

SUMMARY
This review focuses on the era of antibiosis that led to a better understanding of bacterial morphology, in particlar the cell wall component peptidoglycan. This is an effort to take readers on a tour de force from the concept of antibiosis, to the serepidity of antibiotics, evolution of beta-lactam development, and the molecular biology of antibiotic resistance. These areas of research have culminated in a deeper understanding of microbiology, particularly in the area of bacterial cell wall synthesis and recycling. In spite of this knowledge, which has enabled design of new even more effective therapeutics to combat bacterial infection and has provided new research tools, antibiotic resistance remains a worldwide health care problem.
Keywords: Penicillin, beta-lactam, penicillin binding protein, Alexander Fleming, beta-lactamases, amp genes
INTRODUCTION
The concept of using of chemicals to alleviate diseases, particularly infectious diseases, dates back to the Ancient Egypt, Babylon, the Far East and the Incas (1). These early examples of remedies include applying molds to opened cuts and wounds, and eating radishes, leeks, garlic and onions that are now known to be anti-bacterial.
The possibility of one organism interfering with the growth of another has been the subject of intense investigation since the early days of microbiology, In 1871, Sir John Burdon-Sanderson observed that media exposed to air rapidly became turbid with bacteria, but if a Penicillium mold happened to grow on the surface of the broth, less turbidity ensued (2). Others, including William Roberts, John Tyndall and Joseph Lister reported similar observations with Penicillium (3–5). Although perhaps only Roberts and Lister realized that their findings could be due to the action of an antibacterial compound, these observations are noteworthy nonetheless (6). One of the earliest references to the therapeutic potential of the inhibition of bacterial growth by other microorganisms was in Pasteur and Joubert’s descripton of the inhibition of anthrax by air-borne organisms (7). Further evidence that Lister understood the clinical relevance of the antagonism is his reported treatment of infected wounds with Penicillium in 1884 (6).
PRE-PENICILLIN ERA
Although many observed microbial antagonism, the concept that the phenomenon was caused by the production of a compound by one microorganism that could kill another was not immediately apparent. For example, in a later work, Tyndall surmised that the bacterial growth inhibition observed in the presence of Penicillium was due to limiting oxygen conditions (8). Initial work by Corneil and Babes and substantiated by experiments by Garre, firmly established that microbial antagonism was caused by the action of a diffusible substance produced by one organism on another (9, 10). By the end of the 19th century, not only was this phenomenon well accepted, but it was also given a name – antibiosis (11).
A few researches showing the contradictory effects between Penicillium and bacteria predate the now famous studies of Alexander Fleming and received little attention until after the clinical importance of penicillin was established. While completing his doctoral degree in 1897, Ernest Duschesne found antagonism between Penicilium and Escherichia coli in vitro. He then found that co-injection of a normally lethal dose of Salmonella typhi, the causative agent of typhoid fever, with Penicillium glaucum prevented the animals from contracting typhoid (12).
Another notable pioneer was Gratia, a Belgian scientist who did ground-breaking research on the phenomenon of bacteriolysis and its application to the defeat of bacteria pathogens (13). In classic experiments performed in 1925–1926, Gratia and his assistants exposed a 2 % water agar plate containing dead Staphylococcus to the laboratory air. A culture of white actinomycete grew on the plate surrounded by a clear zone of dissolved bacteria. This airborne contaminant was next demonstrated to attack killed cultures of Pseudomonas aeruginosa, Mycobacterium tuberculosis and Escherichia coli. Gratia called the lytic agent “mycolysate”. The mycolysate was non-toxic when introduced into guinea pigs and rabbits. In addition, he used the mycolysate on human subjects in 1930 and obtained considerable success (13).
From these accounts, Fleming was therefore not the first to observe antibiosis between the fungal and bacteria in a vessel, but without controversy, he was the first to study the substance he termed “penicillin”.
BETA-LACTAM DISCOVERY AND DEVELOPMENT FOR CLINICAL USE
Discovery of penicillin
The story began in 1921. Alexander Fleming (Fig. 1), a Scottish bacteriologist, suffering from a common cold, inoculated agar plates with his own nasal secretion to determine the change of his nasal bacterial flora (14). No colonies appeared for several days. This suggested the presence of a diffusible substance in the nasal secretion that affected the ability of bacteria to grow at all. The substance, a protein Fleming named lysozyme, was found to be a potent enzyme capable of dissolving the cell wall and causing lysis in many Gram-positive bacteria. The discovery of lysozyme paved the path for the later serendipitous discovery of penicillin.
Fig. 1.

Antibiotic hall of fame. Courtesy of Nobel e-Museum (http://www.nobel.se/medicine/laureates/1945/index.html).
In the summer and early fall of 1928, Fleming was studying the relationship between colony morphology of Staphylococcus and their virulence (14). Before leaving for vacation, he inoculated culture plates with Staphylococcus colonies and stacked the plates on the corner of his laboratory bench. When he returned, he found several cultures contaminated with molds. He discarded the contaminated plates into a Lysol basin. He worked with many cultures that day, and a few culture plates rested above the level of the liquid antiseptic, escaping the disinfectant. The next day, in the process of describing his experiments to a colleague, Fleming dug up some previously discarded culture plates (14). Upon re-examination, one of the plates contained a contaminating mold whose presence seemed to be influencing the morphology of the surrounding Staphylococcus colonies: colonies in proximity to the mold were transparent and seemed to be undergoing lysis. This observation was reminiscent of what Fleming had previously seen with lysozyme, led him to realize that the mold contained a microbial antagonistic property.
Fleming carefully sub-cultured and preserved the Penicillium notatum, which he originally described as Penicillium rubrum. He described the growth property of the mold, elaborated an extraction procedure and designed a rapid antimicrobial assay (15). Fleming, together with his assistants Dr. Stuart Craddock and Mr. Frederick Ridley, set out to purify the the lytic agent, which he dubbed “penicillin”, released into the broth by the mold (16). According to Craddock’s notebook, Craddock and Ridley found that activity could be recovered from broth and concentrated by evaporation as long as the pH was kept at 6.9 or below and that the activity could be extracted with ether, acetone, or alcohol (16). Serial dilutions of these extracts inhibited growth of Staphylococci, Streptococcus pyrogenes and Pneumococcus cultures. However, the active ingredient did not harm Escherichia coli, Haemophilus influenzae, Salmonella typhi, Pseudomonas aeruginosa, Bacillus proteus or Vibrio chloerae and thus was found to be useful in culturing these organisms. The extract was also applied directly to animal tissues, specifically wounds and human eyes, and found to be neither toxic nor irritating (15). These legendary experiments formed the foundation for the subsequent purification of penicillin and an easy antimicrobial testing technique which is being widely used today. However, Fleming did not extend his work to clinical study because he was not able to purify enough penicillin for the experiments. So, while the discovery was made in 1928, the use of penicillin as a therapeutic agent to treat infections did not happen until the 1940s.
Bringing penicillin to bedside
In 1939, 12 years after the Fleming’s ground-breaking discovery, Howard Walter Florey (Fig. 1), Head of the Sir William Dunn School of Pathology, Oxford University, England, obtained a $25,000 research grant for a period of five years from the Rockefeller Foundation to investigate the chemical and biologic properties of antibacterial substances produced by molds and other bacteria (17). He recruited Ernst Chain to develop the biochemical methodologies (Fig. 1). Among the molds, they selected Fleming’s strain of Penicillium notatum for further study because of its potent antimicrobial activity reported by Fleming (16). Florey, Chain and eight coworkers, became legendary for the progress made on Fleming’s penicillin in a surprisingly short period. In August 1940, they reported successful use of penicillin to cure infections in mice, rats and cats (18). A year later, the first clinical use of penicillin in 10 human subjects suffering from S. aureus infection was described extensively (19).
With the initial success, the Oxford group was prepared to enter the next phase, a large-scale clinical trial involving 100 patients. However, these experiments required an enormous amount of penicillin, which was impossible to be manufactured in a small laboratory. Hence, Florey and Heatley, another group member, crossed the Atlantic Ocean to the United States of America in June 1941 to seek aid in mass producing penicillin (20). Through several contacts, they were introduced into the new large scale fermentation laboratory, the Northern Regional Research Laboratory of the Department of Agriculture at Peoria, Illinois. (20). In Peoria, they were able to isolate a novel Penicillium strain, Penicillium chrysogenum that produced substantially more penicillin than previously used strains. This work began to stimulate the interest of several pharmaceutical firms, among others, Charles Pfizer & Co., E.R. Squibb & Sons, and Merck & Co. as well as the government (17, 21). The strong monetary investment from these firms enabled the mass production of penicillin. This led to multiple large-scale clinical trials in England and in the United States in late 1942 that continued into 1943 (22–27). By 1946, a year after the Nobel Prize for Medicine was awarded to Fleming, Florey and Chain, penicillin was finally available in the open market.
Penicillin as β-lactam
The work at the Northern Regional Research Laboratory also revealed that different strains, culture conditions, and media result in the production of different penicillin compounds. Penicillin from the United States strain was successfully crystallized and analyzed at E.R. Squibb & Sons (17) and Florey’s group subsequently crystallized the compound from the English strain (28). Florey and colleagues demonstrated that their strain used for the English clinical trials was primarily 2-pentenylpenicillin (Penicillin F or I) while the United States penicillin (Penicillin G or Penicillin II) was mainly benzyl-penicillin (29). It was later shown that both forms of penicillin contained β-lactam rings (Fig. 2).
Fig. 2.
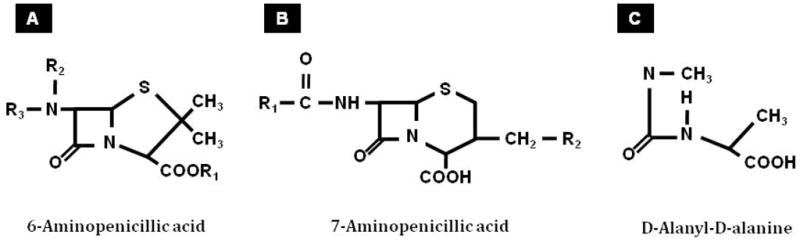
Molecular structure of 6-aminopenicillic acid, 7-aminopenicillic acid and D-alanine D-alanine
β-lactam derivatives
With the diverse chemical structure of β-lactam molecules and their respective antibacterial potency, chemists were eager to look beyond what nature had supplied, and how science could contribute and substitute. Early work using different fermentation conditions were not very successful (30). The β-lactam nucleus, 6-aminopenicillanic acid (6-APA; Fig. 2) proved to be the key in penicillin synthesis and modification. J.C. Sheehan produced Penicillin V by acylation of chemically synthesized 6-APA (31). This important piece of work opened up the floodgate where novel beta-lactam agents could be mass-produced by adding unusual side chains to 6-APA. Thereafter, semi-synthetic β-lactam compounds have been developed continuously and systematically. For example, methicillin, approved for clinical use in the United States in 1960, was the first semi-synthetic penicillin introduced to resist hydrolysis by penicillinase (32). More significantly, a semi-synthetic compound effective against Pseudomonas aeruginosa, carbenicillin, a penicillin analogue with a carboxyl group in place of amino group of ampicillin, was introduced in 1967 (33).
Though the generation of semi-synthetic compounds presented a great opportunity, natural sources continued to be explored. Abraham and Newton isolated cephalosporin C from a strain of Cephalosporium acremonium (34). This compound generated an entirely new family of β-lactam antibiotics because instead of 6-APA, it possesses a nucleus of 7-aminocephalosporinic acid (7-ACA; Fig. 2). Using 7-ACA as the precursor, several generations of cephalosporins with potent broad-spectrum activity have been synthesized. Many related naturally occurring compounds with important antibiotic activity were isolated from bacteria. These include carbapenem from Streptomyces spp.(35), and monobactam from Pseudomonas acidophila (36), Chromobacterium violaceum (37) and Agrobacterium radiobacter (38). Penem and monobactam contained different β-lactam nuclei from 6-APA and 7-ACA. Derivatives of these compounds, either naturally-occurring or semi-synthetic, further expanded the spectrum of β-lactam agents.
β-LACTAM MECHANISM OF ACTION
Interest quickly developed, after the introduction of penicillin, as to the mechanism of penicillin action on bacteria. Investigations of the mode of action of β-lactam antibiotics had progressed through several seemingly independent routes. These routes included (1) physiological analysis of the β-lactam effects on growth, viability, shape division and integrity of bacterial cells; (2) biochemical analysis of the cell wall structure and the enzymes involved in its biosynthesis; (3) biophysical analysis of the cellular components that tightly bind penicillin and attempts at correlating this binding with its physiological effects; and (4) genetic analysis of mutants with altered response to the β-lactam antibiotics. These lines of research ultimately converged and became coherent. Readers are encouraged to seek other recent reviews for more comprehensive overviews of specific topics of interest (39–41).
Physiological analysis
Gardner observed that at low concentrations of penicillin, bacteria formed filaments (42). This provided an early indication that penicillin interferes with the maintenance of cell shape. In a subsequent study, using different concentration of penicillin, Duguid showed that penicillin interfered with cell division and maintenance of the cell structures (43). These morphological changes suggested penicillin inhibited the synthesis of an unknown outer supporting cell wall structure (the nature of the supporting wall was not known at that time).
Research on the bacterial cell wall, particularly the murein, was revived when the link between β-lactam antibiotic and cell wall was established. The earliest documentation of the presence of bacterial cell wall was traced as early as the age of Anthony van Leeuwonhoek. Under his microscope, he observed that the bacteria were bonded by some sort of structure and expressed his hope to resolve the question of what hold them together (44). This turned out to be a nontrivial problem. Hans Christian Gram, a Danish bacteriologist, devised a staining procedure that allowed him to observe colored bacteria under light microscope (45). Since then, his technique has been widely adopted and used to classify bacteria into two categories: the Gram-positive and the Gram-negative (45). Although a variety of distinguishing features or chemical differences in eubacteria have been suggested as being important for differentiating Gram-positive versus Gram-negative organisms (45), it is apparent that the murein content of the cell wall is the primary differentiating architecture (46). The breathtaking first thin-section images of Bacillus cereus vividly demonstrated the bacterial surfaces and architecture of the cell wall making it apparent, for the first time, that the cell wall of Gram-positive bacteria was predominately single-layered (47). On the other hand, the cell wall of Gram-negative bacteria was controversial. It was not until Umeda’s group in Japan used the freeze-substitution cryotechnique on Escherichia coli. The resulting Gram-negative images were so remarkable that microbiologists consequently re-evaluated their previous analysis (48). The periplasmic term was coined when Kellenberger’s group from Switzerland produced tangible microscopic images of a rich, dense periplasmic space of Escherichia coli’s cell envelope (49).
Biochemical analysis
Bacterial cell wall biology was a relative newcomer to the scene of biochemical research in the 1950s. The first biochemical clue to the site of penicillin action was provided by Park and Johnson (50, 51) who demonstrated that several novel uridine peptides accumulated in the cytoplasm of penicillin-treated S. aurues. Park and Strominger subsequently observed that the sugar and amino acid compositions of those uridine peptides were similar to those of the bacterial cell wall, suggesting that they were cell wall precursors which accumulated as a result of penicillin inhibition of cell wall biosynthesis (52). Originally, molecules comprised of approximately equal proportions of protein and sugar components were called peptidoglycans and the peptidoglycan making up the bacterial cell envelope were referred to as murein. Over time, these terms became used interchangeably as they are in this review. Murein sacculus refers exclusively to the rigid murein polymer that is present in the cell envelope.
Ironically, prior to 1951, virtually nothing was known about the chemical composition of bacterial envelopes beyond the fact that such well-known structural polysaccharides of plant cell walls as cellulose and chitin were absent. In 1951, Salton and Horne described a large-scale method for cell wall isolation (53). This technique was based on the fact that cell walls are physically quite resistant to mechanical stress. Electron microscopic illustrations accompanying every step of purification, showed that there was no gross physical contamination from other cellular compounds (53). Qualitative examinations of the purified cell walls from a large number of Gram-positive eubacteria demonstrated the presence of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) in all these cellular extracts (54, 55). In addition, the amino acids glycine, D-glutamic acid, D- and L-alanine and L-lysine were detected in each cell wall preparation (Table 1; (56)). Four amino acids were present in the form of a tetrapepide whose sequence was determined to be L-alanyl-D-glutamyl-L-lysyl-D-alanine (Fig. 3). Glycine was present as an interpeptide cross-bridge. These data correlated well with the first general hypothesis about the basal component of cell wall structure, namely mucopeptides, consisting of a hexosamine which is either GlcNAc and MurNAc and a peptide component made up of alanine, glutamic acid, glycine and either diaminopimelic acid or lysine (Fig. 3; (55, 57)). In fact, this compound was exceedingly similar to the nucleotides Park obvserved accumulating in S. aureus as a result of penicillin attack resulting in the convergence of these two lines of investigation (52).
Table 1.
The chemical composition of Gram-positive and Gram-negative bacterial murein.
| Gram positive | Gram negative | |
|---|---|---|
| Sugar | Glucosamine | Glucosamine |
| Acetylmuramic | Acetylmuramic | |
| Amino acid | D-Glutamic acid | D-Glutamic acid |
| D- and L-Alanine | D- and L-Alanine | |
| L-Lysine or L- Diaminopimelic acid Glycine | L-Diaminopimelic acid | |
Fig. 3.
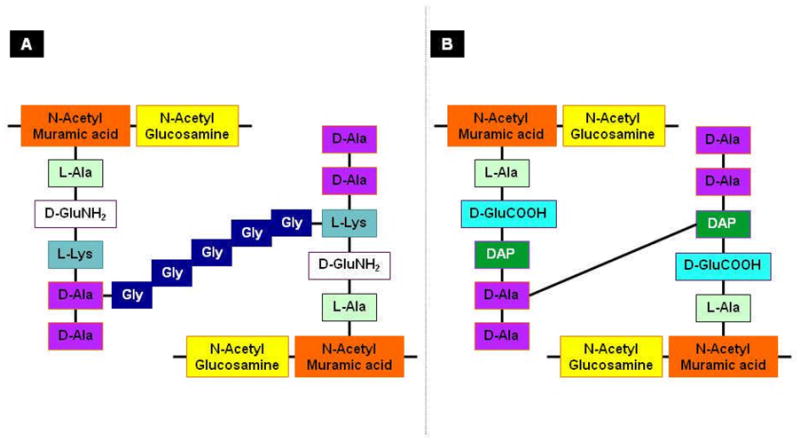
Schematic representation of the (A) S. aureus and (B) Escherichia coli murein monomers. (A) In S. aureus, N-acetylglucosamine and N-acetylmuramic acid form the backbone of the murein. It is attached to pentapeptides containing L-alanine, D-glutamc acid, L-lysine and D-alanyl-D-alanine. The two pentapeptides are linked with a penta-glycine cross bridge. (B) In Escherichia coli, the murein backbone is composed of the same sugar moieties as S. aureus, N-acetylglucosamine and N-acetylmuramic acid. The backbone is attached to pentapeptides containing L-alanine, D-glutamic acid, L-diaminopimelic acid and D-alanyl-D-alanine. Cross-linking occurs between diaminopimelic acid and D-alanine. The D-alanines in red boxes are usually liberated upon transpeptidation.
Though early investigation on Gram-positive organisms successfully elucidated the chemical structure of bacterial cell wall, it became apparent that this was not the case in Gram-negative bacteria when the purified Gram-negative cell wall was subjected to chemical examination. Specifically, the Gram-negative cell wall preparation contained more lipid and less amino sugar than the Gram-positive cell wall (58). The detection of a lipid component in the Gram-negative’s cell wall was not surprising because the presence of lipopolysaccharide was known from the 18th century as the substance that was associated with fever and diseases (59). Furthermore, a full range of amino acids, presumably corresponding to normal protein structures, were identified (58). However, the observations that in Escherichia coli, lysis occurred by treatment with lysozyme, and that penicillin produced osmotic fragility, suggested the presence of peptidoglycan (60).
Although the basic biochemical compositions were known, the architecture of the Gram-negative bacterial cell wall remained elusive. Elegant work by Weidel and Primosigh indicated that phenol could remove the lipopolysaccharide layer (80 % of the whole) from Escherichia coli, leaving behind a layer, consisting of sugars, lipids and amino acids. This layer retained the shape of the intact wall and was, therefore, assumed to be responsible for its rigidity (61). Subsequently, modified purification procedures allowed them to show that the ‘wall’ remaining after phenol treatment contained glucosamine, muramic acid, and a preponderance of alanine, glutamic acid, and diaminopimelic acid (62). Quantitative dissection revealed that various Escherichia coli strains gave essentially the same molar proportions as S. aureus: 1.0 glutamic acid, 1.5 alanine, 0.2 lysine, 0.65 diaminopimelic acid, 0.8 glucosamine and 0.5 muramic acid (63). Essentially, the same mucopeptide composition is found in the cell walls of a wide range of Gram-negative species. The absence of glycine in the Gram-negative cell wall strongly indicated the absence of interpeptide cross-bridges (Fig. 3).
Murein biosynthesis
Murein is a macromolecule of enormous size. It completely envelopes and maintains the structure of bacteria. The realization of the mode of β-lactam agents on the murein layer is an essential prelude to the study of its biosynthesis. Ten years were devoted to elucidating the enzymatic reactions involved in the biosynthesis of the complex murein molecules. Key enzymes involved in the murein synthesis and cell wall recycling and the E. coli and P. aeruginosa genes encoding them are listed in Table 2.
Table 2.
List of genes involved in cell wall biogenesis
| Proteins | Genes in E. colia | Genes in P. aeruginosab | Functional Annotation |
|---|---|---|---|
| Penicilin Binding Proteins | |||
| PBP1a | ponA/mrcA | PA5045 | Membrane carboxypeptidase; Catalyzes the polymerization of the glycan subunit and crosslinking of muropeptides |
| PBP1b | ponB/mrcB | PA4700 | Membrane carboxypeptidase |
| PBP1c | pbpC | PA0378 | Transglycosylase |
| PBP2 | pbpA/mrdA | PA4003 | Chain-length regulation; Murein-elongation initiating enzyme |
| PBP3 | ftsI/pbpB | PA4418 | Transpeptidation of muropeptides exclusively for septal murein synthesis; septal murein-synthesizing enzyme; Important for cell dividion |
| PBP3a | None | PA2272 | Transpeptidase |
| PBP4 | dacB | PA3047 | D-alanyl-D-alanine endopepidase |
| PBP5 | dacA | PA3999? | D-alanyl-D-alaninecarboxypeptidase |
| PBP6 | dacC | D-alanyl-D-alanine carboxypeptidase; Stabilization of the murein at the stationary phase | |
| PBP6b | dacD | D-alanyl-D-alanine carboxypeptidase | |
| PBP7/8 | pbpG | PA0869 | D-alanyl-D-alanine carboxypeptidase; |
| Other proteins involved in bacterial cell shape | |||
| EnvA | envA/asmB/lpxC | PA4406 | UDP-3-O-acyl-N-acetylglucosamine deacetylase |
| RodA | rodA/mrdB | PA4002 | Rod shape-determining protein |
| RodX | rodX | None | |
| RodY/MreB/EnvB | rodY/mreB/envB | PA4481 | Rod shape-determining protein |
| MreC | mreC | PA4480 | Rod shape-determining protein |
| MreD | mreD | PA4479 | Rod shape-determining protein |
| Other proteins involved in bacterial cell wall metabolism | |||
| MpaA | mpaA | None | Gamma-D-glutamyl-meso-diaminopimelate hydrolase |
| MtgA | mtgA (mgt) | PA0378 | Glycan strand polymerization |
| Mpl | mpl | PA4020 | UDP-N-acetylmuramate:L-alanyl-gamma-D-glutamyl-meso-diaminopimelate ligase |
| SltY | sltY | PA3020? | Lytic transglycosylase |
| MltA | mltA | PA1222 | Lytic transglycosylase |
| MltB | mltB |
PA4444/mltB1/sltB PA4001/sltB1/mltB2 |
Membrane-bound lytic murein transglycosylase B Lytic transglycosylase |
| MltC | mltC | PA3020? | Lytic transglycosylase |
| MltD | mltD | PA1812 | Lytic transglycosylase |
| MltE | mltE/emtA | PA3764 | Lytic transglycosylase |
| AmiA | amiA | PA5538 | N-acetylmuramoyl-L-alanine amidase; Cell separation |
| AmiB | amiB | PA4947 | Cell separationN-acetylmuramoyl-L-alanine amidase; Cell separation |
| AmiC | amiC | PA4947?/PA5538? | Cell separation |
| MepA | mepA | None | LD-endopeptidase |
| NagZ | nagZ | PA3005 | β-N-acetyl-D-glucosaminidase |
| LdcA | ldcA | PA5198 | LD-carboxypeptidase |
| EnvC | envC | PA5133 | Endopeptidase |
| FlgJ | flgJ | PA1085 | Muramidase (flagellum-specific) |
| Amp and Pox proteins | |||
| AmpC | ampC | PA4110 | AmpC beta-lactamase |
| AmpR | None | PA4109 | Transcriptional regulator AmpR |
| AmpD | ampD | PA4522 | 1,6-anhydro-N-acetylmuramyl-L-alanine amidase |
| ampDh2 | PA5485 | N-acetyl glucosamidase | |
| ampDh3 | PA0807 | N-acetyl glucosamidase | |
| AmpE | ampE | PA4521 | Hypothetical membrane protein |
| AmpG | ampG | PA4218 | Transport of degraded muropeptides: GlcNAc- anhMurNAc |
| AmpF | None | PA4219 | Hypothetical membrane protein |
| AmpP | None | PA4393 | Permease |
| AmpO | None | PA4219 | Hypothetical membrane protein |
| PoxA | None | PA5513 | Hypothetical hydrolase |
| PoxB | None | PA5514 | Class D beta-lactamase |
| Mur & Mra proteins | |||
| MurA | murA | PA4450 | UDP-N-acetylglucosamine enolpyruvyl transferase |
| MurB | murB | PA2977 | UDP-N-acetylpyruvoylglucosamine reductase |
| MurC | murC | PA4411 | UDP-N-acetylmuramate--alanine ligase |
| MurD | murD | PA4414 | UDP-N-acetylmuramoylalanine--D-glutamate ligase |
| MurE | murE | PA4417 | UDP-N-acetylmuramoylalanyl-D-glutamate-2, 6-diaminopimelate ligase |
| MurF | murF | PA4416 | UDP-N-acetylmuramoylalanyl-D-glutamyl-2, 6-diaminopimelate--D-alanyl-D-alanyl ligase |
| MurG | murG | PA4412 | UDP-N-acetylglucosamine--N-acetylmuramyl- (pentapeptide) pyrophosphoryl-undecaprenol N- acetylglucosamine transferase |
| MurI | murI | PA4662 | Glutamate racemase |
| MraY | mraY | PA4415 | Phospho-N-acetylmuramoyl-pentapeptide-transferase |
| MraW | mraW | PA4420 | Peptidoglycan biosynthetic process with methyl transferase activity |
| MraZ | mraZ | PA4421 | Role in cell-wall biosynthesis |
| MraR | mraR/ftsL | None | Cell division |
Biosynthesis of the cell wall is comprised of three distinct stages occurring at three distinct sites in bacterial cells (64). The first site is in the cytoplasm. This chain of reactions is initiated with the committed step in which a three-carbon phosphoenolpyruvic acid (PEP) is attached to UDP-N-acetylglucosamine (UDP-GlcNAc), leading to the formation of UDP-N-acetylmuramic acid (UDP-MurNAc; Fig. 4). This is followed by an enzymatic addition of amino acids in a stepwise manner, and finally to the production of UDP-MurNAc pentapeptide, the compound first isolated by Park and Johnson (Fig. 4; (50)). The second stage of the bacterial murein synthesis occurs at the cytoplasmic membrane of the cell. At this site, the intracellular precursors, UDP-GlcNAc and UDP-MurNAc pentapeptide, are joined at the sugar ends to form a linear murein with protruding short peptide chains at every N-acetylmuramic acid sugar (Fig. 4). In addition, depending on the organism, the murein peptides are modified. For the genus Staphylococcus, the α-carboxyl group of D-glutamic acid in the pentapeptide is amidated to yield D-isoglutamic acid (65).
Fig. 4.
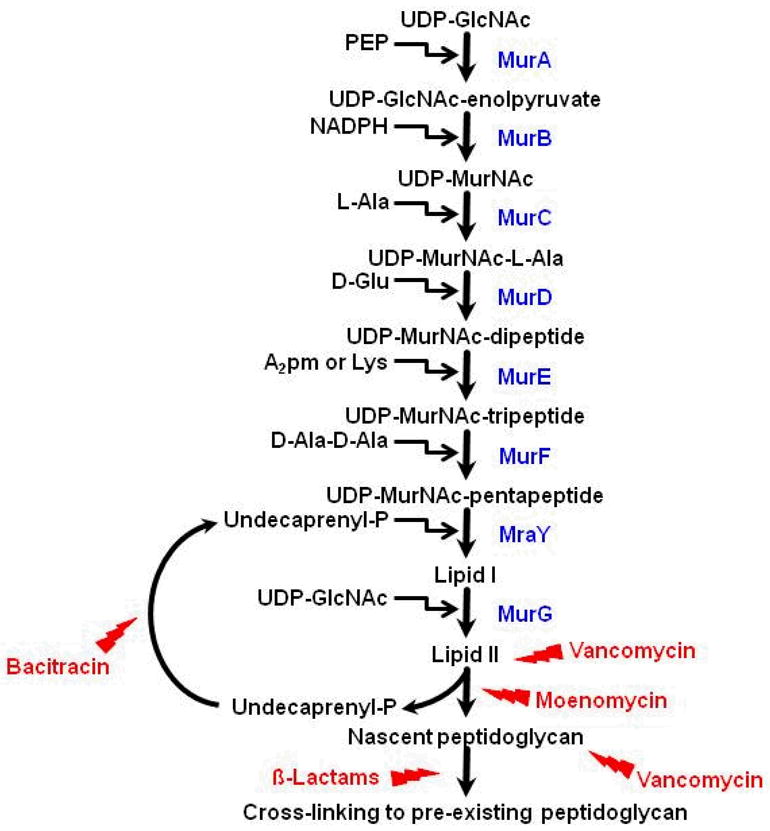
The biosynthetic pathway of bacterial murein. The pathway initiates with committing steps, generating UDP-acetylmuramic acid (UDP-MurNAc). L-alanine, D-glutamic acid, lysine or diaminophenolic acid, and D-alanyl-D-alanine are enzymatically added to UDP-MurNAc in a stepwise manner. The formation of UDP-acetylmuramyl-pentapeptide (UDP-MurNAc-pentapeptide) occurs in the cytoplasm. Then, the UDP-MurNAc-pentapeptide is transferred to the cytoplasmic membrane where two lipid carriers (Lipid I and Lipid II) are involved in the joining of UDP-MurNAc-pentapeptide and UDP-acetylglucosamine (UDP-GlcNAc) to form a linear murein with protruding short peptide chains. Finally, the linear murein chains are cross-linked with each other and to pre-existing murein. A few chemical compounds are known to block this pathway. β-lactams are known to inhibit the cross-linking, whereas bacitracin inhibits the recycling of undecaprenyl. Moenomycin and vancomycin inhibit the function of Lipid II that carries the disaccharide peptide monomer units.
The final transpeptidation step does not require any input of energy. In vivo investigation of crude protein preparations of Escherichia coli showed that there are in fact two concomitant transpeptidation reactions (66, 67). Cross-linking between two murein strands is introduced by transpeptidase, followed by a second reaction catalyzed by a D-alanine carboxypeptidase in which the terminal D-alanine residue of the pentapeptide in the other strand is removed (68–70). Among the enzymatic reactions, only the transpeptidase and D-alanine carboxypeptidease are sensitive to β-lactam antibiotics (66). However, the inhibition of the D-alanine carboxypeptidase enzyme occurs at a much lower MIC than that for the organism (66, 67). Thus, it is proposed that the inhibition of this activity is not lethal. In sharp contrast, the concentration required to inhibit growth was virtually similar to the concentration required to halt the activity of transpeptidase, the enzyme carrying out the actual cross-linking reaction, suggesting that this is the crucial step inhibited by β-lactam antibiotics (66, 67, 71).
Penicillin is a cyclic dipeptide of two amino acids, L-cysteine and D-valine. The conformation of one edge of the penicillin molecule is nearly identical as the conformation of the backbone of the D-alanyl-D-alanine (Fig. 2). A crucial feature in this case is the fact that the highly reactive CO-N bond in the β-lactam ring of the penicillin molecule lies in exactly the same position as the CO-N bond in D-alanyl-D-alanine which is the target of transpeptidation (Fig. 2). Owing to the striking structural similarity, it was thus hypothesized that penicillin acts as a substrate analog, binding to the substrate-anchoring site normally occupied by D-alanyl-D-alanine (69).
Biophysical analysis
Transpeptidases have been characterized as penicillin-sensitive proteins since their activity is specifically prevented by the covalent binding of β-lactam antibiotics to their active site (72). The inference that transpeptidase is a penicillin-binding protein (PBP) converges yet another line of independent research to the overall understanding of the penicillin action.
As early as 1949, ingenious work had employed radioactive penicillin to locate the site of its action on bacterial cells (73–76). These studies clearly indicated that penicillin bound a definite trace target, termed penicillin-binding component (PBC (74)). It appeared that the entire penicillin molecule reacted with PBC. Once bound, radioactive penicillin was unable to be removed by neutral buffers, acids or detergent, but treatment with dilute alkali released it as penicilloic acid (74, 76, 77). In addition, the PBC was destroyed at high temperature and treatment with strong solvents indicative of protein components (78). Taken together, this suggested penicillin is covalently bound to PBC. When the PBC-penicillin complex was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the PBC resolved into several proteins ranging in molecular weight from 40 to 90 kDa (79). These proteins were named penicillin-binding proteins (PBP) and numbered according to their descending molecular weights. The number of PBPs, their molecular weight, their concentration (molecule per cell) and their sensitivity to β-lactam antibiotics of these proteins varied widely between bacterial species (e.g. Table 2; (80)). Some of the low-molecular-weight PBPs, PBP5, PBP6 and PBP7, were found abundantly in bacilli but not in cocci (80). Gram-positive cocci, for example the two cousins, Staphylococcus aureus and Staphylococcus epididermis, express only four penicillin-binding enzymes, whereas Bacillus subtilis and Bacillus cereus harbor seven to eight PBPs (80, 81). In Escherichia coli, PBP1–6 were identified using radioactive benzyl-penicillin (79). Similar numbers of PBP were also found in Pseudomonas aeruginosa, Entaerobacter cloacae, Salmonella enteric serovar Typhimurium (S. Typhimurium) and Serratia marcescens (80).
Genetic analysis
Functional and morphological correlations of the PBPs strongly relied on their binding specificity of β-lactam antibiotics and isolation of temperature-sensitive mutants. Since PBPs were named based on their SDS-PAGE migration, PBP nomenclature is organism dependent. For example, Escherichia coli PBP2 is most similar to Staphylococcus aureus PBP3. The numbering system used below is based upon Escherichia coli PBPs. Much has been elucidated about the structure and biochemical properties of PBPs. Readers are referred to recent reviews focusing on PBPs (82–85).
PBP1
Cephaloridine, a cephalosporin agent which blocks cell elongation at sub-inhibitory concentration, has the highest affinity for Escherichia coli PBP1, suggesting that PBP1 might be involved in cell elongation (86). The high-molecular-weight PBP1 band on SDS-PAGE was actually composed of two polypeptides (79). Spratt first observed the resolution of these two closely related proteins into two fragments, but this was inconclusive as the separation was inconsistent (79). Subsequent modification of the SDS-PAGE technique allowed resolution of PBP1 into two distinct components, PBP1a and PBP1b (87).
The gene coding for PBP1a was mapped to ponA or mrcA using N-methyl-N-nitro-N-nitrosoguanidine chemical treatment (87, 88). Strains containing mutations in ponA/mrcA appeared to grow normally, suggesting that that PBP1a might have a subtle role in cell growth and morphology (87, 88). However, PBP1a mutants underwent a slow rate of β-lactam induced lysis, suggesting involvement in murein metabolism (87). Subsequent studies showed that PBP1a catalyzed the polymerization of the glycan subunit and crosslinking of muropeptides (88).
PBP1b could be further resolved into at least three bands (88); however, chemical mutagenesis of a single gene, ponB or mrcB, resulted in the loss of all three fragments. The three could be recovered simultaneously by P1 transduction of the wild-type gene located at 3.3 minute of the Escherichia coli chromosome, suggesting that PBP1b could exist in three isoforms (88). By examining the in vitro incorporation of radioactive UDP-MurNAc-pentapeptide into the murein, membrane fractions isolated from ponB/mrcB mutants had significantly decreased murein biosynthetic activity, indicating that PBP1b catalyzed the majority of this activity in vitro (88). Subsequently, PBP1b was functionally indicated to be responsible for transglycosylase and DD-transpeptidase enzymatic reactions (89–91).
PBP1a and PBP1b differ markedly in their affinities for several β-lactams. For example, cephalothin and cephaloridine both bind PBP1a strongly, while PBP1b has high affinity only for cephaloridine (87). In agreement with this, Escherichia coli cells lacking PBP1b are hypersensitive to cephalothin, owing to the binding of this β-lactam antibiotic to the PBP1a (88). Although individual loss of PBP1a or PBP1b did not confer thermosensitive growth, the loss of both proteins was lethal, indicative of redundancy in the system (90). Inhibition of both PBPs was necessary to prevent cell elongation and thereby induce lysis (90).
PBP1c, a third penicillin-binding protein related to PBP1a and PBP1b, has also been identified in Escherichia coli (92). Unlike PBP1a and PBP1b which function as both transglycosidases and transpeptidases, PBP1c appears to only function as a transglycosylase. Overexpression of PBP1c, which is encoded by pbp1c located at 56.9 min on the Escherichia coli chromosome, cannot suppress the effect of mutating both PBP1a and PBP1b. Mutation in PBP1c does affect cell murein synthesis (92).
PBP2
PBP2 was the first penicillin-binding protein to be dissected functionally due to the ability of this enzyme to specifically bind to mecillinam (79). Covalent binding of mecillinam to PBP2 caused alteration in Escherichia coli shape from rod into ovoid (93). This morphologic alteration allowed the isolation of mutants lacking PBP2 after an intensive chemical mutagenesis (94). These thermolabile mutants grew slowly as rod-shaped cells at 30°C and became round cells upon temperature shift to 42°C (94). Hence, PBP2 was thought to be the major protein involved in maintenance of cell shape. Absence of PBP2 also caused inhibition of cell division and delayed muropeptide synthesis (94, 95).
The actual functions of PBP2 attracted a lot of attention. In vitro, upon the presence of mecillinam, the carboxypeptidase, transpeptidase and endopeptidase activities were unaffected, suggesting that PBP2 might catalyze a novel reaction (93, 96). However, it was also postulated that PBP2 mediated a topologically restricted transpeptidase reaction, perhaps during a specific phase of the cell cycle. Several lines of evidence agreed with this postulation:
Inactivation of PBP2 resulted in a doubling of the Escherichia coli cell wall anhydromuramic acid content, thus implicating PBP in cell-wall glycan chain-length regulation (97).
In the presence of cefsulodin and cefmetazole, cephalosporins that bind to all PBPs but PBP2, the murein synthesized acquired a degree of cross-linkage even higher than the control cells, demonstrating that PBP2, or other proteins, might have a transpeptidase activity in vivo able to produce a highly cross-linked peptidoglycan (95).
A mutation in ponB or mrcB, encoding PBP1b, resulted in the over-production of PBP1a and PBP2. This was consistent with the suggestion that PBP2 might act to ensure that new initiation sites for elongation were introduced with the correct rod orientation (86).
These data argued that in the absence of active PBP2, novel glycan chains might be utilized preferentially by the septum-forming enzyme system resulting in growth as round cells. (98).
Because the strains lacking PBP2 were isolated upon intensive chemical treatments, mapping the gene encoding PBP2 was non-trivial. Several genes were found to be involved in round-shaped morphology and mecillinam resistance. In fact, the earliest report that associated the appearance spherical Escherichia coli with antibiotic resistance, interestingly, pre-dated the discovery of PBPs (99). Normark, investigating the effect of ultraviolet light tolerance, observed round-shaped Escherichia coli under the electron microscope when the strains harbored a mutation in envB (99). This envB gene was subsequently mapped to the 64 minute of the chromosome (99). Half a decade later, a second gene, rodA, responsible for coccal-shaped cells, was mapped to 15 min between the purE and pyrC loci (100). Escherichia coli strains with mutation in rodA were also found to be tolerant to ultraviolet irradiation (100). Moreover, intensive chemical mutagenesis analysis mapped two other genes at minute 14 (rodX) and 70 (rodY) that were involved in the maintenance of the rod-shape (101). However, since the round-shaped morphology could be due to the mutation of any of these genes, these reports did not correlate the morphological change to the loss of PBP2 protein. Eventually, the gene encoding PBP2, with the concomitant loss of PBP2 protein, was mapped to a region at 14.4 minute (88, 102). Using a series of defective lambda transducing phage, the region was mapped to a 12-kb DNA containing the lip-dacA-rodA-pbpA-leuS region (103). This cluster of genes was involved in cell shape determination and murein synthesis (103). Analysis of the proteins produced from lambda transducing phage carrying a well-defined DNA fragment showed that pbpA encoded PBP2, whereas dacA coded for PBP5 (103). Although mutation in the rodA gene resulted in spherical cells, loss of RodA did not affect the PBP2 activity (103). RodA was later found to act synergistically with PbpA as transglycosylase and transpeptidase (104). Elegant genetic analysis of pbpA demonstrated that pbpA spherical mutants could be isolated that retained penicillin binding activity (98). Further analysis showed that deletion of pbpA is lethal, but may be suppressed by overexpression of ftsZ, either directly or by increasing the intracellular ppGpp pool (105, 106). Together, this suggests that PBP2 plays an essential role in cell division.
PBP3
Escherichia coli strains expressing temperature-sensitive PBP3 mutated proteins were isolated from nitrosoguanine mutagenesis (79). At the permissive temperature of 30 °C, these mutants appeared slightly longer than the parental strain (102). On shifting to the restrictive temperature of 42 °C, cell division ceased. But, the continuous increase in cell density and the incorporation of 3H-thymine suggested that cell growth and DNA replication were not affected in the absence of PBP3 at restrictive temperature. As a result of continuous replication without division, long filamentous polynucleated Escherichia coli was observed (102). This observation strongly argued that PBP3 was an essential cell-division protein. Supporting evidence for its role in cell division came from the use of PBP3-specific β-lactam antibiotics, furazlocillin and piperacillin (107). PBP3 is recruited to the septal ring where it functions as a transpeptidase to crosslink the peptidoglycan at the division septum (108, 109). Due to its function in cell division, PBP3 is also known as FtsI.
Biochemically, at the permissive temperature, both [14C]-diaminopimelic acid incorporation into murein of intact cells and [14C] N-acetylglucosamine incorporation into murein were reduced in septating PBP3 mutants, as compared to the wild-type while no inhibition was detected in filaments growing at the nonpermissive temperature (108). The authors suggested that PBP3 is involved in transpeptidation of muropeptides exclusively for septal murein synthesis but not for the elongating murein production (107, 108).
Low molecular weight PBPs
The lower molecular weight PBPs 4, 5 and 6 were shown as the major DD-carboxypeptidases (67). These enzymes displace the terminal D-alanine residue from one of the two peptide chains to form crosslinked murein (67). It was argued that DD-carboxypeptidase regulated the extent of cross-linking in murein by removing the terminal D-alanine from a proportion of the pentapeptide side chains of nascent peptidoglycan, thus preventing their involvement in cross-links (67). This DD-carboxypeptidase activity was divided into two enzymatic reactions, namely IA and IB, each catalyzed by different PBPs (67, 110).
Enzyme IA was shown to catalyze DD-carboxypeptidase and transpeptidase reactions, bind to [14C]-penicillin G, confer a weak penicillinase activity, but was devoid of endopeptidase activity (110).
Enzyme IB was demonstrated to catalyze DD-carboxypeptidase and endopeptidase activities, confer weak penicillinase activity, but not bind [14C]-penicillin G and had poor transpeptidase activity (110).
PBP4
Escherichia coli strains lacking the highly penicillin-sensitive activities of DD-carboxypeptidase IB and DD-endopeptidase showed a concomitant loss of PBP4 (111, 112). This mutation was mapped to the dacB gene located at 68 minute on the Escherichia coli map (112). These mutants grew normally under a wide range of growth conditions, suggesting that PBP4 was not essential for the growth of Escherichia coli under laboratory conditions (88, 111). However, overexpression of PBP4 showed an increase of DD-carboxypeptidase and DD-endopeptidase, which did not alter the transpeptidation reaction (113). PBP4 was demonstrated as the only PBP of Escherichia coli that possessed DD-endopeptidase activity. This enzyme was able to cleave the D-alanyl-γ-meso-2,6-diaminopimelyl peptide bond in the crosslinks of murein (114).
PBP5
Purified DD-alanine carboxypeptidase IA activity in Escherichia coli was attributed to two polypeptides (115). Both proteins bound strongly to benyzlpenicillin and were identified as PBP5 and PBP6 (79). These membrane-bound proteins were shown to catalyze a transpeptidase reaction and had a weak penicillinase activity (110). Loss of PBP5 proteins could be complemented with P1 phage that carried the 14 minute region of the chromosome, indicating that PBP5 gene was located within the lip-leuS region (116). Using lambda transducing phage carrying well-defined DNA fragments, the gene encoding PBP5 was mapped to dacA (103). Mutation in dacA abolished the DD-alanine carboxypeptidase IA activity, rendered the cells sensitive to penicillin, though they exhibited normal morphology (117). A dacA dacB double mutant demonstrated a negligible carboxypeptidase activity, suggesting that PBP4 and PBP5 contributed at least 90 % of the DD-carboxypeptidase activity (117).
PBP6
The dacC-encoded PBP6 protein shares 62 % sequence identity with PBP5 (118). Both purified PBP5 and PBP6 were able to catalyze identical reactions but PBP5 had a three- to fourfold higher specific activity towards uncross-linked peptidoglycan than PBP6 (119). Deletion of dacC had no effect on cell morphology and growth rate (120). However, strains lacking PBP6 showed a very slight increase in antibiotic sensitivity (120). However, comparison of the expression of PBP5 and PBP6 at different phases of growth exposed the temporally differential functions of PBP5 and PBP6. The concentration of PBP5 remained fairly constant throughout all phases, whereas the amount of PBP6 increased two- to ten-fold in stationary phase as compared to exponential phases (121). This growth-phase dependent expression of PBP6 strongly suggested that it might be involved in stabilizing the murein at stationary phase.
Another low molecular weight PBP that is similar to PBP5 and PBP6 was identified in Escherichia coli, PBP6b (122). PBP6b has DD-carboxypeptidase activity and binds [3H] benzylpenicillin. The gene encoding PBP6b, dacD, is located at minute 44 on the Escherichia coli chromosome and was identified using the Kohara lambda phage library (122). Like PBP5 and PBP6, PBP6b is non-essential for viability (122).
PBP7/8
PBP7 and PBP8 were characterized more recently than most of the other PBPs in part due to their irreproducible presence in bacterial preparations. PBP8 is an OmpT proteolytic product of PBP7 (123). PBP8 has been shown to be a D-alanyl-DAP-endopeptidase (124) and increased expression is related to increased cephaloridine and ceftazidime resistance (125). Both are soluble perplasmic proteins that are peripherally associated with the membrane. Use of the Kohara lambda phage miniset library narrowed the genomic location of the PBP7-encoding gene to 47.8 and 48 min on the Escherichia coli chromosome (126). Subsequent subcloning of one open reading frame, yohB now renamed pbpG, and insertional mutagenesis confirmed that pbpG encoded PBP7.
HIDDEN BLACKBOX: ANTIBIOTIC RESISTANCE
The introduction of penicillin from laboratory to bedside heralded a new era in the clinical settings. Physicians had a landslide victory in treating many dominating lethal infectious diseases such as meningitis, tuberculosis, pneumonia and so forth. The development of other classes of antibiotics, such as erythromycin, tetracyclines, and chloramphenicol, fully strengthened the foundation of infection chemotherapy. Antimicrobial therapy provided physicians with the ability to prevent some infections, to cure others and to curtail the transmission of certain diseases. At the beginning of the twentieth century, infectious diseases were the leading cause of death worldwide. The success of vaccination and the discovery of antibiotics changed this trend. Between 1900 and 1980, in U.S., mortality from infectious diseases fell from 797 to 36 per 100,000 individuals (127). In 1969, amid with the success of vaccination and the development of antibiotics, the Surgeon General told the United States Congress that it was time to ‘close the book on infectious diseases’ (128). The availability and success of antibiotics resulted in a confidence that technology and modern medicine would be victorious against infectious diseases. The emergence of outbreaks and epidemics of antimicrobial resistant infections resulted in a re-evaluation of this optimism.
At the very beginning, resistance to antibacterial agents was recognized as a potential problem. Resistance to penicillin was actually the focus of Alexander Fleming’s 1929 ground-breaking masterpiece where he exploited the advantage of the substance to eliminate Staphylococcus and Streptococcus contamination and thus, isolated pure cultures of Escherichia coli, Salmonella enterica serovar Typhi and Haemophilus influenzae (15). Yet, the potential of penicillin as therapeutic agent overshadowed the resistance issue.
All was not lost as this report intrigued some scientists, particularly Edward Abraham and Ernst Chain. They setup to answer the question of why some bacteria are more resistant than others. In 1940, they found penicillinase enzyme in the culture fluid of a penicillin-resistant Gram-negative rod that had contaminated Penicillium cultures and in an extract of Escherichia coli, but detected no such activity in a penicillin-sensitive strain of Staphylococcus aureus (18). The purified enzyme from Staphylococcus was able to inactivate penicillin effectively (129). Thus, it was known from the beginning of the human-pathogen battle that bacteria have the ability to counteract the antibiotics. Furthermore, Edward Abraham and his colleagues also demonstrated that cultures of staphylococci could be made resistant by continuous subculture in the presence of the antibiotic in vitro (19).
Within a year of introduction, four penicillin-resistant staphylococci strains were isolated from patients during the course of treatment of localized infections (130). This was followed by a number of reports showing the emergence of penicillin-resistant staphylococci (131–133). This phenomenon did not raise an immediate alarm because the initial increase in penicillin resistance could be compensated with an elevated dosage of the drug, as the antibiotic was relatively safe and became more widely available and inexpensive (134). By 1947, just five years after its introduction, the majority of hospital isolates of Staphylococcus aureus were resistant to penicillin G and six other common antibacterial agents, including streptomycin, tetracycline and erythromycin (135). This prevalent inter-hospital spreading Staphylococcus aureus strain 80/81 complex was the first encounter of multiple-resistant pathogens (135). Barber in England was the first to call attention to the problem of the increasing occurrence of penicillin-resistant staphylococci in hospitals (136–139). Worldwide alarm of Staphylococcus aureus resistance quickly followed as reports emerged from many hospitals and laboratories in the United States (135), Canada, Australia, Norway, Sweden, Denmark, France, India and Chile (134).
In the 1960s, analysis of the organisms associated with nosocomial infections shifted away from Gram-positive species to Gram-negative species such as Escherichia coli, K. pneumonia and Proteus mirabilis, a group of bacteria which scientists and bedside physicians had little understanding (140). According to Finland’s longitudinal survey of the panorama of infections, the introduction of antimicrobial agents was accompanied with negative rather than positive overall impact to the patients (140). In fact, the potential therapeutic effect was nullified by a shift towards more drug-resistant organism and serious-infections by more virulent organisms. In addition, organisms challenged with antibiotics increased their number, and enhanced their pathogenicity and invasiveness (141).
On the other hand, the medicine renaissance increased the life expectancy, but not the life quality, of the human race with sophisticated treatments, including intensive care for terminal patients, artificial instrument insertions and chemotherapy of cancer patients. As a result, there was a sudden surge of patients who were immuno-compromised or required frequent medical attention. These events directly augmented the opportunity of the infections of a group of pathogens such as Pseudomonas aeruginosa, Staphylococcus epidermidis, Klebsiella pneumoniae and Serratia marcescens (140). Unlike Staphylococcus aureus, Escherichia coli and Salmonella Typhi, this group of bacteria are successful environmental organisms, evolutionarily equipped with an armory to counteract naturally occurring growth inhibitors including naturally occurring antibiotics, antiseptics and heavy metals (140).
The failure of penicillin to be the magic bullet and the shift in infection pattern pushed investigators to seek alternative routes. The age of ‘drug rejuvenation’ began with the artificial synthesis of 6-Aminopenicillanic acid (31). This discovery allowed the rapid introduction of β-lactam antibiotics by changing the chemical structures in vitro. For a short period of time, drug company chemists managed to keep ahead in the race against antibiotic resistance by making slight changes in the structures of their antibiotics. The synthesis of semi-synthetic methicillin, which was β-lactamase stable, temporarily rescued the failure of the penicillin (142). Meanwhile, the production of ampicillin, another semi-synthetic β-lactam antibiotic, inhibited a wide range of Gram-negative organisms, including Escherichia coli, Haemophilus influenzae, Salmonella Typhi and Shigella. As a result of the explosive antibiotic development, the market was crowded with more than one hundred antibacterial agents (143). Thinking that they had won a total victory, many pharmaceutical firms started to withdraw the efforts to develop new antibiotics in the 1980s, instead focusing on drugs for chronic illnesses such as heart disease, cancer and diabetes, the leading causes of mortality and morbidity in developed countries (128). Thus far, antibiotics have been used with unrestrained passion far surpassing the needs of management and infection control. This is owing to an unorthodox belief that antibiotics play a life-saving role in all debilitating diseases (144). Over the years, antibiotic resistance issues frequently come under the spotlight in the media.
As the gruesome antibiotic resistance alarm became inescapable, international conferences, workshops and other actions were organized to persuade funding agencies to take notice and bring the issue to the forefront (145). In 1990, Gail Cassell, President of the American Society of Microbiology (ASM) and a member of the advisory council of the National Institutes of Allergy and Infectious Diseases (NIAID) warned, ‘we are at a very critical crossroads in terms of readiness to deal with infectious diseases and antibiotic resistance from a funding standpoint’ (145). Even though better monitoring or a surveillance program could help prevent and manage outbreaks of antibiotic resistant disease, most scientists argued that basic research on how bacteria defy antibiotics was equally pressing and seriously lacking.
BETA-LACTAMASES
β-lactam was the first antibiotic to be described (28). Consequently, resistance to β-lactam antibiotics is the first to be understood. The most effective way for bacteria to counteract these chemicals has been by producing β-lactamases, enzymes that inactivate the drugs by hydrolyzing the β-lactam ring (18). Based on the sequence analysis, β-lactamases and the PBPs are believed to diverge from a common ancestor. All PBPs possess β-lactam catalyzing capability to a smaller extent. For instance, PBP5 was demonstrated to have the highest β-lactamase activity. At pH 7.0 and 30°C, the half-life of penicilloyl-PBP5 was averaged at 10 min (119). In addition, the monofunctional penicillin-binding DD-peptidases and penicillin-hydrolyzing serine beta-lactamases retain the same tertiary folding, three-motif amino acid (Lys-Thr-Gly) sequence signature, serine-assisted catalytic mechanism and active-site topology (Fig. 5 (72)).
Fig. 5.
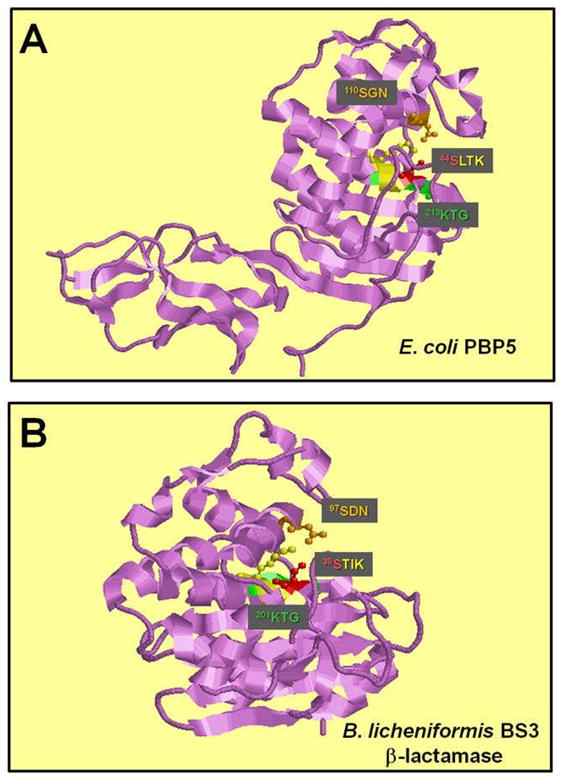
Structure of the active site of DD-carboxypeptidase and β-lactamase. A. The X-ray crystal structure of E. coli PBP5 (240) and Bacillus licheniformis BS3 beta-lactamase (241). The conserved SerXXLys, Ser[Tyr]XAsn, and Lys[His]Thr[Ser]Gly motifs (242) are colored in yellow, orange and green, respectively. The catalytic serine of the SerXXLys motif is colored in red.
Thus far, more than 300 enzymes have been documented (146). Because of the diversity of enzymatic characteristics of the many β-lactamases discovered, multiple attempts have been made to categorize them since the late 1960s (147–150). Theseclassifications involve two major approaches: the first and older one is based on the biochemical and functional characteristics of the enzyme, whereas the second approach is based on the molecular structure of the enzyme. In the former classification scheme, several criteria are used, including the spectrum of antimicrobialsubstrate profile, enzyme inhibition profile, hydrolysis rate (Vmax), binding affinity (Km), isoelectricfocusing (pI), protein molecular weight, and amino acid composition (149, 150). The molecular classification of β-lactamases is based on the nucleotide and amino acid sequences in these enzymes. To date, four classes are recognized (A-D), correlating with the functional classification. Classes A, C, and D act by a serine based mechanism, whereas class B or metallo β-lactamasesneed zinc for their action (147, 148).
Ambler Class A
Ambler molecular class A was initially described in Gram-positive plasmids. However, plasmid-, transposon- and chromosomally-mediated β-lactamases (VHS, PER, TEM and SHV) in Gram-negative bacteria have also been reported (151). This class of enzymes, dubbed penicillinases, exhibits the highest degree of sequence variability and kinetic properties (152). Plasmid-encoded Ambler class A β-lactamases identified in Pseudomonas aeruginosa are active against carbenicillin hence referred to as CARB or Pseudomonas-specific β-lactamases (PSE) (153, 154). In addition, VHS-, PER- and TEM-derived β-lactamases have also been reported in Pseudomonas aeruginosa (155).
Ambler Class B
The Class B enzymes contain a small number of Zn2+ metallo-β-lactamases, whereby their activities could be inhibited by EDTA (156). IMP-1 is the first metallo-β-lactamase described in Pseudomonas aeruginosa (157). Its gene, blaIMP, appears to be dispersed among Pseudomonas aeruginosa and other Gram-negative rods in Japan (158). An integron-borne metallo-β-lactamase gene, blaVIM, which was originally described in Pseudomonas aeruginosa isolated in Italy and is prevalently found in Greece, gives rise to the resistance of meropenem and imipenem (159, 160).
Ambler Class C
The Ambler Class C enzymes are active against cephalosporins, hence dubbed cephalosporinases (161). They are chromosome-encoded and synthesized by most Gram-negative bacteria. The known sequences of these enzymes are highly conserved (162). The Pseudomonas aeruginosa Class C cephalosporin-hydrolyzing chromosomal β-lactamase, encoded by ampC (PA4110), has been cloned and sequenced (163).
Ambler Class D
The enzymes belonging to the Class D group of β-lactamases are called oxacillinases as they are able to degrade isoxazolyl β-lactams such as oxacillin and methicillin (164), while clavulanic acid serves as a good inhibitor for these enzymes (165). Due to the structural similarity between Class A and Class D enzymes, Couture et al suggested the use of the 26 conserved amino acid residues as the class D standard numbering scheme (DBL numeration) (151). Plasmid- and transposon-mediated oxacillin-hydrolyzing β-lactamases in Pseudomonas aeruginosa are common and complex. The characterization of a Pseudomonas aeruginosa ampC mutant led to the discovery of a novel chromosomally-encoded Class D β-lactamase, encoded by poxB [PA4511; (166)]. This Class D β-lactamase was independently reported by Girlich et al., and the enzyme was named OXA-50 (167). However, sequence analysis argues that PoxB is distinct from OXA enzymes (166). Nosocomial outbreak with Pseudomonas aeruginosa extended-spectrum of Class D β-lactamases (ESBLs) was reported (168) and more episodes are expected to arise in the near future.
THE CHROMOSOMAL β-LACTAMASE INDUCTION MECHANISM
There are two kinds of chromosomal-encoded β-lactamases: constitutive and inducible. The former is present at a predictable level under all circumstances, whereas the latter is repressed in the absence of antibiotics and induced in the presence of β-lactam agents (169). The analysis of chromosomal mediated β-lactamases revealed that in bacteria species that possess a specific transcriptional regulatory system, synthesis of β-lactamase is inducible, whereas in strains without this system, synthesis is usually constitutive (170, 171).
The Gram-negative organisms possess chromosomally determined β-lactamases and the types of β-lactamase produced are often species-specific (171). The inducible expression of β-lactamases of Gram-negative bacteria suggests that this mode of bacterial resistance does not involve a single-protein one-step mechanism. Indeed, genetic studies performed on Enterobacter cloacae, Citrobacter freundii and Escherichia coli revealed at least five genes found in three loci directly participate in β-lactam resistance. They are ampC, ampR, ampD, ampE and ampG. Many human pathogens harbor AmpR and AmpC homologs, namely, Burkholderia cepacia (172), Yersinia enterocolitica (173), Citrobacter freundii (174), Enterobacter cloacae (175), Morganella morganii (176), Serratia marcescens (177), Laribacter hongkongensis (178) and Ochrobactrum anthropi (179).
ampC
The chromosomally encoded β-lactamases have been a subject of intense study since the 1980s (161). Most of these enzymes are cephalosporinases that are capable of hydrolyzing third generation cephalosporins with low Vmax value and high Km values (161). They are classified as the type I β-lactamases in the Richmond and Sykes scheme, and Class C enzymes in Ambler’s classification (147, 150). These are relatively large enzymes (30–42 kDa), usually with alkaline isoelectric points (150).
The chromosomal β-lactam hydrolyzing gene, ampC, of Escherichia coli K-12 was first to be cloned and sequenced (180). This 1536-bp gene encodes a protein of 377 amino acids. The first 19 amino acids form a signal peptide, suggesting that this protein is translated as a precursor and transported into the periplasm as a mature 39.6 kDa enzyme (180). The AmpC β-lactamase with substrate specificity for cephalosporins showed no significant sequence homologies with β-lactamases of the Ambler’s Class A penicillinase or with the D-alanine carboxypeptidases (161).
The expression of AmpC in Escherichia coli K-12 was constitutively low in the absence of β-lactam antibiotics (181). However, the amount of AmpC protein increased proportionately with the growth rate and the nutrient availability (181). The checkpoint for the limited expression of this enzyme was due to the presence of an attenuator located between the promoter and the structural gene (182). This attenuator formed a stem-loop structure followed by a string of U’s similar to a ρ-independent transcriptional terminator. Using an in vitro transcription system, Jaurin et al. determined that less than one fourth of the transcripts initiated at the promoter escape attenuation (182). This mechanism suggested the low expression level of Escherichia coli β-lactamase.
Unlike the ampC of Escherichia coli K-12, the expression of Enterobacter cloacae ampC was repressed in the absence of β-lactam antibiotics, and induced in the presence of β-lactam agents (183, 184). Intriguingly, the Enterobacter cloacae AmpC did not hydrolyze the third generation cephalosporins (185). However, the authors concluded that β-lactamase expression was solely responsible for resistance towards cefoperazone, cefotaxime, ceftriaxone, ceftizoxime, ceftazidime and moxalactam because the cloning of the ampC gene into Escherichia coli conferred an identical resistant pattern as Enterobacter cloacae (185).
The β-lactamase gene of Citrobacter freundii was first cloned and transferred to Escherichia coli as a 1.5-kb DNA fragment (186). The presence of this fragment in Escherichia coli conferred a similar resistance profile to third generation of cephalosporins as the Citrobacter freundii AmpC. Subsequently, the ampC of Citrobacter freundii strain OS60 was sequenced and demonstrated to encode a 380 amino acid polypeptide with a 19 residual signal peptide (187). The mature protein had a molecular weight of 39.8 kDa. It shared 77 % amino acid identity with Escherichia coli K-12 AmpC (187).
The ampC of Pseudomonas aeruginosa PAO1 was cloned and sequenced in the 1990’s (163). Sequence analysis of the cloned 1.3 kb Pseudomonas aeruginosa chromosomal DNA showed the presence of an ampC gene with a good homology to Enterobacter cloacae and Citrobacter freundii AmpC proteins (163). When the putative ampC gene was transformed into a non-β-lactamase-producing Pseudomonas aeruginosa strain, there was detectable chromogenic activity (163). Western blotting using anti-Pseudomonas aeruginosa AmpC antibody indicated that Pseudomonas aeruginosa AmpC had an approximate molecular weight of 35.5 kDa (188). However, using crude protein extracts, the isoelectric point (pI) of Pseudomonas aeruginosa β-lactamases was shown to be heterogenous (189). Multi-step purification of the β-lactamase enzymes followed by protein sequencing suggests that the C-terminal truncations of AmpC might cause the variations in isoelectric point (190). However, Gates et al. proposed that multiple β-lactamase genes might be present in Pseudomonas aeruginosa (191).
Similar to Citrobacter freundii and Enterobacter cloacae, the expression of Pseudomonas aeruginosa chromosomal β-lactamases could be induced in the presence of β-lactam antibiotics (191). Examination of a large number of Pseudomonas aeruginosa clinical strains revealed four patterns of β-lactamase expression: low-basal, inducible; moderate-basal, inducible; moderate-basal, constitutive; and high-basal, constitutive (189). Since the existence of PoxB was not known, and the biochemical assays used were not sensitive enough to distinguish these two β-lactamases, it was presumed that AmpC was overexpressed in the hyperproducering clinical isolates (189). A high constitutive expression of β-lactamase seen in Pseudomonas aeruginosa strains with ampR mutation suggests that the clinical Pseudomonas aeruginosa isolates that are hyperproducing β-lactamase may harbor poxB and a mutation in ampR (166). A Southern blot analysis of a few of these clinical Pseudomonas aeruginosa isolates with ampR-and ampC-specific probes showed no gross changes in the ampR-ampC intergenic DNA (192). This led the authors to rule out any changes in ampR and ampC genes, although this technique is not sensitive enough to detect minor deletions or point mutations (192). In fact, one of these Pseudomonas aeruginosa β-lactamase hyperproducers was shown to have a point mutation in ampR (193). It is not clear if this clinical isolate harbors the poxB gene, though it has been demonstrated that poxB is fairly ubiquitous and is found in chromosomes of both clinical and environmental Pseudomonas aeruginosa isolates (166).
ampR
Unlike Escherichia coli, the AmpC β-lactamases of Citrobacter freundii, Enterobacter cloacae and Pseudomonas aeruginosa show differential expression in the absence and presence of β-lactam antibiotics, suggesting that the regulatory systems governing ampC gene expression are very different from that of Escherichia coli. Bergstrom et al. showed that in Citrobacter freundii, the ampC gene was in close proximity to the fumarate reductase operon (frd) as in Escherichia coli (194). However, whereas the frd operon and ampC overlapped in Escherichia coli, they were separated by a 1100-bp region in Citrobacter freundii (194). This region appeared to contain the regulatory element required for the inducibility of AmpC (194). Indeed, when this 1100-bp region was cloned independently from the ampC gene, it repressed the expression of ampC, suggesting that it encoded a trans regulatory element (174). Using a minicell system, the small 1-kb region was also demonstrated to express a 31-kDa protein, known as AmpR (174). Notably, as compared to the absence of ampR, the presence of Citrobacter freundii ampR resulted in more than two-fold increase in Citrobacter freundii AmpC β-lactamase activity in the presence of β-lactam agents in Escherichia coli (174). This indicated that AmpR might have a dual role in β-lactamase expression, as a repressor in the absence of an inducer, and as an activator in the presence of the inducer (174).
Similar to Citrobacter freundii, in Enterobacter cloacae, the frd operon and ampC gene do not overlap (194). Using a minicell system, Honore et al. and Lindberg et al. demonstrated that the region between the frd operon and ampC gene encoded for a polypeptide of 31.5 kDa (175, 195). Consistent with the nomenclature in Citrobacter freundii, this gene was called the ampR. Under non-induced condition, Enterobacter cloacae ampC expression was constitutively low in Escherichia coli, albeit the presence of ampR (195). This observation suggested that unlike Citrobacter freundii AmpR, Enterobacter cloacae AmpR might not act as a repressor of ampC in the absence of an inducer. The repressive function of Citrobacter freundii AmpR was further confirmed using Citrobacter freundii ampC in an Escherichia coli host, in which the synthesis of Citrobacter freundii AmpC β-lactamase was eleven-fold lower in the presence of the homologous ampR gene than when complementing with ampR from Enterobacter cloacae (195).
However, the inductive role of Citrobacter freundii and Enterobacter cloacae ampR gene product was indistinguishable because the Enterobacter cloacae AmpC β-lactamase was elevated approximately 42-fold and 40-fold, respectively, when the Enterobacter cloacae or Citrobacter freundii ampR was present in trans (195). So, though it is highly homologous, Citrobacter freundii and Enterobacter cloacae AmpR exhibited different properties in the regulation of cognate ampC gene.
The ampR gene encodes a DNA-binding protein that belongs to the LysR family of regulatory proteins (196). The LysR family comprises of small sized, autoregulatory transcriptional regulators. They have a helix-turn-helix motif at the N-terminus and substrate-binding domain(s) at the C-terminus. The function of the DNA-binding motif depends upon binding of the substrate-binding domain to small molecules as co-inducers or co-repressors (196). Mutational studies and amino acid sequence similarities identify: (a) a DNA-binding domain employing a helix-turn-helix motif (residues 1–65), (b) domains involved in co-inducer recognition and/or response (residues 100–173 and 196–206), (c) a domain required for both DNA binding and co-inducer response (residues 227–253;(196)).
Gel-mobility-shift assay is a common way to examine the interaction of a DNA-binding molecule to a DNA fragment. Crude cellular extracts of Escherichia coli harboring the Citrobacter freundii ampR in trans was demonstrated to retard the radioactively-labeled ampR-ampC intercistronic region (197). No significant difference in mobility shift was detected when extracts from the wild type Escherichia coli was used. This retardation in gel mobility clearly indicated that the AmpR is a DNA-binding protein (197). It interacts specifically with the ampR-ampC intercistronic region and thus regulates the expression of ampC (197).
In order to the determine the DNA binding region more precisely, a more refined technique, DNase I footprinting, is often used. DNase I footprinting using cellular extracts from Escherichia coli expressing Citrobacter freundii ampR, a DNA segment of 38-bp was indicated to bind to AmpR (197). This 38-bp region was located immediately upstream of the ampC promoter. The addition of β-lactam antibiotics did not change the footprinting properties of AmpR (197). However, a slight change the footprinting of Citrobacter freundii AmpR with muramyl pentapeptide has been observed and discussed in detail below (198).
The 38-bp AmpR binding site overlaps the AmpR promoter (197). This led to the postulation that AmpR binding represses its own expression, an auto-regulatory mechanism frequently seen with other LysR transcriptional factors (196). However, the auto-regulatory mechanism of ampR was not consistent. In an in vivo system using an Escherichia coli heterologous host, the PampR-lacZ activity was repressed three-fold in the presence of Citrobacter freundii ampR. (174). However, this mode of regulation was lost in a minicell system with Enterobacter cloacae AmpR (195). The latter concurred with studies of Citrobacter freundii and Pseudomonas aeruginosa ampR: the amount of Citrobacter freundii AmpR expressed in Escherichia coli minicells remains constant in the absence and presence of inducer (174), constitutive ampR expression is low with a short half-life mRNA (175), and that the ampR transcription (199), (197) and the level of AmpR remains unaffected in the absence and presence of antibiotics (197).
The sequence of the 5′-end of Pseudomonas aeruginosa ampR was initially reported along with the ampC gene and subsequently, the whole sequence was described (163, 200). Based on the sequence analysis, the Pseudomonas aeruginosa ampR gene product was predicted to be a polypeptide of 323 amino acids with a helix-turn-helix motif at the N-terminus (200). By aligning sequences with those of Enterobacter cloacae and Citrobacter freundii, Lodge et al. proposed that the translation initiation codon of the Pseudomonas aeruginosa ampR was a rare TTG codon, rather than the usual ATG start codon (163). Overall, this rare TTG codon accounts for only 1 % of the known initiator sequences (201). The derived amino acid sequence showed a high degree of identity with Enterobacter cloacae AmpR (62.5 %) and Citrobacter freundii AmpR (58.3 %; (200)). The binding motif of Citrobacter freundii AmpR was also present in the ampR-ampC intercistronic region of Pseudomonas aeruginosa (200). In a gel mobility-shift assay, crude cell extracts of Escherichia coli containing the Pseudomonas aeruginosa ampR was shown to bind to the region ~50 bp upstream of ampR, but not to the promoter region of ampC (200).
Recent work on Pseudomonas aeruginosa AmpR shows that it is a global regulator because the strain carrying the ampR mutation produced higher levels of pyocyanin and LasA protease, and lower levels of LasB elastase (199). The production of LasA protease has been correlated with the LasI-LasR quoromone system (202, 203). In the Pseudomonas aeruginosa ampR mutant, the increase in LasA levels positively correlated with the lasI/R quorum sensing gene expression (199). The reduction in the LasB activity was positively correlated with the rhlI/R quorum sensing gene expression. Thus, AmpR plays a dual role, positively regulating ampC, lasB, and rhlR expression, and negatively regulating poxB, lasA, lasI, and lasR expression (199).
ampD and ampE
When Escherichia coli K-12 harboring Citrobacter freundii ampR-ampC was grown with high concentration of cefotaxime, chromosomal mutants producing high level of β-lactamase constitutively arose at a considerable rate (10−6 to 10−7; (174). Analyses of these mutants showed the presence of an intact functional ampC-ampR genetic region, and the phenotype could not be complemented using a Citrobacter freundii frd-ampC region, suggesting the presence of other regulatory genes (174). In addition, when the frd-ampC region of a β-lactamase overproducing Enterobacter cloacae strain was transformed into Escherichia coli, an inducible expression of AmpC β-lactamase was observed, ruling out any loss of integrity of the frd-ampC region (204). These led to the hypothesis that the constitutive β-lactamase overproduction was due to loss of function in a gene required for inducibility that resided outside of ampR-ampC in the chromosome of Escherichia coli (205). In addition, the constitutive overproduction of β-lactamase was lost in the absence of ampR, suggesting that the function of the gene was ampR-dependent (174).
The mapping of this novel mutation was a tedious task. Using a series of Tn10 transposon mutageneses and Hfr strains, the mutation in Escherichia coli was localized to the nadC-lpd region (205). An Escherichia coli strain with a deletion at nadC-aroP region also showed constitutive production of Citrobacter freundii AmpC β-lactamase, suggesting that a gene necessary for inducibility was located within this region (205). By supplementing the nadC-aroP region in trans, the inducible β-lactamase phenotype could be restored from Escherichia coli strains that overproduced the enzyme (205). Using a similar approach as Lindberg et al. (205), Honore et al. identified that a loss of Escherichia coli nadC-aroP region also caused an overproduction of Enterobacter cloacae AmpC (206).
Dideoxy-sequencing of the nadC-aroP region of Escherichia coli revealed the presence of two ORFs (206, 207). The upstream ORF was designated ampD, overlapping the downstream ampE. A S1 nuclease protection assay confirmed that the two ORFs constituted an operon (206). Primer extension analysis performed on ampDE transcript showed that the 5′-end of the ampD mRNA was localized to an adenosine residue 29 nucleotides upstream from the translational start codon (206). This knowledge of the transcript start site enabled the promoter to be pinpointed. The promoter of the ampDE operon did not possess a canonical -35 region, recognized by the σ70 RNA polymerase holoenzyme. The -10 region (5′-TACTCT-3′) resembled the consensus sequence and was preceded by a TG motif at -12 (206). The TG motif has been reported to greatly strengthen promoters lacking the consensus -35 element (208, 209).
The Escherichia coli ampD gene product is 183 amino acids long with an apparent molecular weight of 25 kDa. The Escherichia coli AmpE, with 284 amino acids, has its putative initiation codon overlapping the ampD stop codon. Escherichia coli AmpE has an apparent molecular weight of 28 kDa (207). Using a minicell fractionation technique, AmpD was demonstrated to localize in the cytoplasm. This was in agreement with its hydopathy profile that is indicative of a hydrophilic protein (207).
Fractionation of minicell harboring Escherichia coli ampE showed that AmpE was located in the inner membrane fraction. The AmpE hydrophobicity plot suggested the presence of four transmembrane regions: residues 44–60, 68–84, 195–211 and 268–284 (207). The primary sequence of Escherichia coli AmpE possessed two of the three motifs found in β-lactam binding proteins (210). These are the Ser-Arg-Asn corresponding to the Ser-X-Asn motif, and the Ser-Met-Ala-Lys corresponding to the Ser-X-X-Lys motif (206). However, Escherichia coli AmpE failed to bind to any β-lactam antibiotics (207). In addition, AmpE could be an ATP-binding protein because the Walker’s ATP-binding consensus is found at residues 74–87 and 184–198 (207). The function of AmpE in Escherichia coli is not clear, however, unpublished data suggests that it plays a role in D-Ala D-Ala murein recycling (40).
Phenotypically, the loss of Escherichia coli ampD caused a constitutive overproduction of AmpC β-lactamase that could be further induced by the addition of an inducer, suggesting that ampD acts as a negative regulator of ampC. This was in contrast to the loss of Escherichia coli ampD and ampE that caused high level of β-lactamase production constitutively, and the level could not be increased any further, albeit the addition of an inducer (207). Moreover, the complementation of Escherichia coli ampE in an ampDE mutant background restored only 50 % of the Enterobacter cloacae AmpC β-lactamase induction (206). These data strongly suggest that ampE also played a role in the inducibility of ampC. However, intriguingly, the inducible phenotype of the ampDE mutant could be complemented with ampD alone (207). This could have been due to overproduction of AmpD from a multicopy plasmid.
At this point, it is noteworthy that all the extensive molecular studies were performed using Escherichia coli ampD and ampE, which complemented the constitutive expression of Citrobacter freundii and Enterobacter cloacae β-lactamases (205–207). Thus far, no homologous system has been developed to understand the mechanism. Hence, little is known about the Citrobacter freundii and Enterobacter cloacae ampDE genes, except for the presence of ampDE homologs in these organisms (205, 211). Sequencing of the wild-type ampD gene of Citrobacter freundii and Enterobacter cloacae revealed that they are 564 bp, corresponding to a gene product of 187 aminoacid long (212). Sequence comparison revealed that they share 74 to 83 % homology to Escherichia coli ampD at the nucleotide level (212). This level of homology is similar to those found for their respective ampC and ampR genes, reflecting that these molecules evolved simultaneously (162, 175, 212). More importantly, the three AmpD proteins are highly homologous except for their carboxyl termini with a loss of four amino acids in Escherichia coli AmpD. As compared to Escherichia coli, Enterobacter cloacae and Citrobacter freundii AmpD carry four additional amino acids yielding a Ser-X-X-Lys motif typically found in β-lactamases and penicillin-binding proteins (210). However, AmpD is unlikely to belong to this family of proteins because other signature motifs of β-lactam binding proteins are not present (212). To determine the importance of the carboxyl terminus of Enterobacter cloacae AmpD, a stop codon was introduced, giving rise to a truncated ampD gene product lacking the 16 carboxyl-terminal residues (212). Though the truncated AmpD protein is stable, the truncated protein failed to complement the Escherichia coli ampD mutation, indicating that the carboxyl terminus of Citrobacter freundii and Enterobacter cloacae AmpD was crucial for its function (212). On the other hand, so far, the identity of Citrobacter freundii and Entrobacter cloacae ampE remains unreported, however, our analysis of the sequenced Citrobacter rodentium genome revealed the presence of an AmpE homolog.
Mutation of ampD gene caused an uncontrolled expression of AmpC β-lactamase, leading to the hypothesis that AmpD was most likely a negative regulator of ampC transcription. However, the primary protein structure of AmpD had no characteristic helix-turn-helix motif or Zn-binding finger, two well-known DNA-binding motifs (206). So, it was postulated that AmpD might not affect the transcription of ampC directly, but it might act through the transcription factor AmpR since the constitutive expression required ampR (174). In addition, it has been speculated that AmpD played a role in murein metabolism since Escherichia coli strains with defective ampD lost up to 40 % of their cell wall per generation (213, 214). The function of AmpD was elucidated when a highly purified functional AmpD protein was incubated with Escherichia coli sacculi (215). The presence of AmpD decreased the relative amount of all anhydromuropeptides, suggesting that AmpD was involved in the metabolism of murein (215). Using a more refined isolate of muropeptides, AmpD was shown to degrade anhydro-N-acetylmuramyl-L-Ala-D-Glu-DAP (anhMurNAc-tripeptide) over time, giving rise to anhMurNAc and tripeptide (216). Hence, AmpD protein cleaved the amide bond between the carboxyl group of anhydro muramic acid and the α-amino group of L-Ala-D-Glu-DAP (216). AmpD appeared to be specifically cleaving the 1,6 anhydro bond on the muramic acid since MurNAc-pepides, and the cytoplasmic precursors of murein, UDP-MurNAc-peptides, were not cleaved by AmpD (216). This unique 1,6 anhydro bond was the results from an intramolecular transglycosylation reaction that occurred when the peptidoglycan was degraded by the periplasmic Escherichia coli lytic transglycosylases (217). Murein precursors did not contain the 1,6 anhydro bond, allowing AmpD to distinguish the precursors from the murein degraded products. This functional specificity of AmpD was similar to the membrane-bound N-acetyl-anhydromuramyl-L-alanine amidase of Escherichia coli (218). However, instead of interacting with the cytoplsmic membrane, AmpD is a cytosolic N-acetyl-anhydromuramyl-L-alanine amidase (216).
Besides Citrobacter freundii, Escherichia coli and Enterobacter cloacae, the ampDE region had also been determined in Pseudomonas aeruginosa. The Pseudomonas aeruginosa ampDE region was detected using a degenerate probe against a phage genomic library of PAO1 (219). This strategy located ampD gene to a 2.6-kb genomic region. Sequencing of this 2.6-kb DNA fragment revealed two ORFs (219). The Pseudomonas aeruginosa ampD gene was 567-bp long, corresponding to a gene product of 188 amino acids with a predicted molecular mass of 21 kDa. The deduced Pseudomonas aeruginosa AmpD protein exhibited 65, 63 and 62 % similarity to the Escherichia coli, Citrobacter freundii and Enterobacter cloacae AmpD proteins, respectively (219). However, our analysis shows that although the carboxyl terminus of Citrobacter freundii, E. cloacace and Pseudomonas aeruginosa AmpD proteins are highly conserved, the Ser-X-X-Lys motif found at the carboxyl terminus of Citrobacter freundii and Enterobacter cloacae was not found in Pseudomonas aeruginosa AmpD.
Similar to the gene organization in Escherichia coli, the Pseudomonas aeruginosa ampE start codon overlapped the translational termination codon of ampD. Pseudomonas aeruginosa ampE encoded a 278-aminoacid polypeptide with predicted molecular weight of 31 kDa (219). The predicted Pseudomonas aeruginosa AmpE gene product had the characteristic of a cytoplasmic membrane protein with five predicted transmembrane domains. However, it showed a relatively low degree of homology (33 %) to the counterpart in Escherichia coli (219).
In spite of the sequence, virtually nothing is known about the molecular biology of Pseudomonas aeruginosa ampDE. Similar to Escherichia coli, Pseudomonas aeruginosa ampDE was believed to form an operon and transcript simultaneously. Unfortunately, without accompanying primer extension analysis or any techniques to determine the 5′-end of the ampDE transcript, Langaee et al. claimed that the putative -10 and -35 promoter consensus were 14 and 36 bp, respectively, upstream of a putative Shine-Dalgarno sequence of ampD (219). However, without any conventional techniques, the authentic promoter of Pseudomonas aeruginosa ampDE remains elusive. In addition, our analysis shows the absence of any strong ρ-independent terminator at the 3′-end of ampE, suggesting that the termination of the transcript may be ρ-dependent.
To determine the function of Pseudomonas aeruginosa ampD and ampE gene product, two Escherichia coli strains, ampD and ampDE mutants, harboring Enterobacter cloacae ampR-ampC genes in trans were used for complementation analysis (219). In a heterologous system using components from three bacterial species, Pseudomonas aeruginosa ampD alone and ampDE were exhibited to restore the wild-type expression of Enterobacter cloacae AmpC β-lactamase, low basal level and inducible expression in the presence of β-lactam antibiotics, in the Escherichia coli background regardless of nature of the mutations (219). However, the β-lactamase induction was reduced two-fold and 1.5-fold in the presence of Pseudomonas aeruginosa ampE, as compared to the absence of ampE and Escherichia coli ampE, respectively (219).
In a subsequent analysis of Pseudomonas aeruginosa ampDE, a polar gentamycin cassette was inserted within the ampD gene, generating an isogenic Pseudomonas aeruginosa ampDE mutant, notwithstanding the author’s claim that it was an ampD-defective strain (220). The ampDE mutation of Pseudomonas aeruginosa led to six- and 32-fold increases in the minimum inhibitory concentration (MIC) of ampicillin and cefotaxime, respectively, as well as a 37-fold elevation of the basal non-induced β-lactamase level (220). Unlike Escherichia coli where the loss of both ampD and ampE caused a non-inducible high level of β-lactamase synthesis (207), the β-lactamase production of Pseudomonas aeruginosa ampDE mutant was induced 22-fold in the presence of an inducer (220). This discrepancy suggested that Pseudomonas aeruginosa ampD and/or ampE might play a different role than those of Escherichia coli. However, no complementation analysis was done to confirm their finding (220). Some of these discrepancies could be explained by the recent realization that Pseudomonas aeruginosa encodes three ampD homologs, ampD, ampDh1, and ampDh2, and these are responsible for step-wise upregulationof β-lactamase expression (221). Although single or double mutants of the ampD homologs do not affect Pseudomonas aeruginosa fitness in vitro, deletion of all three genes does (222). This cost of fitness is due to upregulation of ampC because ampC deletion restores fitness (222). Interestingly, in a mouse model, ampD ampDh2 or ampD ampDh3 double mutants as well as the triple mutant affect fitness. This effect of the triple mutant is ampC independent. Of the three, ampDh3 plays the greatest role in virulence in a mouse model system; an ampD ampDh2 double mutant also has reduced virulence (222). This study also looked at the ampD homologue sequences of β-lactamase wild type and overproducing pairs of clinical isolates from ten different subjects (222). Although six of the overproducing strains contained mutations in ampD, loss of function mutations were not identified in ampDh2 or ampDh3, consistent with a high biological fitness cost of the double mutants (222). These findings also suggest that the four groups of β-lactamase producers identified by Campbell et al (189) may harbor mutations in the various ampD genes.
ampG
In comparison to other amp genes, the ampG gene was the least characterized. The ampG was discovered when an Enterobacter cloacae strain 55M-L was reported to produce constitutive low-levels of β-lactamase (223). This intriguing strain was derived from an ampD parental strain, Enterobacter cloacae 55M, which synthesized a high basal level of β-lactamase constitutively (223). Complementation analyses with wild-type ampR-ampC and ampD failed to recover the parental phenotype (224). Furthermore, the 55M-L ampR-ampC and ampD regions retained the activities of the parental strain, suggesting that a novel locus, known as ampG, which was unlinked to ampR-ampC or ampD, was responsible for the low-basal β-lactamase level (224). Based on the assumption that low-level constitutive expression of β-lactamase in Enterobacter cloacae was due to a novel mutation in ampG, it was proposed that a wild-type ampG allele of the gene should be dominant over the mutated allele (224). If this was correct, the introduction of the wild-type ampG allele should change Enterobacter cloacae 55M-L into a resistant strain since it was mutated in the ampD gene (224). A random genomic library was constructed using a wild-type Enterobacter cloacae chromosome and transformed into Enterobacter cloacae strain55M-L. Selection of the resistant transformants showed that a 2.9-kb fragment was able to transform Enterobacter cloacae 55M-L into a β-lactam resistant strain that produced β-lactamase as high as the parental strain, 55M (224). The presence of this 2.9-kb fragment along with the ampD gene in Enterobacter cloacae M55 re-established the inducibility of the AmpC β-lactamase (224). Since a mutation in ampG led to an effect that was dominant over the phenotypes of ampR and ampD, it was proposed that AmpG might play a role upstream of AmpR and AmpD in the induction of AmpC β-lactamase (224).
The presence of an ampG locus in Escherichia coli was confirmed through Southern blotting using the Enterobacter cloacae 2.9-kb ampG fragment (225). A 3.2-kb fragment containing ampG from Escherichia coli was able to complement Enterobacter cloacae 55M-L (225). An ampG knockout mutant was generated in Escherichia coli using allelic replacement via homologous recombination (225). The resulting ampG mutant was unable to induce Citrobacter freundii AmpC β-lactamase in the presence of an inducer, confirming that the induction of Citrobacter freundii AmpC required a functional ampG gene (225).
Sequencing of the Escherichia coli 3.2-kb fragment showed the presence of two ORFs flanked by an upstream bloA gene and a downstream cyoA gene, which encoded a morphogene that affected carboxypeptidase activity, and the cytochrome o ubiquinol oxidase, respectively (225). The deduced AmpG protein sequence yielded a 491 amino acid polypeptide with a predicted molecular mass of 53.2 kDa. However, minicells carrying Escherichia coli ampG could not detect the presence of AmpG in the cytoplasmic fraction (225). Bioinformatic analysis and fusion analysis suggested the existence of 10 membrane-spanning regions in the ampG gene product (225, 226).
Upstream of ampG, there was an ORF of 579-bp in the same orientation. This preceding ORF might encode a putative lipoprotein since it contains a strong lipoprotein signal of Leu-X-Gly-Cys (225). A polar insertional inactivation of this ORF showed a non-inducible phenotype of Escherichia coli harboring Citrobacter freundii ampR-ampC, similar to an ampG mutant (225). But, the β-lactamase inducibility was regained when the strain was complemented with ampG alone or orf-ampG, suggesting that the lipoprotein played no role in β-lactamase induction (225).
When the Escherichia coli ampG gene was cloned with the upstream intergenic region, a promoter must be inserted upstream in order to complement a chromosomal ampG mutation. This strongly suggested that the ampG promoter was not immediately upstream (225). Furthermore, the short 43-bp intracistronic sequence did not contain any Escherichia coliσ70 promoter consensus, suggesting that ampG was co-transcribed with an upstream ORF of 495 bp (225). A northern blotting analysis using an ampG probe showed a band of 2.4 kb corresponding to the transcript size covering both the ampG and ORF, confirming the hypothesis that ampG and its upstream ORF constituted an operon (225). Using primer extension analysis, the 5′-end of the transcript was mapped to a G residue, 43 bp upstream of the proposed AUG start codon of the upstream ORF (225). With this information in hand, the determined promoter of the ampG operon showed high homology to the consensus sequence for RpoD promoters (225).
However, interestingly, when a probe hybridizing to the upstream ORF was used in the northern blot, two transcripts of 0.7-kb and 2.4-kb in size were found (225). The 2.4-kb band corresponded to the transcript of both the ampG and the upstream ORF, whereas the 0.7-kb transcript coincided with the expected size of the upstream ORF alone (225). This suggests that although the two genes were transcribed in an operonic manner, expression of Escherichia coli ampG was post-transcriptionally regulated. Examination of the intergenic region between the ORF and ampG revealed the presence of a short inverted repeat of 11 bp upstream of the start codon of ampG (225). This inverted repeat was believed to function as a transcriptional terminator. In order to further determine the regulation of the ampG gene, two promoter fusions, Porf-lacZ and PampG-lacZ, were constructed (225). It was indicated that the β-galactosidase activity of Porf-lacZ was always 1.5-fold higher than the transcription of PampG-lacZ, confirming the northern blot data that there was down regulation of ampG transcription (225). However, the ampG transcription was not affected by AmpD or different growth phases (225).
Although AmpG protein appeared to be a transmembrane protein, the function of AmpG remains elusive. The fact that Escherichia coli contains ampG and ampD genes, even though this organism lacks the ampR gene and an inducible ampC gene, provided clues that the primary functions of AmpG and AmpD proteins were not involved in AmpC β-lactamase induction (175, 205, 225). The initial clue of AmpG function came from Jacobs et al., who demonstrated that Escherichia coli strains with a defective ampG gene lost 20–40 % of the labeled muropeptides from their cell wall in each generation, whereas the control strain lost only 4–8 % per generation (213). In addition, a large proportion of the labeled muropeptides was released into the medium (213). These data indicated that Escherichia coli strains with a defective ampG gene were unable to reuse the cell wall components during cell regeneration. This data together with the fact that AmpG was a transmembrane protein suggest that AmpG protein acted as a permease that allowed the transport of peptidoglycan recycling intermediates from the bacterial periplasm to the cytoplasm (213).
To ascertain the specific function of AmpG, hot water extracts of the DAP-labeled wild-type Escherichia coli and a Escherichia coli strain with defective ampG were analyzed using high pressure liquid chromatography (HPLC) (213). It was demonstrated that the ampG mutant failed to retain anhMurNAc-peptides in the cytoplasm. However, the presence of anhMurNAc-peptides, the AmpD substrates, remained peculiar because the unprocessed enzymatic products of cell wall hydrolyases were GlcNAc-anhMurNAc-peptides (213). Thus, the ampG gene product could be transporting either the GlcNAc-anhMurNAc-peptides or anhMurNAc-peptides into the cytoplasm. To determine whether GlcNAc-anhMurNAc-peptides were cleaved in the periplasm or cytoplasm, the culture medium of the ampG mutant was assayed. A large amount of GlcNAc-anhMurNAc-peptides was found in the culture medium of Escherichia coli ampG and not in the wild-type, suggesting that AmpG protein was a transporter of GlcNAc-anhMurNAc-peptides from the periplasm to the cytoplasm (213). The GlcNAc-anhMurNAc-peptides were then cleaved in the cytoplasm by an intracellular N-acteyl-glucosaminidase (227, 228).
In a different experiment, the substrate specificity of AmpG was investigated (229). Using different GlcNAc-anhMurNAc-peptides of various lengths, it was shown that the principal requirement of AmpG was the presence of GlcNAc-anhMurNAc disaccharide, independent of the length of the peptide side chain (229). Muropeptides lacking either GlcNAc or anhMurNAc were not imported into the cytosol. In addition, the function of AmpG was sensitive to carbonyl cyanide m-chlorophenylhydrazone (CCCP), an H+ ionophore that dissipates the H+ gradient and uncouples electron transport from ATP synthesis (230), suggesting that AmpG transport is dependent on the proton motive force.
Although the ampG gene was first detected in Enterobacter cloacae (224), all the molecular and biochemical work were performed on the Escherichia coli ampG gene and its gene product (213, 225). The ampG homolog of Enterobacter cloacae remains to be characterized. Analysis of sequenced Pseudomonas aeruginosa PAO1 genome shows the presence of two copies of ampG, currently designated as ampG and ampP (Kong et al. manuscript submitted). Both the permeases are essential for β-lactamase induction.
MUREIN RECYCLING AND β-LACTAMASE INDUCTION
The most important issue in the study of β-lactamase induction is the coordinate regulation of the presence of β-lactam antibiotics in the periplasm, and the intracellular activation of β-lactamase. β-lactam antibiotics are known to act on the murein localized in the periplasm, thus these chemicals do not enter the cytoplasm to mediate their effects. Yet, the presence of β-lactam agents is able to induce β-lactamase, suggesting that an efficient communication must exist between the extracellular site of β-lactam actions and the intracellular site of β-lactamase transcription. Because of structural homology between β-lactam ring and D-alanyl-D-alanine, Tuomanen et al. proposed that the pathway that leads to the induction of β-lactamase expression may share regulatory components with the pathway of cell wall metabolism (214). If this is true, the authors postulated that mutants with altered β-lactamase induction must have aberrant cell wall metabolism. To test the hypothesis, the difference in peptidoglycan composition between wild-type Escherichia coli and an isogenic mutant with defective ampDE was determined using HPLC (214). Interestingly, it was shown that the muropeptide composition in ampDE mutant was profoundly altered as compared to the wild-type. However, this alteration could be recovered by complementing the ampD alone in trans, suggesting that ampE played a subtle role in this event (214). Thus, it is proposed that the ampD gene product repressed the β-lactamase activity by retaining the concentration of GlcMurNAc-L-Ala-D-Glu-meso-DAP-D-Ala-D-Ala (214). Two important phenomena converged in this study: murein recycling and β-lactamase induction.
The current molecular model of β-lactamase induction was proposed (Fig. 6). According to this model, during cell growth, specific cell wall hydrolyases degrade the peptidoglycan to yield murein-peptides with different lengths. This muropeptides (Glc-anhMurNAc-peptides) are transported into the cytoplasm by the permease AmpG. They are then directly cleaved by AmpD, an N-acetylmuramyl amidase, into the N-acetylmuramic acids and peptides (Fig. 6). During normal physiologic growth, ampC expression is repressed by AmpR binding to the promoter region (Fig. 6; (198)). Jacobs et al. demonstrated that in the presence of β-lactam antibiotics, there was a relative decrease in the murein precursors, UDP-MurNAc-L-Ala-D-Glu-meso-DAP-Ala-Ala (213). It was postulated that the Citrobacter freundii AmpR was maintained in the repressive form by the murein precursors. This was indeed demonstrated with purified AmpR in an in vitro transcription assay that the ampC transcript diminished with an increase of UDP-MurNAc-pentapeptide (198). In addition, the negative effect of UDP-MurNAc-pentapeptide in β-lactamase induction was also investigated by using an AmpR mutant, AmpRGly102Glu (198). Expression of this AmpR variant resulted in a high basal-level constitutive β-lactamase expression in the absence of β-lactam agents in wild-type as well as in an ampG mutant (231). In the transcription assay, purified AmpRGly102Glu behaved like the wild-type protein in the activation of ampC transcription. However, increasing concentrations of UDP-MurNAc-pentapeptide failed to reduce the level of AmpRGly102Glu-mediated ampC transcription (198). This result clearly indicated that an UDP-MurNAc-pentapeptide binding-site was present in AmpR around amino acid 102.
Fig. 6.

Mechanism of β-lactamase induction in Enterobacteriaceae. In the absence of β-lactam antibiotics, the murein is constantly being re-modeled for cell elongation and cell division. The breakdown products, muramyl peptides, are released into the periplasm and transported to the cytoplasm via AmpG. These muramyl peptides are then processed by ampD for the murein recycling. In this instance, the ampC gene is kept at basal-level by a suppressed AmpR transcriptional regulator. In the presence of β-lactam antibiotics, the homeostasis is disrupted because these antibiotics target the murein resulting in an excessive breakdown of the murein and an accumulation of the muramyl peptides both in the periplasm and cytoplasm. However, the AmpD protein is the limiting factor. An accumulation of the muramyl peptides renders the activation of AmpR, which then induces the production of AmpC β-lactamase. It is not clear which of the muramyl products directly interact with AmpR. AmpE has been implicated in the transport of free pentapeptide to the periplasm (40). The transport of the muramyl peptides into the periplasm may involve AmpE or some yet to be unidentified protein. Abbreviations, CM, IM, PP, PG and OM refer to cytoplasm, inner membrane, periplasm, peptidoglycan and outer membrane, respectively.
On the other hand, gel mobility-shift assay showed no difference in the DNA binding property between the wild-type AmpR and AmpRGly102Glu (198). Detailed study carried out using DNaseI footprinting also gave rise to an identical footprint (198). However, the presence of UDP-MurNAc-pentapeptide resulted in a small but consistent alteration in the protection pattern of wild-type AmpR, but not AmpRGly102Glu (198). Specifically, the base in position -49 (relative to the +1 site of ampC) was not protected by the wild-type AmpR in the presence of UDP-MurNAc-pentapeptide, though the significance of this nucleotide was not discussed (198).
The identification of a repressing ligand of AmpR was not sufficient to explain the induction of ampC transcription because the increased pool of UDP-MurNAc-pentapeptide in an ampD mutant exhibited a high basal-level of β-lactamase expression constitutively (213). It was then proposed that a different ligand may be responsible for the co-induction of ampC transcription. In this case, anhMurNAc-tripeptide served as a potential candidate since it dramatically accumulated in an ampD mutant (213). In an in vitro transcription experiment, anhMurNAc-tripeptide consistently exerted the negative effect of UDP-MurNAc-pentapeptide on ampC transcription (198). Thus, in the presence of β-lactam antibiotics, as a result of increased murein breakdown, high concentration of anhMurNAc-tripeptide displaced the repressing UDP-MurNAc-pentapeptide from its AmpR-binding site (Fig. 5; (198)). However, it was not known if the footprint of AmpR changed with the binding to anhMurNAc-tripeptide.
The delicate coordination of murein breakdown and β-lactamase expression was elegantly demonstrated using a complicated heterologous system (Fig. 5; (198)). Most of the experiments were performed in an Escherichia coli background which carried Citrobacter freundii or Enterobacter cloacae ampR-ampC system. Escherichia coli does not have an inducible AmpC β-lactamase, albeit the presence of ampD and ampG. The Escherichia coli ampC was regulated through an attenuator present at the 5′-end of the ampC transcript (182). β-lactam resistance in Escherichia coli clinical isolates was brought upon by various mechanisms, including tandem amplification of the ampC gene caused by recombination between short homologies (232), mutations to the promoter region that up-regulated ampC expression (233), insertion of an IS element at the promoter that created a strong σ70 promoter (234), and horizontal chromosomal DNA transfer from Shigella (235). Thus far, no data has demonstrated the importance of Escherichia coli ampD and ampG in the transcriptional up-regulation of its cognate ampC. Moreover, Citrobacter freundii and Enterobacter cloacae ampD and ampG have not been sufficiently demonstrated to involve AmpC β-lactamase induction or cell wall metabolism in a homologous system. The Citrobacter freundii ampR gene product is believed to bind to different murein breakdown products and murien precursors present in Escherichia coli. The identity and the physiological concentration of these molecules in Citrobacter freundii, Enterobacter cloacae, Pseudomonas aeruginosa or any other organisms remain to be defined.
CLOSING REMARKS
It is conceivable that the recognition of antibiotics marks one of the most important milestones in human history. The use of antibiotics has brought upon immeasurable benefits to medicine, physiology, and biology. Over the decades, the understanding of antibiotics and the resistance to them has facilitated a deeper understanding of the microbial physiology and pathogenesis. In addition, we have also exploited this knowledge as tools to facilitate the search for new knowledge. For example, the ampicillin resistance gene is routinely used as a selectable marker in recombinant DNA technology. However, as a community, we should also proceed with caution because the misuse and abuse of these substances will lead to unwanted consequences including the return of multi-drug resistant bacteria.
Acknowledgments
The authors would like to thank members of the Mathee lab for thoughtful comments on the manuscript. The work was supported by NIH S06 GM08205 and 5SC1AI081376 (K.M.) and a Florida International University Teaching Assistantship (K.-F.K.).
ABBREVIATIONS
- 6-APA
6-aminopenicillanic acid
- 7-ACA
7-aminocephalospoinic acid
- anhMurNAc
anhydro-N-acetylmuramyl
- ASM
American Society of Microbiology
- ATP
adenosine triphosphate
- CARB
Carbenicillin β-lactamase
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- DAP
diaminopimelic acid
- DNA
deoxyribonucleic acid
- EDTA
ethylenediaminetetraacetic acid
- ESBL
extended spectrum β-lactamase
- frd
fumerate reductase operon
- GlcNAc
N-acetylglucosamine
- HPLC
High performance liquid chromatography
- kb
kilobase
- kDa
kiloDalton
- MIC
minimum inhibitory concentration
- MurNAc
N-acetylmuramic acid
- NIAID
National Institutes of Allergy and Infectious Diseases
- ORF
open reading frame
- PampR
ampR promoter
- PBC
penicillin binding component
- PBP
penicillin binding protein
- PEP
phosphoenolpyruvic acid
- PSE
Pseudomonas-specific β-lactamase
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- UDP-GlcNAc
Uridine diphosphate-N- acetylglucosamine
- UDP-MurNAc
Uridine diphosphate-N-acetylmuramic acid
References
- 1.Bottcher HM. A history of antibiotics. Philadelphia: JB Lippincott Company; 1964. [Google Scholar]
- 2.Burdon S. Memoirs: The origin and distribution of microzymes (bacteria) in water, and the circumstances which determine their existence in the tissues and liquids of the living body. 1871:323–52. [Google Scholar]
- 3.Lister J. A contribution to the germ theory of putrefaction and other fermentative changes, and to the natural history of torulae and bacteria. Trans R Soc Edinburg. 1875;27:313. [Google Scholar]
- 4.Roberts W. Studies of biogenesis. Philos Trans R Soc Lond. 1874;164:457–77. [Google Scholar]
- 5.Tyndall J. The optical deportment of the atmosphere in relation to the henomena of putrefaction and infection. Philos Trans R Soc Lond. 1876;166:27–63. doi: 10.1136/bmj.1.787.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selwyn S. Pioneer work on the “penicillin phenomenon,” 1870–1876. J Antimicrob Chemother. 1979;5:248–55. doi: 10.1093/jac/5.3.249. [DOI] [PubMed] [Google Scholar]
- 7.Pasteur L, Joubert J. Charbon et septicemia. CR Acad Sci Paris. 1877;85:101–15. [Google Scholar]
- 8.Tyndall J. Essays on the Floating Matter of the Air in Relation to Putrefactin and Infection. Appleton; New York: 1881. [Google Scholar]
- 9.Cornil CV, Babes V. Concurrence vitale des bacteries: attenuation de leurs proprietes dan de milieux nutritifs modifies par d’autres bacteries: Tentative de therapeautique. J Conaiss Med Prat Pharmacol. 1885;7:321–3. [Google Scholar]
- 10.Garre C. Uber Antagonisten unter den Bacterien. Corresp Bl Schweiz Aerzte. 1887;17:384–92. [Google Scholar]
- 11.Vuillemin P. Antibiose et symbiose. CR Assoc Fr Av Sci. 1889;2:525–43. [Google Scholar]
- 12.Duchesne E. Contribution à l’étude de la concurrence vitale chez les micro-organismes: antagonisme entre les moisissures et les microbes. Lyon: l’Ecole du Service de Santé Militaire de Lyon; 1897. [Google Scholar]
- 13.Wainwright M. Andre Gratia (1893–1950): Forgotten pioneer of research into antimicrobial agents. J Med Biography. 2000;8:39–42. [PubMed] [Google Scholar]
- 14.Steffee CH. Alexander Fleming and penicillin. The chance of a lifetime? NCMJ. 1992;53:308–10. [PubMed] [Google Scholar]
- 15.Fleming A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Br J Exp Pathol. 1929;10:226–36. [Google Scholar]
- 16.Hare R. New light on the history of penicillin. Med His. 1982;26:1–24. doi: 10.1017/s0025727300040758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chain E. The early years of the penicillin discovery. Trends Pharmacol Sci J. 1979;1:6–11. [Google Scholar]
- 18.Abraham EP, Chain E. An enzyme from bacteria able to destroy penicillin. Nature. 1940;146:837–8. [PubMed] [Google Scholar]
- 19.Abraham EP, Chain E, Fletcher CM, Florey HW, Gardner AD, Heatley NG, et al. Further observations on penicillin. Eur J Clin Pharmacol. 1941;42:3–9. [PubMed] [Google Scholar]
- 20.Henderson JW. The yellow brick road to penicillin: a story of serendipity. Mayo Clin Proc. 1997;72:683–7. doi: 10.1016/S0025-6196(11)63577-5. [DOI] [PubMed] [Google Scholar]
- 21.Richards AN. Statement released by the Committee on Medical Research. J Am Med Assoc. 1943:122. [Google Scholar]
- 22.Fleming A. Streptococcal meningitis treated with penicillin. Measurement of bacteriostatic power of blood and cerebrospinal fluid. Lancet. 1943:434–8. [Google Scholar]
- 23.Florey HW, Florey ME. General and local administration of penicillin. JAMA. 1943;1:387–97. [Google Scholar]
- 24.Hobby GL, Meyer K, Chaffee E. Chemotherapeutic activity of penicillin. Proc Soc Exp Biol Med. 1942;50:285–8. [Google Scholar]
- 25.Keefer CS, Blake FG, Marshall ER, Lockwood JS, Wood WB. Penicillin in the treatment of infections. A report of 500 cases. Statement by the Committee on Chemotherapeutic and other agents, Division of Medical Sciences, National Research Council. JAMA. 1943;122:1217–44. [Google Scholar]
- 26.Rammelkamp CH, Keefer CS. The absorption, excretion, and distribution of penicillin. J Clin Invest. 1943;22:425–37. doi: 10.1172/JCI101412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rammelkamp CH, Keefer CS. Penicillin: Its antibacterial effect in whole blood and serum for the hemolytic Streptococcus and Staphylococcus aureus. J Clin Invest. 1943;22:649–57. doi: 10.1172/JCI101437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Queener SF. History and Origins of Beta-Lactam Antibiotics. In: Queener SF, Webber JA, Queener SW, editors. Beta-Lactam Antibiotics for Clinical Use. New York: Marcel Dekker, Inc; 1986. [Google Scholar]
- 29.Abraham EP, Chain E, Florey HW, Florey ME, Heatley NG, Jennings MA, et al. Historical introduction. In: Florey HW, Heatley CE, Jennings NG, Sanders MA, Abraham AG, Florey EPME, editors. Antibiotics. London: Oxford University Press; 1949. [Google Scholar]
- 30.Behrens OK, Corse J, Huff DE, Jones RG, Soper QF, Whitehead CW. Biosynthesis of penicillins III. Preparation and evaluation of precursors of new penicillins. J Biol Chem. 1948;175:771–92. [PubMed] [Google Scholar]
- 31.Sheehan JC, Henery-Logan KR. A general synthesis of the penicillins. J Am Chem Soc. 1959;81:5838–9. [Google Scholar]
- 32.Fairbrother RW, Taylor G. Sodium methicillin in routine therapy. Lancet. 1961;1:473–6. doi: 10.1016/s0140-6736(61)90058-7. [DOI] [PubMed] [Google Scholar]
- 33.Acred P, Brown DM, Knudsen ET, Rolinson GN, Sutherland R. New semi-synthetic penicillin against Pseudomonas pyocyanea. Nature. 1967;215:25–30. doi: 10.1038/215025a0. [DOI] [PubMed] [Google Scholar]
- 34.Abraham EP, Newton GGF. The structure of cephalosporin C. Biochem J. 1961;79:377–93. doi: 10.1042/bj0790377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagarajan R, Boeck LD, Gorman M, Hamill RL, Higgens CE, Hoehn MM, et al. Beta-lactam antibiotics from Streptomyces. J Am Chem Soc. 1971;93:2308–10. doi: 10.1021/ja00738a035. [DOI] [PubMed] [Google Scholar]
- 36.Imada A, Kitano K, Kintaka K, Muroi M, Asai M. Sulfazecin and isosulfazecin, novel beta-lactam antibiotics of bacterial origin. Nature. 1981;289:590–1. doi: 10.1038/289590a0. [DOI] [PubMed] [Google Scholar]
- 37.Sykes RB, Cimarusti CM, Bonner DP, Bush K, Floyd DM, Georgopapadakou NH, et al. Monocyclic beta-lactam antibiotics produced by bacteria. Nature. 1981;291:489–91. doi: 10.1038/291489a0. [DOI] [PubMed] [Google Scholar]
- 38.Well JS, Trejo WH, Bush K, Georpapadakou NH, Bonner DP, Sykes RB. EM5400, a family of monobactam antibiotics produced by Agrobacterium radiobacter. I. Taxonomy, fermentation and biological properties. J Antibiotics. 1982;35:295–9. doi: 10.7164/antibiotics.35.295. [DOI] [PubMed] [Google Scholar]
- 39.Jacoby GA. AmpC beta-lactamases. Clinical microbiology reviews. 2009;22:161–82. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park JT, Uehara T. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan) Microbiol Mol Biol Rev. 2008;72:211–27. doi: 10.1128/MMBR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS microbiology reviews. 2008;32:361–85. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 42.Gardner AD. Morphological effects of penicillin on bacteria. Nature. 1940;146:837–8. [Google Scholar]
- 43.Duguid JP. The sensitivity of bacteria to the action of penicillin. Edinburg Med J. 1946;53:401–12. [PMC free article] [PubMed] [Google Scholar]
- 44.Salton MRJ. Specific detection of glucose on paper chromatograms. Nature. 1960;186:966–7. doi: 10.1038/186966b0. [DOI] [PubMed] [Google Scholar]
- 45.Bartholomew JW, Mittwer T. The Gram Stain. Bacteriological Reviews. 1952;16:1–29. doi: 10.1128/br.16.1.1-29.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salton MRJ. The relationship between the nature of the cell wall and the Gram stain. J Gen Microbiol. 1963;30:223–35. doi: 10.1099/00221287-30-2-223. [DOI] [PubMed] [Google Scholar]
- 47.Chapman GB, Hiller J. Electron microscopy of ultra-thin sections of bacteria. I. Cellular division in Bacillus cereus. J Bacteriol. 1953;66:362–73. doi: 10.1128/jb.66.3.362-373.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amako K, Murata K, Umeda A. Structure of the envelope of Escherichia coli observed by the rapid freezing and substitution fixation method. Microbiol Immunol. 1983;27:95–9. doi: 10.1111/j.1348-0421.1983.tb03571.x. [DOI] [PubMed] [Google Scholar]
- 49.Hobot JA, Carlemalm E, Villiger W, Kellenberger E. Periplasmic gel: new concept resulting from the reinvestigation of bacterial cell envelope ultra-structure by new methods. J Bacteriol. 1984;160:143–52. doi: 10.1128/jb.160.1.143-152.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park JT, Johnson MJ. Accumulation of labile phosphate in Staphylococcus aureus grown in the presence of penicillin. J Biol Chem. 1949;179:585–92. [PubMed] [Google Scholar]
- 51.Park JT. Uridine-5′-pyrophosphate derivatives. III. Amino acid-containing derivatives. J Biol Chem. 1952;194:897–904. [PubMed] [Google Scholar]
- 52.Park JT, Strominger JL. Mode of action of penicillin. Science. 1957;125:99–101. doi: 10.1126/science.125.3238.99. [DOI] [PubMed] [Google Scholar]
- 53.Salton MRJ, Horne RW. Studies of the bacterial cell wall. II. Methods of preparation and some properties of the cell wall. Biochim Biophys Acta. 1951;7:177–97. doi: 10.1016/0006-3002(51)90017-0. [DOI] [PubMed] [Google Scholar]
- 54.Strange RE, Dark FA. An unidentified amino-sugar present in cell walls and spores of various bacteria. Nature. 1956;177:186–8. [Google Scholar]
- 55.Work E. Biochemistry of the bacterial cell wall. Nature. 1957;179:841–7. doi: 10.1038/179841a0. [DOI] [PubMed] [Google Scholar]
- 56.Cummins C. The chemical composition of the bacteria cell wall. Int Rev Cytol. 1956;5:25–50. [Google Scholar]
- 57.Brumfitt W, Wardlaw AC, Park JT. Development of lysozyme-resistance in Micrococcus lysodiekticus and its association with an increased O-acetyl content of the cell wall. Nature. 1958;181:1783–4. doi: 10.1038/1811783a0. [DOI] [PubMed] [Google Scholar]
- 58.Salton MRJ. Bacterial cell wall. IV. Composition of the cell wall of Gram-positive and Gram-negative bacteria. Biochim Biophys Acta. 1953;10:512–23. doi: 10.1016/0006-3002(53)90296-0. [DOI] [PubMed] [Google Scholar]
- 59.Rietschel ET, Westphal O. History of endotoxins. In: Brade H, Vogel SMOSN, Morrison DC, editors. Endotoxins in Health and Disease. New York: Marcel Dekker; 1999. pp. 1–30. [Google Scholar]
- 60.Repaske R. Lysis of Gram-negative organisms and the role of versene. Biochim Biophys Acta. 1958;30:225–32. doi: 10.1016/0006-3002(58)90044-1. [DOI] [PubMed] [Google Scholar]
- 61.Weidel W, Primosigh J. Biochemical parallels between lysis by virulent phage and lysis by penicillin. J Gen Microbiol. 1958;18:513–7. doi: 10.1099/00221287-18-2-513. [DOI] [PubMed] [Google Scholar]
- 62.Weidel W, Frank H, Martin HH. The rigid layer of the cell wall of Escherichia coli strain B. J Gen Microbiol. 1960;22:158–66. doi: 10.1099/00221287-22-1-158. [DOI] [PubMed] [Google Scholar]
- 63.Mandelson J. Isolation of lysozyme-soluble mucopeptides from the cells wall of Escherichia coli. Nature. 1961;189:855–6. doi: 10.1038/189855a0. [DOI] [PubMed] [Google Scholar]
- 64.van Heijenoort J. Formation of the glycan chains in the synthesis of bacterial peptidoglycan. Glycobiology. 2001;11:25R–36R. doi: 10.1093/glycob/11.3.25r. [DOI] [PubMed] [Google Scholar]
- 65.Siewert G, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls. XI. Formation of the isoglutamine amide group in the cell walls of Staphylococcus aureus. J Biol Chem. 1968;243:783–90. [PubMed] [Google Scholar]
- 66.Araki Y, Shimada A, Ito E. Effect of penicillin on cell wall mucopeptide synthesis in a Escherichia coli particulate system. Biochim Biophys Res Commun. 1966;23:518–25. doi: 10.1016/0006-291x(66)90760-1. [DOI] [PubMed] [Google Scholar]
- 67.Izaki K, Matsuhashi M, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls. VIII. Peptidoglycan transpeptidase and D-alanine carboxypeptidase: penicillin-sensitive enzymatic reaction in strains of Escherichia coli. J Biol Chem. 1968;243:3180–92. [PubMed] [Google Scholar]
- 68.Tipper DJ, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls. XII. Inhibition of cross-linking by penicillins and cephalosporins: studies in Staphylococcus aureus in vivo. J Biol Chem. 1968;243:3169–79. [PubMed] [Google Scholar]
- 69.Tipper DJ, Strominger JL. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc Natl Acad Sci U S A. 1965;54:113–1141. doi: 10.1073/pnas.54.4.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wise EM, Park JT. Penicillin: its basic site of action as an inhibitor of a peptide cross-linking reaction in cell wall mucopeptide synthesis. Proc Natl Acad Sci U S A. 1965;54:75–81. doi: 10.1073/pnas.54.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Izaki K, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls. XIV. Purification and properties of two D-alanine carboxypeptidases from Escherichia coli. J Biol Chem. 1968;243:3193–201. [PubMed] [Google Scholar]
- 72.Goffin C, Ghuysen JM. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol Mol Biol Rev. 1998;62:1079–93. doi: 10.1128/mmbr.62.4.1079-1093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cooper PD, Rowley D. Investigations with radioactive penicillin. Nature. 1949;163:480–1. [Google Scholar]
- 74.Cooper PD, Rowley D, Dawson IM. Location of radioactive penicillin in Staphylococcus aureus after contact with the drug. Nature. 1949;164:842. doi: 10.1038/164842a0. [DOI] [PubMed] [Google Scholar]
- 75.Maass EA, Johnson MJ. Penicillin uptake by bacterial cells. J Bacteriol. 1949;57:415–22. doi: 10.1128/jb.57.4.415-422.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maass EA, Johnson MJ. The relations between bound penicillin and growth in Staphylococcus aureus. J Bacteriol. 1949;58:361–6. doi: 10.1128/jb.58.3.361-366.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schepartz SA, Johnson MJ. The nature of the binding of penicillin by bacterial cells. J Bacteriol. 1956;71:84–90. doi: 10.1002/path.1700710112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cooper PD. The site of action of penicillin: some properties of the penicillin-binding component of Staphylococcus aureus. J Gen Microbiol. 1955;12:100–6. doi: 10.1099/00221287-12-1-100. [DOI] [PubMed] [Google Scholar]
- 79.Spratt BG, Pardee AB. Penicillin-binding proteins and cell shape in Escherichia coli. Nature. 1975;254:516–7. doi: 10.1038/254516a0. [DOI] [PubMed] [Google Scholar]
- 80.Georgopapadakou NH, Liu FY. Penicillin-binding proteins in bacteria. Antimicrob Agents Chemother. 1980;18:148–57. doi: 10.1128/aac.18.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Suginaka H, Blumberg PM, Strominger JL. Multiple penicillin-binding components in Bacillus subtilis, Bacillus cereus, Staphylococcus aureus and Escherichia coli. J Biol Chem. 1972;247:5279–88. [PubMed] [Google Scholar]
- 82.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS microbiology reviews. 2008;32:234–58. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 83.Ghosh AS, Chowdhury C, Nelson DE. Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends In Microbiology. 2008;16:309–17. doi: 10.1016/j.tim.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 84.Vollmer W, Bertsche U. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta. 2008;1778:1714–34. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 85.Vollmer W, Joris B, Charlier P, Foster S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiology Reviews. 2008;32:259–86. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 86.Spratt BG. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci USA. 1975;72:2999–3003. doi: 10.1073/pnas.72.8.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Spratt BG, Jobanputra V, Schwarz U. Mutants of Escherichia coli which lack a component of penicillin-binding protein 1 are viable. FEBS Letters. 1977;79:374–8. doi: 10.1016/0014-5793(77)80824-7. [DOI] [PubMed] [Google Scholar]
- 88.Suzuki H, Nishimura Y, Hirota Y. On the process of cellular division in Escherichia coli: a series of mutants of E. coli altered in the penicillin-binding proteins. Proc Natl Acad Sci USA. 1978;75:664–8. doi: 10.1073/pnas.75.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nakagawa J, Tamaki S, Tomioka S, Matsuhashi M. Functional biosynthesis of cell wall peptidoglycan by polymorphic bifunctional polypeptides. Penicillin-binding protein 1Bs of Escherichia coli with activities of transglycosylase and transpeptidase. J Biol Chem. 1984;259:13937–46. [PubMed] [Google Scholar]
- 90.Tamaki S, Nakajima S, Matsuhashi M. Thermosensitive mutation in Escherichia coli simultaneously causing defects in penicillin-binding protein-1Bs and in enzyme activity for peptidoglycan synthesis in vitro. Proc Natl Acad Sci USA. 1977;74:5472–6. doi: 10.1073/pnas.74.12.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tamura T, Suzuki H, Nishimura Y, Mizoguchi J, Hirota Y. On the process of cellular division in Escherichia coli: isolation and characterization of penicillin-binding proteins 1a, 1b, and 3. Proc Natl Acad Sci USA. 1980;77:4499–503. doi: 10.1073/pnas.77.8.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schiffer G, Holtje JV. Cloning and characterization of PBP 1C, a third member of the multimodular class A penicillin-binding proteins of Escherichia coli. J Biol Chem. 1999;274:32031–9. doi: 10.1074/jbc.274.45.32031. [DOI] [PubMed] [Google Scholar]
- 93.Park JT, Burman L. FL-1600: A new penicillin with a unique mode of action. J Bacteriol. 1973;51:863–8. doi: 10.1016/0006-291x(73)90006-5. [DOI] [PubMed] [Google Scholar]
- 94.Iwaya M, Goldman R, Tipper DJ, Feingold B, Strominger JL. Morphology of an Escherichia coli mutant with a temperature-dependent round cell shape. J Bacteriol. 1978;136:1143–58. doi: 10.1128/jb.136.3.1143-1158.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de la Rosa EJ, de Pedro MA, Vazquez D. Penicillin binding proteins: role in initiation of murein synthesis in Escherichia coli. J Bacteriol. 1985;82:5632–5. doi: 10.1073/pnas.82.17.5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Matsuhashi S, Kamiryo T, Blumberg PM, Linnett P, Willoughby E, Strominger JL. Mechanism of action and development of resistance to a new amidino penicillin. J Bacteriol. 1974;117:578–87. doi: 10.1128/jb.117.2.578-587.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Essig P, Martin HH, Gmeiner J. Murein and lipopolysaccharide biosynthesis in synchronized cells of Escherichia coli K 12 and the effect of penicillin G, mecillinam and nalidixic acid. Arch Microbiol. 1982;132:245–50. doi: 10.1007/BF00407959. [DOI] [PubMed] [Google Scholar]
- 98.Ogura T, Bouloc P, Niki H, D’Ari R, Hiraga S, Jaffe A. Penicillin-binding protein 2 is essential in wild-type Escherichia coli but not in lov or cya mutants. J Bacteriol. 1989;171:3025–30. doi: 10.1128/jb.171.6.3025-3030.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Normark S. Mutation in Escherichia coli K-12 mediating spherelike envelopes and changed tolerance to ultraviolet irradiation and some antibiotics. J Bacteriol. 1969;98:1274–7. doi: 10.1128/jb.98.3.1274-1277.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Matsuzawa H, Hayakawa K, Sato T, Imahori K. Characterization and genetic analysis of a mutant of Escherichia coli K-12 with rounded morphology. J Bacteriol. 1973;115:436–42. doi: 10.1128/jb.115.1.436-442.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iwaya M, Jones CW, Khorana J, Strominger JL. Mapping of the mecillinam-resistant, round morphological mutants of Escherichia coli. J Bacteriol. 1978;133:196–202. doi: 10.1128/jb.133.1.196-202.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Spratt BG. Temperature-sensitive cell division mutants of Escherichia coli with thermolabile penicillin-binding proteins. J Bacteriol. 1977;131:293–305. doi: 10.1128/jb.131.1.293-305.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spratt BG, Boyd A, Stoker N. Defective and plaque-forming lambda transducing bacteriophage carrying penicillin-binding protein-cell shape genes: genetic and physical mapping and identification of gene products from the lip-dacA-rodA-pbpA-leuS region of the Escherichia coli chromosome. J Bacteriol. 1980;143:569–81. doi: 10.1128/jb.143.2.569-581.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ishino F, Park W, Tomioka S, Tamaki S, Takase I, Kunugita K, et al. Peptidoglycan synthetic activities in membranes of Escherichia coli caused by overproduction of penicillin-binding protein 2 and RodA protein. J Biol Chem. 1986;261:7024–31. [PubMed] [Google Scholar]
- 105.Vinella D, D’Ari R, Jaffe A, Bouloc P. Penicillin binding protein 2 is dispensable in Escherichia coli when ppGpp synthesis is induced. Embo J. 1992;11:1493–501. doi: 10.1002/j.1460-2075.1992.tb05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vinella D, Joseleau-Petit D, Thevenet D, Bouloc P, D’Ari R. Penicillin-binding protein 2 inactivation in Escherichia coli results in cell division inhibition, which is relieved by FtsZ overexpression. J Bacteriol. 1993;175:6704–10. doi: 10.1128/jb.175.20.6704-6710.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schmidt LS, Botta G, Park JT. Effects of furazlocillin, a beta-lactam antibiotic which binds selectively to penicillin-binding protein 3, on Escherichia coli mutants deficient in other penicillin-binding proteins. J Bacteriol. 1981;145:632–7. doi: 10.1128/jb.145.1.632-637.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Botta GA, Park JT. Evidence for involvement of penicillin-binding protein 3 in murein synthesis during septation but not during cell elongation. J Bacteriol. 1981;145:333–40. doi: 10.1128/jb.145.1.333-340.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spratt BG. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci U S A. 1975;72:2999–3003. doi: 10.1073/pnas.72.8.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tamura T, Imae Y, Strominger JL. Purification to homogeneity and properties of two D-alanine carboxypeptidases I From Escherichia coli. J Biol Chem. 1976;251:414–23. [PubMed] [Google Scholar]
- 111.Iwaya M, Strominger JL. Simultaneous deletion of D-alanine carboxypeptidase IB-C and penicillin-binding component IV in a mutant of Escherichia coli K12. Proc Natl Acad Sci USA. 1977;74:2980–4. doi: 10.1073/pnas.74.7.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Matsuhashi M, Takagaki Y, Maruyama IN, Tamaki S, Nishimura Y, Suzuki H, et al. Mutants of Escherichia coli lacking in highly penicillin-sensitive D-alanine carboxypeptidase activity. Proc Natl Acad Sci USA. 1977;74:2976–9. doi: 10.1073/pnas.74.7.2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Korat B, Mottl H, Keck W. Penicillin-binding protein 4 of Escherichia coli: molecular cloning of the dacB gene, controlled overexpression, and alterations in murein composition. Mol Microbiol. 1991;5:675–84. doi: 10.1111/j.1365-2958.1991.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 114.de Pedro MA, Schwarz U. Heterogeneity of newly inserted and preexisting murein in the sacculus of Escherichia coli. Proc Natl Acad Sci U S A. 1981;78:5856–60. doi: 10.1073/pnas.78.9.5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Spratt BG, Strominger JL. Identification of the major penicillin-binding proteins of Escherichia coli as D-alanine carboxypeptidase IA. J Bacteriol. 1976;127:660–3. doi: 10.1128/jb.127.1.660-663.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Spratt BG. Deletion of the penicillin-binding protein 5 gene of Escherichia coli. J Bacteriol. 1980;144:1190–2. doi: 10.1128/jb.144.3.1190-1192.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Matsuhashi M, Tamaki S, Curtis SJ, Strominger JL. Mutational evidence for identity of penicillin-binding protein 5 in Escherichia coli with the major D-alanine carboxypeptidase IA activity. J Bacteriol. 1979;137:644–7. doi: 10.1128/jb.137.1.644-647.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Broome-Smith JK, Ioannidis I, Edelman A, Spratt BG. Nucleotide sequences of the penicillin-binding protein 5 and 6 genes of Escherichia coli. Nucleic Acid Res. 1988;16:1617. doi: 10.1093/nar/16.4.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Amanuma H, Strominger JL. Purification and properties of penicillin-binding proteins 5 and 6 from Escherichia coli membranes. J Biol Chem. 1980;255:11173–80. [PubMed] [Google Scholar]
- 120.Broome-Smith JK, Spratt BG. Deletion of the penicillin-binding protein 6 gene of Escherichia coli. J Bacteriol. 1982;152:904–6. doi: 10.1128/jb.152.2.904-906.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Buchanan CE, Sowell MO. Synthesis of penicillin-binding protein 6 by stationary-phase Escherichia coli. J Bacteriol. 1982;151:491–4. doi: 10.1128/jb.151.1.491-494.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Baquero MR, Bouzon M, Quintela JC, Ayala JA, Moreno F. dacD, an Escherichia coli gene encoding a novel penicillin-binding protein (PBP6b) with DD-carboxypeptidase activity. J Bacteriol. 1996;178:7106–11. doi: 10.1128/jb.178.24.7106-7111.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Henderson TA, Dombrosky PM, Young KD. Artifactual processing of penicillin-binding proteins 7 and 1b by the OmpT protease of Escherichia coli. J Bacteriol. 1994;176:256–9. doi: 10.1128/jb.176.1.256-259.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Romeis T, Holtje JV. Penicillin-binding protein 7/8 of Escherichia coli is a DD-endopeptidase. Eur J Biochem. 1994;224:597–604. doi: 10.1111/j.1432-1033.1994.00597.x. [DOI] [PubMed] [Google Scholar]
- 125.Malouin F, Chamberland S, Brochu N, Parr TR., Jr Influence of growth media on Escherichia coli cell composition and ceftazidime susceptibility. Antimicrob Agents Chemother. 1991;35:477–83. doi: 10.1128/aac.35.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Henderson TA, Templin M, Young KD. Identification and cloning of the gene encoding penicillin-binding protein 7 of Escherichia coli. J Bacteriol. 1995;177:2074–9. doi: 10.1128/jb.177.8.2074-2079.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Armstrong GL, Conn LA, Pinner RW. Trends in infectious disease mortality in the United States during the 20th century. JAMA. 1999;281:61–6. doi: 10.1001/jama.281.1.61. [DOI] [PubMed] [Google Scholar]
- 128.Cohen M. Changing patterns of infectious disease. Nature. 2000;406:762–7. doi: 10.1038/35021206. [DOI] [PubMed] [Google Scholar]
- 129.Kirby W. Extraction of a highly potent penicillin inactivator from penicillin-resistant staphylococci. Science. 1944;99:452–3. doi: 10.1126/science.99.2579.452. [DOI] [PubMed] [Google Scholar]
- 130.Rammelkamp CH, Moxon Resistance of Staphylococcus aureus to action of penicillin. Proc Soc Exp Biol and Med. 1942;51:386–9. [Google Scholar]
- 131.Gallardo E. Sensitivity of bacteria from infected wounds to penicillin. II results in one hundred and twelve cases. War Med. 1945;7:100–3. [Google Scholar]
- 132.Rantz LA, Kirby WMM. Action of penicillin on Staplylococcus in vitro. J Immunol. 1944;48:335–43. [Google Scholar]
- 133.Selbie FR, Simon RD, McIntosh J. Bacteriological aspects of penicillin therapy. J Pathol and Bac. 1942;57:47–58. [Google Scholar]
- 134.Finland M. Emergence of antibiotic-resistant bacteria. N Engl J Med. 1955;253:909–22. doi: 10.1056/NEJM195511242532105. [DOI] [PubMed] [Google Scholar]
- 135.Finland M, Frank PF, Wilcox C. In vitro susceptibility of pathogenic staphylococci to seven antibiotics. With a note on the changing resistance of staphylococci to penicillin. Am J Clin Path. 1950;20:325–34. doi: 10.1093/ajcp/20.4.325. [DOI] [PubMed] [Google Scholar]
- 136.Barber M. Staphylococcal infection due to penicillin-resistant strains. Br J Med. 1947;2:863–5. doi: 10.1136/bmj.2.4534.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Barber M. Congaluase-positive staphylococci resistant to penicillin. J Pathol and Bact. 1947;59:373–84. [Google Scholar]
- 138.Barber M, Rozwadowska-Dowzenko M. Infection by penicillin-resistant staphylococci. Lancet. 1948;2:641–4. doi: 10.1016/s0140-6736(48)92166-7. [DOI] [PubMed] [Google Scholar]
- 139.Barber M, Whitehead JEM. Bacteriophage types in penicillin-resistant staphylococcal infection. Br Med J. 1949;2:565–9. doi: 10.1136/bmj.2.4627.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Finland M. Changing ecology of bacterial infections as related to antibacterial therapy. J Infect Dis. 1970;122:419–31. doi: 10.1093/infdis/122.5.419. [DOI] [PubMed] [Google Scholar]
- 141.Rogers DE. The changing pattern of life-threatening microbial disease. N Engl J Med. 1959;261:677–83. doi: 10.1056/NEJM195910012611401. [DOI] [PubMed] [Google Scholar]
- 142.Sneader W. Drug discovery: a history. West Sussex: John Wiley & Sons, Ltd; 2005. [Google Scholar]
- 143.Travis J. Reviving the antibiotic miracle? Science. 1994;264:360–2. doi: 10.1126/science.8153615. [DOI] [PubMed] [Google Scholar]
- 144.Greenwood D. Tarnished gold: sixty years of antimicrobial drug use and misuse. J Med Microbiol. 1995;43:395–6. doi: 10.1099/00222615-43-6-395. [DOI] [PubMed] [Google Scholar]
- 145.Culotta E. Funding crunch hobbles antibiotic resistance research. Science. 1994;264:362–3. doi: 10.1126/science.8153617. [DOI] [PubMed] [Google Scholar]
- 146.Majiduddin FK, Materon IC, Palzkill TG. Molecular analysis of beta-lactamase structure and function. Int J Med Microbiol. 2002;292:127–37. doi: 10.1078/1438-4221-00198. [DOI] [PubMed] [Google Scholar]
- 147.Ambler RP. The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci. 1980;289:321–31. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 148.Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39:1211–33. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mitsuhashi S, Inoue M. Mechanisms of resistance to beta-lactam antibiotics. New York: Springer-Verlag; 1981. [Google Scholar]
- 150.Richmond MH, Sykes RB. The beta-lactamases of Gram-negative bacteria and their possible physiological role. Adv Microb Physiol. 1973;9:31–88. doi: 10.1016/s0065-2911(08)60376-8. [DOI] [PubMed] [Google Scholar]
- 151.Couture F, Lachapelle J, Levesque RC. Phylogeny of LCR-1 and OXA-5 with class A and class D beta-lactamases. Mol Microbiol. 1992;6:1693–703. doi: 10.1111/j.1365-2958.1992.tb00894.x. [DOI] [PubMed] [Google Scholar]
- 152.Matagne A, Misselyn-Bauduin AM, Joris B, Erpicum T, Granier B, Frere JM. The diversity of the catalytic properties of class A beta-lactamases. Biochem J. 1990;265:131–46. doi: 10.1042/bj2650131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Boissinot M, Levesque RC. Nucleotide sequence of the PSE-4 carbenicillinase gene and correlations with the Staphylococcus aureus PC1 beta-lactamase crystal structure. J Biol Chem. 1990;265:1225–30. [PubMed] [Google Scholar]
- 154.Lachapelle J, Dufresne J, Levesque RC. Characterization of the blaCARB-3 gene encoding the carbenicillinase-3 beta-lactamase of Pseudomonas aeruginosa. Gene. 1991;102:7–12. doi: 10.1016/0378-1119(91)90530-o. [DOI] [PubMed] [Google Scholar]
- 155.Naas T, Philippon L, Poirel L, Ronco E, Nordmann P. An SHV-derived extended-spectrum beta-lactamase in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1999;43:1281–4. doi: 10.1128/aac.43.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dufresne J, Vezina G, Levesque RC. Cloning and expression of the imipenem-hydrolyzing beta-lactamase operon from Pseudomonas maltophilia in Escherichia coli. Antimicrob Agents Chemother. 1988;32:819–26. doi: 10.1128/aac.32.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Watanabe M, Iyobe S, Inoue M, Mitsuhashi S. Transferable imipenem resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1991;35:147–51. doi: 10.1128/aac.35.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Senda K, Arakawa Y, Ichiyama S, Nakashima K, Ito H, Ohsuka S, et al. PCR detection of metallo-beta-lactamase gene (blaIMP) in gram-negative rods resistant to broad-spectrum beta-lactams. J Clin Microbiol. 1996;34:2909–13. doi: 10.1128/jcm.34.12.2909-2913.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lauretti L, Riccio ML, Mazzariol A, Cornaglia G, Amicosante G, Fontana R, et al. Cloning and characterization of blaVIM, a new integron-borne metallo-beta-lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob Agents Chemother. 1999;43:1584–90. doi: 10.1128/aac.43.7.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tsakris A, Pournaras S, Woodford N, Palepou MF, Babini GS, Douboyas J, et al. Outbreak of infections caused by Pseudomonas aeruginosa producing VIM-1 carbapenemase in Greece. J Clin Microbiol. 2000;38:1290–2. doi: 10.1128/jcm.38.3.1290-1292.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sanders CC. Chromosomal cephalosporinases responsible for multiple resistance to newer beta-lactam antibiotics. Annu Rev Microbiol. 1987;41:573–93. doi: 10.1146/annurev.mi.41.100187.003041. [DOI] [PubMed] [Google Scholar]
- 162.Galleni M, Lindberg F, Normark S, Cole S, Honore N, Joris B, et al. Sequence and comparative analysis of three Enterobacter cloacae ampC beta-lactamase genes and their products. Biochem J. 1988;250:753–60. doi: 10.1042/bj2500753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lodge JM, Minchin SD, Piddock LJ, Busby JW. Cloning, sequencing and analysis of the structural gene and regulatory region of the Pseudomonas aeruginosa chromosomal ampC beta-lactamase. Biochem J. 1990;272:627–31. doi: 10.1042/bj2720627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dale JW, Smith JT. A direct comparison of two unusual R-factor-mediated beta-lactamases. Biochem J. 1972;128:173–4. doi: 10.1042/bj1280173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Medeiros AA, Cohenford M, Jacoby GA. Five novel plasmid-determined beta-lactamases. Antimicrob Agents Chemother. 1985;27:715–9. doi: 10.1128/aac.27.5.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kong KF, Jayawardena SR, Del Puerto A, Wiehlmann L, Laabs U, Tummler B, et al. Characterization of poxB, a chromosomal-encoded Pseudomonas aeruginosa oxacillinase. Gene. 2005;358:82–92. doi: 10.1016/j.gene.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 167.Girlich D, Naas T, Nordmann P. Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrobial Agents and Chemotherapy. 2004;48:2043–8. doi: 10.1128/AAC.48.6.2043-2048.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Poirel L, Weldhagen GF, De Champs C, Nordmann P. A nosocomial outbreak of Pseudomonas aeruginosa isolates expressing the extended-spectrum beta-lactamase GES-2 in South Africa. J Antimicrob Chemother. 2002;49:561–5. doi: 10.1093/jac/49.3.561. [DOI] [PubMed] [Google Scholar]
- 169.Sanders CC, Sanders WE. Microbial resistance to newer generation beta-lactam antibiotics: clinical and laboratory implications. J Infect Dis. 1985;151:399–405. doi: 10.1093/infdis/151.3.399. [DOI] [PubMed] [Google Scholar]
- 170.Sabath LD, Jago M, Abraham EP. Cephalosporinase and penicillinase activities of a beta-lactamase from Pseudomonas pyocyanea. Biochem J. 1965;96:739–52. doi: 10.1042/bj0960739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sykes RB, Matthew M. The beta-lactamases of Gram-negative bacteria and their role in resistance to beta-lactam antibiotics. J Antimicrob Chemother. 1976;2:115–57. doi: 10.1093/jac/2.2.115. [DOI] [PubMed] [Google Scholar]
- 172.Proenca R, Niu WW, Cacalano G, Prince A. The Pseudomonas cepacia 249 chromosomal penicillinase is a member of the AmpC family of chromosomal beta-lactamases. Antimicrob Agents Chemother. 1993;37:667–74. doi: 10.1128/aac.37.4.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Seoane A, Francia MV, Garcia Lobo JM. Nucleotide sequence of the ampC-ampR region from the chromosome of Yersinia enterocolitica. Antimicrob Agents Chemother. 1992;36:1049–52. doi: 10.1128/aac.36.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lindberg F, Westman L, Normark S. Regulatory components in Citrobacter freundii ampC beta-lactamase induction. Proc Natl Acad Sci USA. 1985;82:4620–4. doi: 10.1073/pnas.82.14.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Honore N, Nicolas MH, Cole ST. Inducible cephalosporinase production in clinical isolates of Enterobacter cloacae is controlled by a regulatory gene that has been deleted from Escherichia coli. EMBO J. 1986;5:3709–14. doi: 10.1002/j.1460-2075.1986.tb04704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Poirel L, Guibert M, Girlich D, Naas T, Nordmann P. Cloning, sequence analyses, expression, and distribution of ampC-ampR from Morganella morganii clinical isolates. Antimicrob Agents Chemother. 1999;43:769–76. doi: 10.1128/aac.43.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mahlen SD, Morrow SS, Abdalhamid B, Hanson ND. Analyses of ampC gene expression in Serratia marcescens reveal new regulatory properties. J Antimicrob Chemother. 2003;51:791–802. doi: 10.1093/jac/dkg133. [DOI] [PubMed] [Google Scholar]
- 178.Lau SK, Ho PL, Li MW, Tsoi HW, Yung RW, Woo PC, et al. Cloning and characterization of a chromosomal class C beta-lactamase and its regulatory gene in Laribacter hongkongensis. Antimicrob Agents Chemother. 2005;49:1957–64. doi: 10.1128/AAC.49.5.1957-1964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nadjar D, Labia R, Cerceau C, Bizet C, Philippon A, Arlet G. Molecular characterization of chromosomal class C beta-lactamase and its regulatory gene in Ochrobactrum anthropi. Antimicrob Agents Chemother. 2001;45:2324–30. doi: 10.1128/AAC.45.8.2324-2330.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jaurin B, Grundstrom T. ampC cephalosporinase of Escherichia coli K-12 has a different evolutionary origin from that of beta-lactamases of the penicillinase type. Proc Natl Acad Sci USA. 1981;78:4897–901. doi: 10.1073/pnas.78.8.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jaurin B, Normark S. In vivo regulation of chromosomal beta-lactamase in Escherichia coli. J Bacteriol. 1979;138:896–902. doi: 10.1128/jb.138.3.896-902.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jaurin B, Grundstrom T, Edlund T, Normark S. The E. coli beta-lactamase attentuator mediates growth rate-dependent regulation. Nature. 1981;290:221–5. doi: 10.1038/290221a0. [DOI] [PubMed] [Google Scholar]
- 183.Gootz TD, Sanders CC. Characterization of beta-lactamase induction in Enterobacter cloacae. Antimicrob Agent Chemother. 1893;23:91–7. doi: 10.1128/aac.23.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hennessey TD. Inducible beta-lactamase in Enterobacter. J Gen Microbiol. 1967;49:277–85. doi: 10.1099/00221287-49-2-277. [DOI] [PubMed] [Google Scholar]
- 185.Seeberg AH, Tolxdorff-Neutzling RM, Wiedemann B. Chromosomal beta-lactamases of Enterobacter cloacae are responsible for resistance to third-generation cephalosporins. Antimicrob Agents Chemother. 1983;23:918–25. doi: 10.1128/aac.23.6.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yamamoto T, Murayama SY, Sawai T. Cloning and expression of the gene(s) for cephalosporinase production of Citrobacter freundii. Mol Gen Genet. 1983;190:85–91. doi: 10.1007/BF00330328. [DOI] [PubMed] [Google Scholar]
- 187.Lindberg F, Normark S. Contribution of chromosomal beta-lactamases to beta-lactam resistance in enterobacteria. Rev Infect Dis. 1986;8:S292–S304. doi: 10.1093/clinids/8.supplement_3.s292. [DOI] [PubMed] [Google Scholar]
- 188.Giwercman B, Rasmussen JW, Ciofu O, Clemmentsen I, Schumacher H, Hoiby N. Antibodies against chromosomal beta-lactamase. Antimicrob Agent Chemother. 1994;38:2306–10. doi: 10.1128/aac.38.10.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Goering RV, Sanders CC, Sanders WEJ, Guay R, Guerin S. Heterogeneity in ampR-ampC gene interaction in Enterobacter cloacae. Rev Infect Dis. 1988;10:786–92. doi: 10.1093/clinids/10.4.786. [DOI] [PubMed] [Google Scholar]
- 190.Walther-Rasmussen J, Johnsen AH, Hoiby N. Terminal truncations in ampC beta-lactamase from a clinical isolate of Pseudomonas aeruginosa. Eur J Biochem. 1999;263:478–85. doi: 10.1046/j.1432-1327.1999.00529.x. [DOI] [PubMed] [Google Scholar]
- 191.Gates ML, Sanders CC, Goering RV, Sanders WE. Evidence for multiple forms of type I chromosomal beta-lactamase in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1986;30:453–7. doi: 10.1128/aac.30.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Campbell JI, Ciofu O, Høiby N. Pseudomonas aeruginosa isolates from patients with cystic fibrosis have different β-lactamase expression phenotypes but are homogeneous in the ampC-ampR genetic region. Antimicrob Agents Chemther. 1997;41:1380–4. doi: 10.1128/aac.41.6.1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bagge N, Ciofu O, Hentzer M, Campbell JI, Givskov M, Hoiby N. Constitutive high expression of chromosomal beta-lactamase in Pseudomonas aeruginosa caused by a new insertion sequence (IS1669) located in ampD. Antimicrobial Agents and Chemotherapy. 2002;46:3406–11. doi: 10.1128/AAC.46.11.3406-3411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bergstrom S, Lindberg FP, Olsson O, Normark S. Comparison of the overlapping frd and ampC operons of Escherichia coli with the corresponding DNA sequences in other Gram-negative bacteria. J Bacteriol. 1983;155:1297–305. doi: 10.1128/jb.155.3.1297-1305.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lindberg F, Normark S. Common mechanism of ampC beta-lactamase induction in enterobacteria: regulation of the cloned Enterobacter cloacae P99 beta-lactamase gene. J Bacteriol. 1987;169:758–63. doi: 10.1128/jb.169.2.758-763.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annual Review of Microbiology. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 197.Lindquist S, Lindberg F, Normark S. Binding of the Citrobacter freundii AmpR regulator to a single DNA site provides both autoregulation and activation of the inducible ampC beta-lactamase gene. J Bacteriol. 1989;171:3746–53. doi: 10.1128/jb.171.7.3746-3753.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jacobs C, Frere JM, Normark S. Cytosolic intermediates for cell wall biosynthesis and degradation control inducible beta-lactam resistance in Gram-negative bacteria. Cell. 1997;88:823–32. doi: 10.1016/s0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- 199.Kong KF, Jayawardena SR, Indulkar SD, Del Puerto A, Koh CL, Hoiby N, et al. Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB beta-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob Agents Chemother. 2005;49:4567–75. doi: 10.1128/AAC.49.11.4567-4575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lodge J, Busby S, Piddock L. Investigation of the Pseudomonas aeruginosa ampR gene and its role at the chromosomal ampC beta-lactamase promoter. FEMS Microbiol Lett. 1993;111:3315–320. doi: 10.1111/j.1574-6968.1993.tb06404.x. [DOI] [PubMed] [Google Scholar]
- 201.Gualerzi CO, Pon CL. Initiation of mRNA translation in prokaryotes. Biochemistry. 1990;29:5881–9. doi: 10.1021/bi00477a001. [DOI] [PubMed] [Google Scholar]
- 202.Preston M, Seed P, Toder D, Iglewski B, Ohman D, Gustin J, et al. Contribution of proteases and LasR to the virulence of Pseudomonas aeruginosa during corneal infections. Infection and Immunity. 1997;65:3086–90. doi: 10.1128/iai.65.8.3086-3090.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Toder DS, Gambello MJ, Iglewski BH. Pseudomonas aeruginosa LasA: a second elastase under the transcriptional control of lasR. Molecular Microbiology. 1991;5:2003–10. doi: 10.1111/j.1365-2958.1991.tb00822.x. [DOI] [PubMed] [Google Scholar]
- 204.Nicolas MH, Honore N, Jarlier V, Philippon A, Cole ST. Molecular genetics analysis of cephalosporinase production and its role in beta-lactam resistance in clinical isolates of Enterobacter cloacae. Antimicrob Agents Chemother. 1987;31:295–9. doi: 10.1128/aac.31.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lindberg F, Lindquist S, Normark S. Inactivation of the ampD gene causes semiconstitutive overproduction of the inducible Citrobacter freundii beta-lactamase. J Bacteriol. 1987;169:1923–8. doi: 10.1128/jb.169.5.1923-1928.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Honore N, Nicolas MH, Cole ST. Regulation of enterobacterial cephalosporinase production: the role of a membrane-bound sensory transducer. Mol Microbiol. 1989;3:1121–30. doi: 10.1111/j.1365-2958.1989.tb00262.x. [DOI] [PubMed] [Google Scholar]
- 207.Lindquist S, Galleni M, Lindberg F, Normark S. Signaling proteins in enterobacterial AmpC beta-lactamase regulation. Mol Microbiol. 1989;3:1091–102. doi: 10.1111/j.1365-2958.1989.tb00259.x. [DOI] [PubMed] [Google Scholar]
- 208.Keilty S, Rosenberg M. Constitutive function of a positively regulated promoter reveals new sequences essential for activity. J Biol Chem. 1987;262:6389–95. [PubMed] [Google Scholar]
- 209.Ponnambalam S, Chan B, Busby S. Functional analysis of different sequence elements in the Escherichia coli galactose operon P2 promoter. Mol Microbiol. 1988;2:165–72. doi: 10.1111/j.1365-2958.1988.tb00018.x. [DOI] [PubMed] [Google Scholar]
- 210.Spratt BG, Cromie KD. Penicillin-binding proteins of Gram-negative bacteria. Rev Infect Dis. 1988;10:699–711. doi: 10.1093/clinids/10.4.699. [DOI] [PubMed] [Google Scholar]
- 211.Korfmann G, Sanders CC, Moland ES. Altered phenotypes associated with ampD mutations in Enterobacter cloacae. Antimicrob Agents Chemother. 1991;35:358–64. doi: 10.1128/aac.35.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kopp U, Wiedemann B, Lindquist S, Normark S. Sequences of wild-type and mutant ampD genes of Citrobacter freundii and Enterobacter cloacae. Antimicrob Agents Chemother. 1993;37:224–8. doi: 10.1128/aac.37.2.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for beta-lactamase induction. EMBO J. 1994;13:4684–94. doi: 10.1002/j.1460-2075.1994.tb06792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tuomanen E, Lindquist S, Sande S, Galleni M, Light K, Gage D, et al. Coordinate regulation of beta-lactamase induction and peptidoglycan composition by the amp operon. Science. 1991;251:201–4. doi: 10.1126/science.1987637. [DOI] [PubMed] [Google Scholar]
- 215.Holtje JV, Kopp U, Ursinus A, Wiedemann B. The negative regulator of beta-lactamase induction AmpD is a N-acetyl-anhydromuramyl-L-alanine amidase. FEMS Microbiol Lett. 1994;122:159–64. doi: 10.1111/j.1574-6968.1994.tb07159.x. [DOI] [PubMed] [Google Scholar]
- 216.Jacobs C, Joris B, Jamin M, Klarsov K, Van Beeumen J, Mengin-Lecreulx D, et al. AmpD, essential for both beta-lactamase regulation and cell wall recycling, is a novel cytosolic N-acetylmuramyl-L-alanine amidase. Mol Microbiol. 1995;15:553–9. doi: 10.1111/j.1365-2958.1995.tb02268.x. [DOI] [PubMed] [Google Scholar]
- 217.Holtje JV, Tuomanen EI. The murein hydrolases of Escherichia coli: properties, functions and impact on the course of infections in vivo. J Gen Microbiol. 1991;137:441–54. doi: 10.1099/00221287-137-3-441. [DOI] [PubMed] [Google Scholar]
- 218.van Heijenoort J, Parquet C, Flouret B, van Heijenoort Y. Envelope-bound N-acetylmuramyl-L-alanine amidase of Escherichia coli K 12. Purification and properties of the enzyme. Eur J Biochem. 1975;58:611–9. doi: 10.1111/j.1432-1033.1975.tb02412.x. [DOI] [PubMed] [Google Scholar]
- 219.Langaee TY, Dargis M, Huletsky A. An ampD gene in Pseudomonas aeruginosa encodes a negative regulator of AmpC beta-lactamase expression. Antimicrob Agents Chemother. 1998;42:3296–300. doi: 10.1128/aac.42.12.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Langaee TY, Gagnon L, Huletsky A. Inactivation of the ampD gene in Pseudomonas aeruginosa leads to moderate-basal-level and hyperinducible AmpC beta-lactamase expression. Antimicrob Agents Chemother. 2000;44:583–9. doi: 10.1128/aac.44.3.583-589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Juan C, Moya B, Perez JL, Oliver A. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level beta-lactam resistance involves three AmpD homologues. Antimicrob Agents Chemother. 2006;50:1780–7. doi: 10.1128/AAC.50.5.1780-1787.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Moya B, Juan C, Alberti S, Perez JL, Oliver A. Benefit of having multiple ampD genes for acquiring beta-lactam resistance without losing fitness and virulence in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2008;52:3694–700. doi: 10.1128/AAC.00172-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Werner V, Sanders CC, Sanders JWE, Goering RV. Role of beta-lactamases and outer membrane proteins in multiple beta-lactam resistance of Enterobacter cloacae. Antimicrob Agents Chemother. 1985;27:455–9. doi: 10.1128/aac.27.4.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Korfmann G, Sanders CC. ampG is essential for high-level expression of AmpC beta-lactamase in Enterobacter cloacae. Antimicrob Agents Chemother. 1989;33:1946–51. doi: 10.1128/aac.33.11.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lindquist S, Weston-Hafer K, Schmidt H, Pul C, Korfmann G, Erickson J, Sanders C, Martin HH, Normark S. AmpG, a signal transducer in chromosomal beta-lactamase induction. Mol Microbiol. 1993;9:703–15. doi: 10.1111/j.1365-2958.1993.tb01731.x. [DOI] [PubMed] [Google Scholar]
- 226.Chahboune A, Decaffmeyer M, Brasseur R, Joris B. Membrane topology of the Escherichia coli AmpG permease required for recycling of cell wall anhydromuropeptides and AmpC beta-lactamase induction. Antimicrob Agents Chemother. 2005;49:1145–9. doi: 10.1128/AAC.49.3.1145-1149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Cheng Q, Li H, Merdek K, Park JT. Molecular characterization of the beta -N-acetylglucosaminidase of Escherichia coli and its role in cell wall recycling. J Bacteriol. 2000;182:4836–40. doi: 10.1128/jb.182.17.4836-4840.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Votsch W, Templin MF. Characterization of a beta -N-acetylglucosaminidase of Escherichia coli and elucidation of its role in muropeptide recycling and beta-lactamase induction. J Biol Chem. 2000;275:39032–8. doi: 10.1074/jbc.M004797200. [DOI] [PubMed] [Google Scholar]
- 229.Cheng Q, Park JT. Substrate specificity of the AmpG permease required for recycling of cell wall anhydro-muropeptides. J Bacteriol. 2002;184:6434–6. doi: 10.1128/JB.184.23.6434-6436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ghoul M, Pommepuy M, Moillo-Batt A, Cormier M. Effect of carbonyl cyanide m-chlorophenylhydrazone on Escherichia coli halotolerance. Appl Environ Microbiol. 1989;55:1040–3. doi: 10.1128/aem.55.4.1040-1043.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bartowsky E, Normark S. Purification and mutant analysis of Citrobacter fruendii AmpR, the regulator for chromosomal AmpC beta-lactamase. Mol Microbiol. 1991;5:1715–25. doi: 10.1111/j.1365-2958.1991.tb01920.x. [DOI] [PubMed] [Google Scholar]
- 232.Edlund T, Normark S. The Escherichia coli beta-lactamase attenuator mediates growth rate-dependent regulation. Nature. 1981;292:269–71. doi: 10.1038/290221a0. [DOI] [PubMed] [Google Scholar]
- 233.Jaurin B, Grundstrom T, Normark S. Sequence elements determining ampC promoter strength in Escherichia coli. EMBO J. 1982;1:875–81. doi: 10.1002/j.1460-2075.1982.tb01263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jaurin B, Normark S. Insertion of IS2 creates a novel ampC promoter in Escherichia coli. Cell. 1983;32:809–16. doi: 10.1016/0092-8674(83)90067-3. [DOI] [PubMed] [Google Scholar]
- 235.Olsson O, Bergstrom S, Lindberg FP, Normark S. ampC beta-lactamase hyperproduction in Escherichia coli: natural ampicillin resistance generated by horizontal chromosomal DNA transfer from Shigella. Proc Natl Acad Sci U S A. 1983;80:7556–60. doi: 10.1073/pnas.80.24.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Keseler IM, Bonavides-Martinez C, Collado-Vides J, Gama-Castro S, Gunsalus RP, Johnson DA, et al. EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Research. 2009;37:D464–70. doi: 10.1093/nar/gkn751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Romero P, Karp P. PseudoCyc, a pathway-genome database for Pseudomonas aeruginosa. Journal of Molecular Microbiology and Biotechnology. 2003;5:230–9. doi: 10.1159/000071075. [DOI] [PubMed] [Google Scholar]
- 238.Winsor GL, Lo R, Sui SJ, Ung KS, Huang S, Cheng D, et al. Pseudomonas aeruginosa Genome Database and PseudoCAP: facilitating community-based, continually updated, genome annotation. Nucleic Acids Research. 2005;33:D338–43. doi: 10.1093/nar/gki047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Winsor GL, Van Rossum T, Lo R, Khaira B, Whiteside MD, Hancock RE, et al. Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Research. 2009;37:D483–8. doi: 10.1093/nar/gkn861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sauvage E, Powell AJ, Heilemann J, Josephine HR, Charlier P, Davies C, et al. Crystal structures of complexes of bacterial DD-peptidases with peptidoglycan-mimetic ligands: the substrate specificity puzzle. Journal of molecular biology. 2008;381:383–93. doi: 10.1016/j.jmb.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Fonze E, Vanhove M, Dive G, Sauvage E, Frere JM, Charlier P. Crystal structures of the Bacillus licheniformis BS3 class A beta-lactamase and of the acyl-enzyme adduct formed with cefoxitin. Biochemistry. 2002;41:1877–85. doi: 10.1021/bi015789k. [DOI] [PubMed] [Google Scholar]
- 242.Ghuysen JM. Serine beta-lactamases and penicillin-binding proteins. Annu Rev Microbiol. 1991;45:37–67. doi: 10.1146/annurev.mi.45.100191.000345. [DOI] [PubMed] [Google Scholar]