice, glial precursor cells overexpress murine soluble TNFα starting at first postnatal week of age. TID clinical manifestations were graded according with a standard clinical severity scale with some modifications (Fife et al., 2000): grade 0, no abnormality; grade 1, weight loss, hunched posture, ruffled fur; grade 2, limb weakness; grade 3, mild ataxia or hind limb paralysis; grade 4, severe ataxia or complete hind limb paralysis or seizure. Mice that were unable to feed themselves or demonstrated more than 20% weight loss were sacrificed. In cases when mice were in stage 3 – 4, powder food and water were easily accessible to them. For some experiments, Wild type (Wt) and Ccr2−/− C57BL/6 mice were backcrossed for more than 10 generations as previously described (Kuziel et al., 1997). All the animal procedures were performed in compliance with the rules of Institutional Animal Care and Use Committee (IACUC) at the University of Texas Health Science Center. All the protocols used were approved by the IACUC.
Reagents
Annexin V-FITC, propidium iodide, and BD Riboquant for RPA (ribonuclease protection assays), were purchased from BD Biosciences (San Jose, CA). L-LME (L-Leucine Methyl Ester), Lipopolysaccharide (LPS), 5-fluoro-2 deoxyuridine, uridine, horse serum (HS), Hoechst 33342, Wortmannin, paraformaldehyde, BSA fraction V, Tween 20 and PBS tablets were acquired from Sigma-Aldrich (St Louis, MO). Trypsin, DMEM, Fetal Bovine Serum, HEPES buffer, PBS and polyacrylamide were from Invitrogen (Carlsbad, CA). Bradford reagent was acquired from Bio-Rad (Hercules, CA). Vectashield and Vectashield + DAPI were obtained from Vector Labs (Burlingame, CA).
Recombinant proteins
Recombinant murine and rat chemokines and cytokines were acquired from Peprotech (Rocky Hill, NJ).
Antibodies for neutralization, immunofluoresence and FACS
Goat antibodies against CCL3 and neutralizing against CCL2 were from R&D systems (Minneapolis, MN). Goat antibodies against CCL5, CCR1 CCR2, CCL2 and rat anti CCR5 were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-microtubule associated protein-2 (MAP-2) antibody was purchased from Sigma-Aldrich (St Louis, MO). Glial fibrillary acid protein (GFAP) antibody was obtained from DAKO (Carpinteria, CA). Donkey antibodies anti-rabbit conjugated with Texas red, anti-goat conjugated with FITC and anti-rat conjugated with FITC were from Jackson ImmunoResearch (West Grove, PA). Primary and secondary antibodies employed for Immunofluorescence were used at a dilution of 1:100
Brain total RNA extraction, tissue lysates preparation and RNase protection assay (RPA)
RPA on RNA from brain samples was performed as previously described (Kalkonde et al., 2007). Briefly, under anesthesia, the arterial system of mice was perfused with a chilled mix of 1:4 of RNAlater (Applied Biosystems, Foster city, CA) in PBS. The brain was dissected and minced in small fragments. The tissue was rinsed with PBS and snap frozen in liquid nitrogen. RNA was isolated and used for RPA as per manufacturer’s instructions and previously described (Rao et al., 2003). Levels of transcripts were quantified by dividing each densitometric reading by that of the GAPDH housekeeping gene using NIH imageJ software. Tissue lysates were prepared using a buffer containing a cocktail of protease inhibitors (without NP-40) that did not interfere with the use of the tissue lysates for ELISA (Quinones et al., 2000) or multiplex bead assays. The concentration of any given biomarker obtained was corrected for the protein concentration of the tissue protein levels (protein levels were quantified using Bradford assay).
Multiplex assay in tissue lysates
Mouse-specific profile of the biomarkers was analyzed by Rules-Based Medicine Inc (Austin, TX; http://www.rulesbasedmedicine.com/services_rodent_antigen.asp) using a multiplex assay. Serum samples and brain tissue lysates (50μl) from 3–5 mice/experiment were pooled for analysis. This multiplex microbead assay measures proteins in similar manner to standard sandwich ELISA. The dynamic range of the assay is 3–4 logs. This range can be extended by diluting samples. Thus, the sensitivity and range of the multiplex assays is comparable to ELISA. Two dilutions (1:1 and 1:10) of each sample were commonly run. The results obtained by multiplex assay were concordant with those obtained using standard ELISA (Quinones et al., 2007)
Astrocyte isolation and culture
Wild type (Wt) and Ccr2−/− C57BL/6j mice were used in these experiments. Primary mixed glial culture were obtained from the cerebra of postnatal day 0–3 as described by McCarthy et al. with few modifications (McCarthy and de Vellis, 1980). Briefly, mice were sacrificed and cerebral hemispheres were dissected. After separating the meninges, the hemispheres were minced finely, digested with 0.5% trypsin for 10 minutes at 37C and the cell suspension was filtered using a 40 um cell strainer (BD Biosciences, Bedford, MA). Cells were cultured in DMEM with 15% fetal bovine serum for 10–14 days. Astrocytes were then purified by shaking off microglia at 200 rpm for 3 hours on a platform shaker followed by 12-hour treatment with 7.5mM L-LME. Astrocytes obtained were >95% pure as confirmed by indirect immunocytochemistry for GFAP. The astrocytes used for assays were in culture for 12–25 days.
Histology
Brains of mice were collected, embedded in OCT (Sakura Finetek, Torrance, CA) and frozen at −70°C. Brains were sectioned axially or coronally at 10μm thickness in a cryotome, sections were air-dried, fixed in 4% paraformaldehyde (PFA), washed in PBS and stained for H&E. To observe loss of myelination, fixed sections were stained with Luxol fast blue (Sigma, St Lois, MO) and counterstained with H&E.
Immunofluoresence and Confocal microscopy
For immunofluorescence staining of brain sections, 8–10 μm OCT embedded sections were air dried and fixed in 4% paraformaldehyde for 30 min. Non specific binding was blocked using 5% BSA × 30 min. Primary antibodies were added, and incubated either for 1h or overnight at 4°C. After washing (PBS-Tween 20 pH 7.4) the sections were incubated for 45 min with FITC and Texas-Red labeled secondary antibodies. Sections were washed with HEPES buffer pH 7.4 and mounted using either Vectashield with or without DAPI. Images were obtained using Zeiss LSM 510 confocal microscope under 40X oil immersion objective NA 1.4. Fluorochromes were excited using a 488 nm argon laser for FITC and a 543 nm HeNe laser for Texas Red and the detector slits were configured to minimize any crosstalk between the channels the detector slits. For comparison between different groups and reproducibility purposes, instrument settings were kept identical for all samples.
Flow cytometry
For CCR2 and GFAP staining of astrocytes these cells were washed with PBS, fixed with 4% paraformaldehyde and permeabilized with methanol. Cells were incubated with anti-mouse CCR2 (rat monoclonal antibody MC21, 1:500 dilution) (Mack et al., 2001) and anti-GFAP antibody followed by incubation with fluorescent labeled secondary antibodies and analyzed by flow cytometry. For Annexin V staining of astrocytes to determine apoptosis, cells were stimulated with CCL2 at various concentrations for 72 hours. After this treatment, cells were washed twice with PBS, detached from the surface of well plates by treating them with 0.25% trypsin. Cells were stained with propidium iodide (PI) and Annexin V-FITC and analyzed within 1 hour of staining. Flow cytometry was done using a Beckton Dickinson FACScalibur cytometer. PI negative/Annexin V positive cells were counted as apoptotic cells. For detecting phosphorylation of Akt, cells were stimulated with CCL2 with and without inhibitors for 15 minute, washed, fixed and permeabilized. Cells were then incubated with primary and secondary antibodies for 30 minutes prior to analysis. A total of 10,000 cells were counted per treatment group. As the optimal effect of CCL2 on astrocyte apoptosis was observed at 100 ng/ml and since this was in agreement with the published literature, we used this concentration of CCL2 for subsequent assays (Bruno et al., 2000; Eugenin et al., 2003).
Glial chemotaxis assays
Chemotaxis assays were performed using 24-well transwell inserts (8um pore size, Corning Costar Corp., Cambridge, MA). Astrocytes were seeded at 4×104 and 2×104 cells per transwell respectively in 100ul of medium. Chemokines were added to the culture medium in the lower chamber. After 16 hours, transwell membranes were fixed for 10 minute with 4% PFA and stained with crystal violet for 4 minutes. Cells that had migrated to the lower side of the membrane were counted by a blinded observer. Chemotaxis index was calculated by dividing the number of cells that had migrated in response to chemokine by the number of cells that had migrated spontaneously in the absence of the chemokine.
SDS-PAGE and Western blots
For western blots, cell extracts were run on 10% polyacrylamide gels. After transferring the proteins to polyvinylidene fluoride (PVDF) membranes, membranes were probed with antibodies against I kappa B alpha (IkBα), I kappa B beta (IkBβ), IKK (IkB kinase) (Sigma-Aldrich, St Louis, MO), pAkt (phosphorylated Akt ser-473) and Akt (Cell Signaling Technology Beverly, MA). Membranes were stripped for probing with multiple antibodies. For inhibitor experiments, cells were pre-treated with Wortmannin (250nM) for 1 hour. Cells were stimulated with CCL2 (100ng/ml) for the durations indicated.
Electrophoretic mobility shift assays (EMSA)
For EMSAs, astrocytes were stimulated with CCL2 or LPS (1μg/ml) for varying time periods and nuclear extracts were prepared as described previously (Mummidi et al., 2000). Nuclear translocation of NF-κB was assayed by EMSAs using a NF-κB gel shift oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGC C-3′) from Santa Cruz biotechnology. For supershift, assays anti NF-κB p50 and anti NF-κB p65 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used. For inhibitor studies, cells were pretreated with inhibitors as described above and then stimulated with CCL2 (100ng/ml) for 2 hours.
Statistical analyses
Significance between animal groups was computed using Mann-Whitney test or t-test depending on whether the data were non-parametric or parametric respectively. Data from chemotaxis assays were analyzed using ANOVA and group comparisons were made using Scheffe’s post hoc test. Analyses were done using Stata™ statistical analysis software.
Results
Clinical manifestations of TID in transgenic mice
In agreement with previous reports (Akassoglou et al., 1998), we observed that TgK21/TNFRI+/+ mice developed acute neurological deterioration usually starting at 4–6 weeks. Commonly, mice with grade 4 paralysis died within 10–12 weeks. This phenotype was prevented by backcrossing the TgK21 mice with TNFRI−/− mice to generate TgK21/TNFRI+/−mice (Akassoglou et al., 1998) (data not shown). In the studies described here, we compared either littermates TgK21/TNFRI−/− (healthy controls) against K21Tg TNFRI+/+ (diseased mice); or TgK21/TNFRI+/+ in presymptomatic stages of disease.
Histopathological findings in mice with acute TID
The classical description of the pathological MS findings in humans includes demyelination within the brain and spinal cord, and at a fine level, the accumulation of inflammatory cells in a perivascular distribution (Hafler, 2004; Noseworthy et al., 2000). Similar to the work of Probert and colleagues (Akassoglou et al., 1998), we observed that TgK21 mice WT for the TNFαR in stages 3–4 of disease develop extensive demyelination as evident by loss of bluish green stain for myelin (Figure 1A–B and data not shown). Loss of myelin was also observed in the pons and medulla (data not shown). Thorough characterization of the histological level phenotype seen in neurologically compromised mice revealed that akin to pathological changes seen in patients with MS, these mice also revealed extensive perivascular inflammatory infiltrates and loss of characteristic oligodendroglial cells (Figure 1C–D).
Figure 1. Demyelination and inflammatory cell infiltration of the brain in TgK21 mice.

(A–B) representative staining of tissue sections of the brains of control (TgK21 TNFRI−/−) and neurologically compromised mice (TgK21 TNFRI+/+). (A–B) Brain sections cut at the level of cerebellum were stained with luxol fast blue that stains myelin light blue. Note severe demyelination, evident by lack of bluish green stain for myelin in B. (C–D) Sections derived from different brain regions were stained with H&E to better identify infiltrating cells. Note marked infiltration of mononuclear cells in the brain tissue of TgK21 TNFRI+/+ (D) mice compared with TgK21 TNFRI−/− (C). Representative sections out of many collected and analyzed in a blinded fashion by the pathologist are shown.
Leukocytic infiltration of the CNS in transgenic mice
In MS the inflammatory cell profile of active lesions is characterized by perivascular infiltration of oligoclonal T cells consisting of CD4+ and CD8+ T cells and monocytes with occasional B cells and infrequent plasma cells (Hafler, 2004). Along with this cellular infiltration, it is very common to observe oligodendrocyte destruction (Hafler, 2004). To determine to what degree this inflammatory pattern was also present in clinically compromised K21Tg mice, we characterized the nature of the inflammatory infiltrate by performing whole brain RNA isolation and RNA protection assays (RPAs) using a probe that detects mRNA transcripts expressed in specific cell types (Quinones et al., 2004). We found that as expected based on the literature in MS and EAE, compared with controls, diseased TgK21 mice had a significant increase in hematopoietic leukocytes (CD45 transcripts), T cells (TCRα chain, CD3ε and CD4 transcripts), and the macrophage lineage markers (F4/80 transcripts) (Figure 2A).
Figure 2.

Infiltrating leukocytes, chemokines and chemokine receptors in the brains of K21 TNFα transgenic mice. mRNA transcripts for (A) cell type specific molecules, (B) chemokines and (C) chemokine receptors were measured in total RNA obtained from the whole brain of mice using RNA protection assay. The data are presented as the ratio of the densitometric signals of the mRNA for the gene of interest and that of a housekeeping gene (GAPDH) and. is shown as Mean ± SD.
Increase levels of mRNA for several chemokines and their receptors in mice with acute TID
A wealth of data supports the notion that chemokine and chemokine receptor interactions mediate processes required for leukocyte recruitment into the sites of inflammation. Thus, we surmised that the robust leukocytic infiltration of the brains of neurologically compromised TgK21 mice might be driven by chemokines. We measured the levels of several chemokines in the brains of TNFα Tg mice. We found that mRNAs for the CCR2 ligand CCL2, as well as the CCR5 ligands, CCL3 and CCL5 were increased in the brains of diseased TgK21 mice. Other chemokines were also up-regulated, including CXCL10 and CCL1, and CXCL2, while Eotaxin was undetectable (Fig. 2B). The data sets shown in Figure 2 were of interest since similar patterns of chemokine up-regulation, i.e., CCL2, CXCL10, CCL5 and CCL3, has been reported in studies of samples from patients with MS, both at the level of the parenchyma, as well as in CNS (data comprehensively reviewed in Trebst and Ransohoff, 2001). Notably, among the chemokine receptors analyzed, there was a significant upregulation of CCR1 and CCR2, receptors for CCL3 and CCL2, respectively. There was no difference in the mRNA levels of CCR5 in the brain of controls and TNFα Tg mice (Fig. 2C)
Protein levels of several chemokines are upregulated in the brains of mice with acute TID
Next, we corroborated our RPA data revealing chemokine-mRNA upregulation, with protein analysis. We used a Multiplex bead assay and measured 57 biomarkers in tissue protein lysates obtained from whole brains of controls and TgK21 mice with active disease. Highlighting a high degree of concordance with our protein data, we found that compared with controls; there was a significant up-regulation of the chemokines CCL2 and CCL5 in the protein tissue lysates of diseased TgK21 mice. Indeed, out of the 57 biomarkers measured in the assay these two chemokines were some of the mediators up-regulated the most (Table 1). The levels of TNFα and IL-1α, other cytokines and chemokines were also up-regulated in the tissue-lysates of diseased TgK21 mice (Table 1).
Table 1.
Brain tissue protein fold increase between TgK21 TNFRI−/− and TNFRI+/+
| Cytokines |
Chemokines |
|---|---|
| -Greater than 50 fold | -Greater than 50 fold |
| TNFα | MCP-1 (CCL2) |
| IL-1α | RANTES (CCL5) |
| -20–50 fold | -20–50 fold |
| FGF-9 | MCP-3 (CCL7) |
| -Less than 20 fold | IP-10 (CXCL10) |
| IL-1β | MIP-2 (CXCL2) |
| IL-10 | -Less than 20 fold |
| IFNγ | MIP1γ (CCL9/10) |
| IL-6 | MIP1β (CCL4) |
| IL-17 | MDC (CCL22) |
| GM-CSF | MIP1α (CCL3) |
| IL-12p70 | MIP3β (CCL19) |
| IL-11 | MCP-5 (CCL12) |
| IL-3 | Eotaxin (CCL11) |
| OSM | Lymphotactin (XCL1) |
| M-CSF | GCP-2 (CXCL6) |
| IL-5 | KC/GROα (CXCL1) |
| TPO | Adhesion molecules and adhesion related molecules |
| EGF | |
| EGF | -20–50 fold |
| IL-7 | TIMP-1 |
| IL-4 | -Less than 20 fold |
| IL-18 | TF |
| CD 40 Ligand | VCAM-1. |
| Endothelin-1 | Fibrinogen |
| IL-2 | Factor VII. vWF |
Cellular source of chemokines and their receptors in mice with acute TID
To determine the source of chemokines, we measured the same array of factors in the sera of control mice or mice with clinically evident TID (stage 3–4). We discovered that none of the biomarkers that we found to be up-regulated in the brain tissue lysates of TgK21 mice with active neurological disease, were increased in the serum (data not shown). These data suggest that local processes, e.g., localized expression of TNFα in the CNS, leading to higher levels of mediators such as chemokines were potentially driving disease pathogenesis.
Activated astrocytes are a source of chemokines in acute TID
We found that there was a clear astrocytosis and cell activation, as determined by abundant large Sulforhodamine 101 positive cells (Supplementary videos 1 and 2) and GFAP+ astrocytes with prominent processes (Fig. 3)
Figure 3.
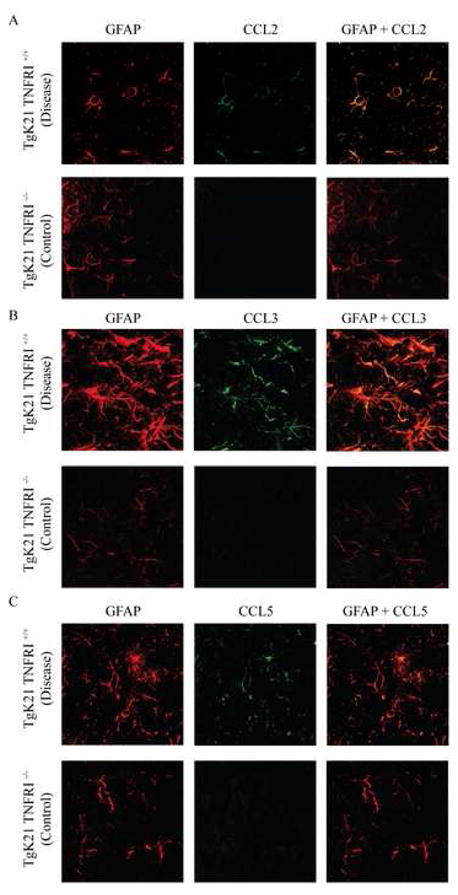
Confocal microscope image of the brains of TgK21 mice immunolabeled with antibodies against the astrocyte marker GFAP and CCL2 (A), CCL3 (B) and CCL5 (C). Note marked increase in GFAP immunoreactivity in TNFαR+/+ mice that were commonly clinically compromised. Coronal brain sections were at the middle portion of the parietal lobes. Photomicrographs are representative of two separate experiments with each experiment having 3 mice per group.
We co-stained brain tissue sections for GFAP plus the chemokines CCL2, CCL3, and CCL5. These chemokines were chosen because we found them to be elevated in the RPA analysis and brain protein lysates of mice with TID. Tissue sections were analyzed using confocal microscopy. We observed that astrocytes were an important source of CCL2 (Fig. 3A), CCL3 (Fig. 3B) and CCL5 (Fig. 3C) mice with TID. Indeed, a rise in chemokine production by astrocytes seemed to herald the development of clinical manifestations, as chemokine positive astrocytes were rarely seen in the brains of TgK21 mice with a functional TNFRI prior to the manifestation of neurological signs (data not shown).
Astrocyte expression of chemokine receptors in mice with acute TID
Having found that astrocytes were the main source of chemokines in TID, we wanted to identify the cellular location of the chemokine receptors that we found to be up-regulated at the mRNA level (Fig. 2C). Notably, we found that similar to the pattern of chemokine expression there was robust expression of chemokine receptors in the brains of mice with stage 3–4 TID that localized to the astrocytes. These cells exhibit strong immunostaining for both CCR2 and CCR1 (Fig 4A–B).
Figure 4.

Confocal microscope image of the brains of TgK21 mice immunolabeled with antibodies against the astrocyte marker GFAP and CCR1 (A) or CCR2 (B). Coronal brain sections were at the middle portion of the parietal lobes. Photomicrographs are representative of two separate experiments with each experiment having 3 mice per group.
Possible role of chemokines in chronic TID
The nature of TID in TgK21 mice is very acute, not only because it develops very early on, but also because once clinical disease is manifested in the mice, it progresses very rapidly (Akassoglou et al., 1999; Akassoglou et al., 1998). Next, we wanted to determine whether a similar pattern of chemokine-chemokine receptor up-regulation will be seen in the Tg6074 TNFRI+/+ transgenic murine model in which clinical manifestations of TID are delayed by several weeks; and in which the mice present a more insidious course of neurological deterioration (Akassoglou et al., 1999; Akassoglou et al., 1998).
A comparison between Tg6074 mice with clinically evident disease (stage 3–4) and those without neurological manifestations due to the lack of expression of the TNFRI demonstrated a similar up-regulation of chemokines to that seen in neurologically compromised TgK21 (Table 2). Importantly, CCL2 levels were also highly detected in cerebral spinal fluid (CSF) of Tg6074 affected mice at the time of sacrifice (22.5 pg/ml vs 2.3 pg/ml in the non-disease mice, P = 0.003). Nevertheless, in regards to chemokine receptors, we found that in diseased Tg6074 there was only a trend for increased mRNA levels of CCR1 and CCR5, while only CCR2’s mRNA was significantly up-regulated.
Table 2.
Chemokine and chemokine receptor expression in the Tg6074 mice model
| TNFRI−/− (n=2) |
TNFRI+/+ (n=2) |
p |
|
|---|---|---|---|
| Chemokine | |||
| Ltn (XCL1) | 0.027 ± 0.002 | 0.056 ± 0.004 | 0.0003 |
| RANTES (CCL5) | 0.043 ± 0.015 | 0.239 ± 0.080 | 0.014 |
| MIP-1β (CCL4) | 0.026 ± 0.000 | 0.091 ± 0.025 | 0.011 |
| MIP-2 (CXCL2) | 0.037 ± 0.007 | 0.505 ± 0.141 | 0.005 |
| IP-10 (CXCL10) | 0.041 ± 0.010 | 0.634 ± 0.069 | 0.0001 |
| MCP-1 (CCL2) | 0.040 ± 0.011 | 0.767 ± 0.160 | 0.001 |
| Receptor | |||
| CCR1 | 0.041 ± 0.008 | 0.086 ± 0.037 | 0.110 |
| CCR5 | 0.076 ± 0.002 | 0.128 ± 0.062 | 0.170 |
| CCR2 | 0.057 ± 0.011 | 0.086 ± 0.008 | 0.040 |
Astrocyte activation in vitro
To better understand, the functional correlates between chemokine up-regulation and astrocytosis, we designed in vitro studies to analyze the role of CCL2-CCR2 axis in astrocyte activation. We focused on this axis for two reasons. First, our mRNA and protein studies revealed up-regulation of this ligand receptor pair in TID. Second, evidence collected in the EAE models points towards the CCL2-CCR2 axis as a critical determinant of disease pathogenesis (Mahad and Ransohoff, 2003). Thus, we sought to determine what could be the functional implications of astrocyte expression of CCR2 that we had seen in the brains of mice with TID. Due to the methodological difficulties associated with establishing astrocyte cultures from adult brains, we resourced to using neonatal mice brains as a source of astrocytes.
Similar to what we found by immunostaining in vivo (Fig 4B), CCR2 was detected on the surface of cultured astrocytes by FACS in vitro (Fig 5A). Next, we determined if CCL2 could induce a functional response such as migration of primary astrocytes. We found that Ccr2+/+ astrocytes migrated in response to CCL2. Interestingly, Ccr2−/− primary astrocytes also migrated in response to CCL2, albeit the chemotatic index of astrocytes derived from Ccr2−/− mice was significantly lower (Fig. 5B). Together, these findings show that CCL2 can induce migration of Ccr2−/− astrocytes and despite genetic inactivation of CCR2, the CCL2-induced migration of astrocytes is not completely abolished; suggesting that, there might be an additional receptor(s) on primary astrocytes that mediate the functions of CCL2.
Figure 5.

CCR2 is expressed on murine astrocytes and evidence for an additional receptor for CCL2. (A), CCR2 expression on GFAP positive murine astrocytes was determined by flow cytometric analysis using monoclonal anti-mouse CCR2 and GFAP antibodies. The plot for Ccr2+/+ cells (black) is shifted to the right relative to the plot for Ccr2−/− cells (grey) and the isotype control (dashed), indicating expression of CCR2 on wild type cells but not on those lacking expression of CCR2. (B), Astrocyte migration in response to CCL2. As a functional readout for CCL2 effects, we determined the migration of Ccr2+/+ and Ccr2−/− astrocytes in response to this chemokine. Astrocyte chemotaxis assays were performed using transwells in the absence or presence of CCL2 for 20 hours. *P < 0.01 for comparison of chemotaxis of Ccr2+/+ astrocytes in the presence and absence of CCL2. #P <0.01 for comparison of chemotaxis of Ccr2+/+ and Ccr2−/− astrocytes in the presence of CCL2. **P < 0.01 for comparison of chemotaxis of Ccr2−/− astrocytes in the presence or absence of CCL2. Data are represented as the chemotaxis index (mean ± SD) derived from three independent experiments conducted in triplicate. (C), CCL2 did not alter astrocyte proliferation. Astrocytes were stimulated with CCL2 (100ng/ml) for 72 hours. During the last 16 hours of culture, cells were pulsed with [3H] thymidine. Thymidine incorporation into the DNA was detected using scintillation scanning. Data is representative of two independent experiments conducted in triplicate (mean ± SEM). (D), CCL2 decreased the spontaneous apoptosis of astrocytes in culture in a dose dependent manner. CCL2 was added to astrocytes maintained in cell culture at the indicated doses, and apoptosis was determined by staining cells with propidium iodide (PI) and Annexin V-FITC. Histograms indicate the PI-negative, Annexin V positive cells. Note, plots for cells treated with CCL2 are shifted to the left relative to plots indicating cells that were untreated. (E). CCL2 (100 ng/ml) did not decrease the spontaneous apoptosis of Ccr2−/− astrocytes, but did so in Ccr2+/+ astrocytes (mean ± SEM). The methods used were similar to those described in panel A.
Inflammation in the nervous system is associated with astrocyte activation and proliferation. We tested if CCL2 would alter astrocyte proliferation. We exposed astrocytes to different levels of CCL2. Astrocyte proliferation was not affected by CCL2 (Fig. 5C). We then evaluated whether CCL2 would alter astrocyte apoptosis. CCL2 was found to inhibit, in a dose-dependent manner, the spontaneous apoptosis of astrocytes in culture (Fig. 5D) We used astrocytes stimulated with LPS, TNFα and IFNγ, stimuli known to induce astrocyte apoptosis (Suk et al., 2001), as positive control for our experiments. Astrocytes stimulated with these inflammatory mediators showed increased astrocyte apoptosis in our in vitro culture system (data not shown).
To determine if CCR2 mediated this pro-survival function, we assayed for astrocyte apoptosis in Ccr2−/− and Ccr2+/+ astrocytes following stimulation with CCL2. We found that CCL2 inhibited the spontaneous apoptosis of Ccr2+/+ astrocytes but had no effect on Ccr2−/− astrocytes (Fig. 5E). Taken together, these findings suggest that CCL2 protects against spontaneous apoptosis of astrocytes in culture in a CCR2 dependent fashion, and that this is unlikely to be due to effects of CCL2 on cell proliferation.
CCL2 induces phosphorylation of Akt
In light of the aforementioned findings, we sought to determine the mechanisms downstream of CCR2 that mediated the pro-survival effects of CCL2 on astrocytes. We hypothesized that CCL2 might recruit specific elements of the anti-apoptotic pathway whose major elements are: phosphatidylinositol 3-OH kinase (PI 3-kinase) → activation of Akt → inhibition of pro-apoptotic factors (Brunet et al., 2001). Recent evidence implicates this pathway in mediating the pro-survival effects of other chemokines in the nervous system. For example, fractalkine/CX3CL1, a member of the CX3C family of chemokines protects neurons from apoptosis by activation of Akt (Meucci et al., 2000).
To test our hypothesis, we used phosphorylation of Ser-473 in Akt as a readout for Akt activation (Alessi et al., 1996). The CCL2-induced phosphorylation of Akt was detected 5 minutes post-stimulation, and the degree of phosphorylation peaked at 15 minutes and declined subsequently (Fig. 6A and B). Wortmannin, a PI 3-kinase inhibitor, inhibited the phosphorylation of Akt (Fig. 6, C and D) as well as the pro-survival effects of CCL2 (Fig. 6E). Taken together, the findings in Fig. 6 support the hypothesis that the pro-survival effects of CCL2 involve a pathway that includes PI 3-kinase and activation of Akt.
Figure 6.
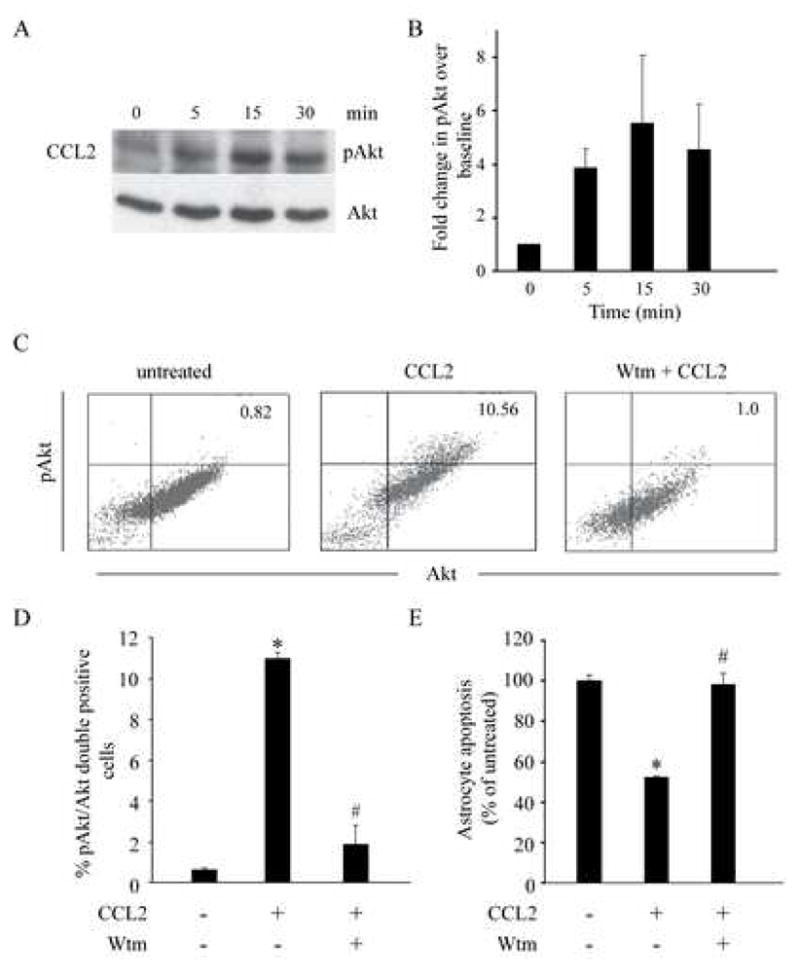
Pro-survival effect of CCL2 in astrocytes was dependent on a PI 3-kinase/Akt pathway. Astrocytes were stimulated with CCL2 (100ng/ml) for the indicated time intervals and cell extracts were then prepared for the analyses. (A). CCL2-induced phosphorylation of Akt in astrocytes (upper panel). Phosphorylation of Akt was detected by western blot using an antibody against phospho-Akt (p-Akt). An antibody against Akt was used to demonstrate equal loading of the samples (lower panel). (B), summary of the kinetics of Akt activation (mean fold change ± SEM) derived from 2 independent experiments. Data are shown as fold increase in Akt phosphorylation relative to untreated cells (time = 0). (C), CCL2-induced phosphorylation of Akt was blocked by wortmannin (Wtm), a PI 3-kinase inhibitor. Astrocytes were treated with wortmannin (250nM) for 1 hour followed by stimulation with CCL2 for 15 minutes. Phosphorylation of Akt was detected using flow cytometry by dual labeling of astrocytes with antibodies against Akt and pAkt. Cells in the upper right quadrant are double positive for Akt and pAkt, indicating that they are astrocytes with phosphorylated Akt. (D), summary data (mean percent change ± SEM) for inhibition of CCL2-induced phosphorylation of Akt by wortmannin from two independent experiments conducted in triplicate. (E), Inhibition of PI 3-kinase reduced the anti-apoptotic effects of CCL2. Apoptosis of astrocytes was detected using double labeling of cells with PI and Annexin V. Data are shown as percentage (%) change (± SEM) in apoptosis relative to untreated cells (100%). For D and E, * P < 0.05 for comparison between cells stimulated vs unstimulated with CCL2, # P < 0.05 for comparison between cells treated with CCL2 vs those treated with wortmannin and CCL2.
CCL2 induces nuclear translocation of NF-κB
Activation of the PI 3-kinase-Akt pathway has been shown to induce several downstream mediators such as NF-κB that, in turn, mediate pro-survival effects (Brunet et al., 2001). We therefore tested the possibility that CCL2 induces nuclear translocation of NF-κB in primary murine astrocytes. Using an EMSA-based approach, we found that CCL2-induced nuclear translocation of NF-κB in murine astrocytes. This effect of CCL2 on NF-κB translocation was (i) time-dependent, peaking at 120 minutes after addition of CCL2 (Fig. 7A); (ii) dose-dependent and for the dose-range tested, the maximal effects were at 100 ng/ml (Fig. 7B); and (iii) dependent predominantly on the p65 component of NF-κB (Fig. 7C, arrow).
Figure 7.
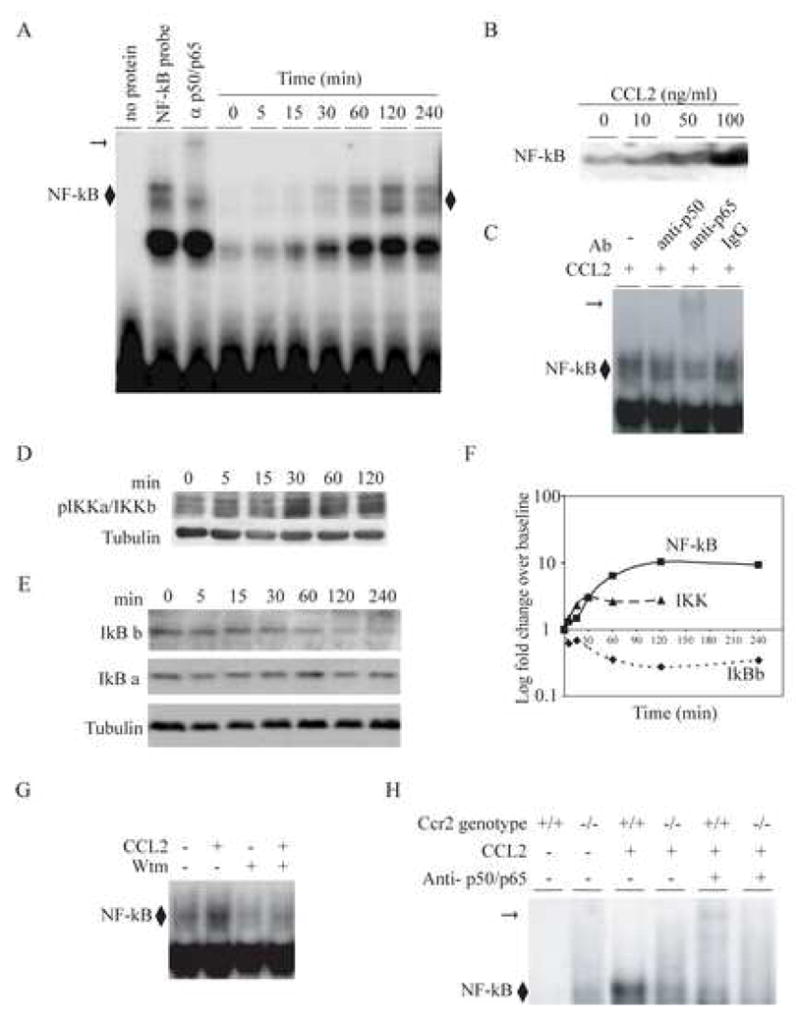
CCL2 induced activation of the NF-κB pathway in primary murine astrocytes. Astrocytes were derived from Ccr2+/+ animals. (A), Time-dependent induction of NF-κB by CCL2 (100ng/ml). Astrocytes were treated with CCL2 for the indicated time-points and nuclear extracts were prepared for EMSAs. EMSAs were conducted using a consensus NF-κB probe and supershift assays were performed using a combination of anti-p50 and -p65 antibodies (third lane). Note, supershift in the third lane indicated by the horizontal arrow. ◆, represents NF-κB bound to the probe. Maximal nuclear translocation of NF-κB was detected at 120 min, and this time interval was therefore used in the experiments shown in panels B and C. (B), CCL2 induced nuclear translocation of NF-κB in a dose-dependent manner. Primary murine astrocytes were treated with the indicated concentrations of CCL2 for 120 min and nuclear translocation of NF-κB was detected by EMSAs. (C), CCL2 induced predominantly the translocation of the p65 component of the NF-κB complex. A supershift was detected with an anti-p65 Ab, but not anti-p50 or isototype (IgG) control Ab. (D), CCL2 activated IKKα/IKKβ in astrocytes. Astrocytes were treated with CCL2 (100ng/ml) for the indicated time intervals. Cytoplasmic extracts were prepared and probed with the indicated Abs using Western blots. Anti-tubulin antibody was used to determine equal protein loading (lower panel). Maximal phosphorylation of IKKα/IKKβ was detected at 30 min. (E), CCL2-induced NF-κB translocation is associated with degradation of IκBβ but not IκBα. Cytoplasmic extracts were prepared from astrocytes treated with CCL2 for the time intervals shown and then probed with the indicated antibodies using Western blots. (F), Summary of time kinetics of signaling events that lead to CCL2 induced NF-κB nuclear translocation. Fold differences shown in panels A to E were log transformed and plotted against time. The CCL2-induced NF-κB nuclear translocation in astrocytes was dependent on PI 3 -kinase and expression of CCR2. (G), CCL2-induced nuclear translocation of NF-κB is inhibited by wortmannin (Wtm). Astrocytes were treated with 250nM wortmannin for 1 hour before stimulation with CCL2 for 2 hours. Nuclear extracts were prepared and used for EMSA. (H), CCL2-induced nuclear translocation of NF-κB was markedly diminished in Ccr2−/− astrocytes. Nuclear extracts were derived from Ccr2+/+ and Ccr2−/− astrocytes treated with CCL2 at 100 ng/ml for 120 min. Supershifts were performed with a combination of anti-p50 and -p65 antibodies.
Additionally, we found that these effects of CCL2 on NF-κB translocation were associated with phosphorylation of IκB kinase (IKK), which causes serine phosphorylation and degradation of IκB (Fig. 7D) (Mattson and Camandola, 2001). Specifically, we found that CCL2 induced degradation of IκBβ but not IkBβ (Fig. 7E). Based on the findings in Fig. 7(A, D and E) we computed the kinetics of these signaling events (Fig. 7F). This analysis revealed the following: Nuclear translocation of NF-κB peaked at 120 minutes after treatment with CCL2; congruent with these findings, phosphorylation of IKK was seen as early as 5 minutes, peaked at 30 minutes; and degradation of IκBβ was maximal at 120 minutes, a time point at which the nuclear translocation of NF-κB was maximal.
CCL2 induced NF-κB translocation is reduced by PI 3-kinase inhibition and absence of CCR2
Having established the induction of NF-κB in astrocytes by CCL2, we tested if this process is dependent on the following pathway: PI 3-kinase → Akt → NF-κB. To test this, astrocytes were pre-treated with the PI 3-kinase inhibitor wortmannin for 1 hour before stimulating them with CCL2. Wortmannin reduced CCL2 induced NF-κB translocation in astrocytes, implying that activation NF-κB by CCL2 in astrocytes is an event downstream of PI 3-kinase activation (Fig. 7G).
As the results presented earlier suggested existence of an alternative receptor/s for CCL2 on astrocytes, we tested if the NF-κB translocation induced by CCL2 was dependent solely on CCR2 expression or could be mediated, in part, via this additional receptor(s). CCL2 induced NF-κB translocation, although not totally absent, was markedly diminished in Ccr2−/−astrocytes, indicating that CCR2 is the major receptor for this effect of CCL2 (Fig. 7H).
Discussion
Here we show that acute and chronic TID is associated with prominent histopathological changes including widespread reactive astrocytosis, recruitment of macrophages, CD4+ and CD8+ T-cells. This cellular phenotype is accompanied by upregulation of several chemokines mRNA levels: CCL2, CCL7, CCL3 and CCL5; and their receptors, mainly CCR1 and CCR2, but not CCR5. Notably, the major source of chemokines, and the cells that more prominently expressed chemokine receptors were reactive astrocytes. Modest production of chemokines and up-regulation of their receptors was seen in other non-astrocyte cell types. Over expression of TNFα restricted to astrocytes (GFAP+ cells) may preferentially activate these cells to release pro-inflammatory factors such as IL-6 or chemokines that favor the disease process. Chemokines such CCL2 acting via CCR2 may promote astrocyte survival, activation and a feedback loop that would maintain and aggravate inflammation. Importanly, we found that the effects of the CCL2-CCR2 axis on murine astrocyte survival via Akt and NF kB mediated pathways.
Robust expression of chemokine and their receptors in astrocytes has been previously described in other experimental models of MS. In rats, induction of EAE has been shown to up-regulate the expression of receptors such as CCR2 (Jee et al., 2002). Expression of CCR2 has also been demonstrated on human astrocytes (Andjelkovic et al., 2002). However the role played by astrocyte derived chemokine and astrocyte expressed chemokine receptors is not clear in the pathogenesis of MS. Although it is widely believed that MS pathogenesis is mainly driven by aberrant immune responses; the contribution of other cells, such as glia cells in disease pathogenesis MS, is now receiving increasing attention (Miller, 2005). An emerging body of literature seems to suggest that abnormal activation of glia itself, may be one of the inciting events in MS (Antony et al., 2004; Mattson and Taub, 2004; Miller, 2005). For instance, a study of tissue from the brainstems and spinal cords of 12 MS patients who died immediately after an outbreak of symptoms, found little evidence of T-cell infiltration into areas of the brain and spinal cord damaged by the disease. Instead, these patients presented widespread signs of oligodendrocyte apoptosis (Barnett and Prineas, 2004).
Supporting our present data, we observed that transplantation of TNFαR−/− or TNFαR+/+ bone marrow (Wild type bone marrow or transgenic bone marrow) into the TNFα transgenics mice prior to the initiation of symptoms was associated with comparable disease course (n=5 recipients per group). Arguably, TNFα transgenics that received TNFαR−/− bone marrow should have been protected to some extent since their leukocytes were rendered irresponsive to the high levels of TNFα in the brain. In this context, activation of astrocytes was suficient to induce leukocyte migration and the progression of the disease.
Importantly, constitutive low CNS expression of CCL2 as engineered by Elhofy et al (Elhofy et al., 2005), may lead to different outcome compared with the significantly high levels of CCL2, in the context of the inflammatory cascade induced by TNFα, that is seen in the TNFα transgenic mice. Moreover, a recent study suggests that the demyelination in the TNFα transgenic mice is mostly T-cell independent (Kassiotis et al., 1999a). In this context, the modulation of T helper Th1/Th2 immune responses by CCL2 may not be a significant factor..
Another recent study of brain tissues collected from autopsies of MS patients revealed that disease was associated with overactivation of astrocytes that was potentially secondary to the overexpression of an endogenous retroviral gene (Antony et al., 2004). These activated astrocytes become toxic to oligodendrocytes, and can induce MS like disease in mice, suggesting that primary activation of astrocytes may ignite neurological damage in MS (Antony et al., 2004). These sequences of events are in consonance with our data in acute TID suggesting that perhaps, mediators released by the astrocyte lead to apoptotic oligodendrocyte death, while perpetuating this effect by prolonging astrocyte cell life through inhibition of astrocyte apoptosis. As a secondary event, these centrally produced chemokines lead to leukocyte recruitment that would be solely necessary to remove the huge quantity of apoptotic membrane (myelin) which is produced by each dying oligodendrocyte. Arguably, this sequence of events may be restricted to the model system that we used.
Taken together the data presented suggests the possibility that astrocytes may play a major role in acute and rapidly progressive disease pathogenesis via release of chemokines, and expression of chemokine receptors, that may promote inflammatory cell recruitment and astrocyte cell survival and activation. However, it remains to be determined whether astrocyte specific-blockade of chemokines/chemokine receptors may prevent the development of TID.
Supplementary Material
Acknowledgments
We are indebted to the people in UTHSCA IACUC (Dr. Kornegay and April Zuniga) for their assistance in the process of acquiring the mice, and to Jose Sigala, Opal Willmon and Molly Dudley for technical assistance. Images were generated in the Core Optical Imaging Facility which is supported by UTHSCSA, NIH-NCI P30 CA54174 (San Antonio Cancer Institute), NIH-NIA P30 AG013319 (Nathan Shock Center) and (NIH-NIA P01AG19316). This work was supported in part by the NIH (AI48644, AR 052755) and VA Merit to S.S.A.; a Veterans Administration Center on AIDS and HIV-1 infection to S.K.A.; a South Texas Health Research Center, San Antonio Area Foundation Grant and Pfizer Scholar Award to M.P.Q. S.K.A. is a recipient of the Elizabeth Glaser Scientist Award (Pediatric AIDS Foundation) and the Burroughs Wellcome Clinical Scientist Award in Translational Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H, Probert L, Strickland S. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci U S A. 2004;101:6698–6703. doi: 10.1073/pnas.0303859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akassoglou K, Bauer J, Kassiotis G, Lassmann H, Kollias G, Probert L. Transgenic models of TNF induced demyelination. Adv Exp Med Biol. 1999;468:245–259. doi: 10.1007/978-1-4615-4685-6_20. [DOI] [PubMed] [Google Scholar]
- Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G, Probert L. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol. 1998;153:801–813. doi: 10.1016/S0002-9440(10)65622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol. 1997;158:438–445. [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic AV, Song L, Dzenko KA, Cong H, Pachter JS. Functional expression of CCR2 by human fetal astrocytes. J Neurosci Res. 2002;70:219–231. doi: 10.1002/jnr.10372. [DOI] [PubMed] [Google Scholar]
- Antony JM, van Marle G, Opii W, Butterfield DA, Mallet F, Yong VW, Wallace JL, Deacon RM, Warren K, Power C. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat Neurosci. 2004;7:1088–1095. doi: 10.1038/nn1319. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Biber K, Zuurman MW, Dijkstra IM, Boddeke HW. Chemokines in the brain: neuroimmunology and beyond. Curr Opin Pharmacol. 2002;2:63–68. doi: 10.1016/s1471-4892(01)00122-9. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Bruno V, Copani A, Besong G, Scoto G, Nicoletti F. Neuroprotective activity of chemokines against N-methyl-D-aspartate or beta-amyloid-induced toxicity in culture. Eur J Pharmacol. 2000;399:117–121. doi: 10.1016/s0014-2999(00)00367-8. [DOI] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Dyment DA, Ebers GC. An array of sunshine in multiple sclerosis. N Engl J Med. 2002;347:1445–1447. doi: 10.1056/NEJMcibr021828. [DOI] [PubMed] [Google Scholar]
- Elhofy A, Wang J, Tani M, Fife BT, Kennedy KJ, Bennett J, Huang D, Ransohoff RM, Karpus WJ. Transgenic expression of CCL2 in the central nervous system prevents experimental autoimmune encephalomyelitis. J Leukoc Biol. 2005;77:229–237. doi: 10.1189/jlb.0804465. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:899–905. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaupp R, Berrios GE. About the limits of psychiatric knowledge. Classic Text No. 51. Hist Psychiatry. 2002;13:327–338. doi: 10.1177/0957154X0201305105. [DOI] [PubMed] [Google Scholar]
- Hafler DA. Multiple sclerosis. J Clin Invest. 2004;113:788–794. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman FM, Hinton DR, Johnson K, Merrill JE. Tumor necrosis factor identified in multiple sclerosis brain. J Exp Med. 1989;170:607–612. doi: 10.1084/jem.170.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Han Y, Rani MR, Glabinski A, Trebst C, Sorensen T, Tani M, Wang J, Chien P, O’Bryan S, Bielecki B, Zhou ZL, Majumder S, Ransohoff RM. Chemokines and chemokine receptors in inflammation of the nervous system: manifold roles and exquisite regulation. Immunol Rev. 2000;177:52–67. doi: 10.1034/j.1600-065x.2000.17709.x. [DOI] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jee Y, Yoon WK, Okura Y, Tanuma N, Matsumoto Y. Upregulation of monocyte chemotactic protein-1 and CC chemokine receptor 2 in the central nervous system is closely associated with relapse of autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol. 2002;128:49–57. doi: 10.1016/s0165-5728(02)00147-9. [DOI] [PubMed] [Google Scholar]
- Kalkonde YV, Morgan WW, Sigala J, Maffi SK, Condello C, Kuziel W, Ahuja SS, Ahuja SK. Chemokines in the MPTP model of Parkinson’s disease: absence of CCL2 and its receptor CCR2 does not protect against striatal neurodegeneration. Brain Res. 2007;1128:1–11. doi: 10.1016/j.brainres.2006.08.041. [DOI] [PubMed] [Google Scholar]
- Kassiotis G, Bauer J, Akassoglou K, Lassmann H, Kollias G, Probert L. A tumor necrosis factor-induced model of human primary demyelinating diseases develops in immunodeficient mice. Eur J Immunol. 1999a;29:912–917. doi: 10.1002/(SICI)1521-4141(199903)29:03<912::AID-IMMU912>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Kassiotis G, Pasparakis M, Kollias G, Probert L. TNF accelerates the onset but does not alter the incidence and severity of myelin basic protein-induced experimental autoimmune encephalomyelitis. Eur J Immunol. 1999b;29:774–780. doi: 10.1002/(SICI)1521-4141(199903)29:03<774::AID-IMMU774>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci U S A. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachy J, Bruhl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlondorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Taub DD. Ancient viral protein enrages astrocytes in multiple sclerosis. Nat Neurosci. 2004;7:1021–1023. doi: 10.1038/nn1004-1021. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. Neuroscience. The dark side of glia. Science. 2005;308:778–781. doi: 10.1126/science.308.5723.778. [DOI] [PubMed] [Google Scholar]
- Mummidi S, Bamshad M, Ahuja SS, Gonzalez E, Feuillet PM, Begum K, Galvis MC, Kostecki V, Valente AJ, Murthy KK, Haro L, Dolan MJ, Allan JS, Ahuja SK. Evolution of human and non-human primate CC chemokine receptor 5 gene and mRNA. Potential roles for haplotype and mRNA diversity, differential haplotype-specific transcriptional activity, and altered transcription factor binding to polymorphic nucleotides in the pathogenesis of HIV-1 and simian immunodeficiency virus. J Biol Chem. 2000;275:18946–18961. doi: 10.1074/jbc.M000169200. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Probert L, Akassoglou K. Glial expression of tumor necrosis factor in transgenic animals: how do these models reflect the “normal situation”? Glia. 2001;36:212–219. doi: 10.1002/glia.1110. [DOI] [PubMed] [Google Scholar]
- Probert L, Akassoglou K, Alexopoulou L, Douni E, Haralambous S, Hill S, Kassiotis G, Kontoyiannis D, Pasparakis M, Plows D, Kollias G. Dissection of the pathologies induced by transmembrane and wild-type tumor necrosis factor in transgenic mice. J Leukoc Biol. 1996;59:518–525. doi: 10.1002/jlb.59.4.518. [DOI] [PubMed] [Google Scholar]
- Probert L, Akassoglou K, Kassiotis G, Pasparakis M, Alexopoulou L, Kollias G. TNF-alpha transgenic and knockout models of CNS inflammation and degeneration. J Neuroimmunol. 1997;72:137–141. doi: 10.1016/s0165-5728(96)00184-1. [DOI] [PubMed] [Google Scholar]
- Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 1995;92:11294–11298. doi: 10.1073/pnas.92.24.11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probert L, Eugster HP, Akassoglou K, Bauer J, Frei K, Lassmann H, Fontana A. TNFR1 signalling is critical for the development of demyelination and the limitation of T-cell responses during immune-mediated CNS disease. Brain. 2000;123 ( Pt 10):2005–2019. doi: 10.1093/brain/123.10.2005. [DOI] [PubMed] [Google Scholar]
- Quinones M, Ahuja SK, Melby PC, Pate L, Reddick RL, Ahuja SS. Preformed membrane-associated stores of interleukin (IL)-12 are a previously unrecognized source of bioactive IL-12 that is mobilized within minutes of contact with an intracellular parasite. J Exp Med. 2000;192:507–16. doi: 10.1084/jem.192.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones MP, Ahuja SK, Jimenez F, Schaefer J, Garavito E, Rao A, Chenaux G, Reddick RL, Kuziel WA, Ahuja SS. Experimental arthritis in CC chemokine receptor 2-null mice closely mimics severe human rheumatoid arthritis. J Clin Invest. 2004;113:856–866. doi: 10.1172/JCI20126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones MP, Martinez HG, Jimenez F, Estrada CA, Dudley M, Willmon O, Kulkarni H, Reddick RL, Fernandes G, Kuziel WA, Ahuja SK, Ahuja SS. CC chemokine receptor 5 influences late-stage atherosclerosis. Atherosclerosis. 2007 doi: 10.1016/j.atherosclerosis.2007.03.026. in press. [DOI] [PubMed] [Google Scholar]
- Rao AR, Quinones MP, Garavito E, Kalkonde Y, Jimenez F, Gibbons C, Perez J, Melby P, Kuziel W, Reddick RL, Ahuja SK, Ahuja SS. CC chemokine receptor 2 expression in donor cells serves an essential role in graft-versus-host-disease. J Immunol. 2003;171:4875–4885. doi: 10.4049/jimmunol.171.9.4875. [DOI] [PubMed] [Google Scholar]
- Sharief MK, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. 1991;325:467–472. doi: 10.1056/NEJM199108153250704. [DOI] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005;26:565–571. doi: 10.1016/j.it.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Suk K, Lee J, Hur J, Kim YS, Lee M, Cha S, Yeou Kim S, Kim H. Activation-induced cell death of rat astrocytes. Brain Res. 2001;900:342–347. doi: 10.1016/s0006-8993(01)02326-5. [DOI] [PubMed] [Google Scholar]
- Trebst C, Ransohoff RM. Investigating chemokines and chemokine receptors in patients with multiple sclerosis: opportunities and challenges. Arch Neurol. 2001;58:1975–1980. doi: 10.1001/archneur.58.12.1975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.