

Abstract
Understanding the epidemiological and evolutionary dynamics of rapidly evolving pathogens is one of the most challenging problems facing disease ecologists today. To date, many mathematical and individual-based models have provided key insights into the factors that may regulate these dynamics. However, in many of these models, abstractions have been made to the simulated sequences that limit an effective interface with empirical data. This is especially the case for rapidly evolving viruses in which de novo mutations result in antigenically novel variants. With this focus, we present a simple two-tiered ‘phylodynamic’ model whose purpose is to simulate, along with case data, sequence data that will allow for a more quantitative interface with observed sequence data. The model differs from previous approaches in that it separates the simulation of the epidemiological dynamics (tier 1) from the molecular evolution of the virus's dominant antigenic protein (tier 2). This separation of phenotypic dynamics from genetic dynamics results in a modular model that is computationally simpler and allows sequences to be simulated with specifications such as sequence length, nucleotide composition and molecular constraints. To illustrate its use, we apply the model to influenza A (H3N2) dynamics in humans, influenza B dynamics in humans and influenza A (H3N8) dynamics in equine hosts. In all three of these illustrative examples, we show that the model can simulate sequences that are quantitatively similar in pattern to those empirically observed. Future work should focus on statistical estimation of model parameters for these examples as well as the possibility of applying this model, or variants thereof, to other host–virus systems.
Keywords: disease dynamics, viral evolution, multi-strain model, influenza, phylodynamics
1. Introduction
The ecological and evolutionary dynamics of many RNA viruses have been increasingly well described over the last several decades, yet the factors driving their dynamics are still only poorly understood. One approach towards identifying key factors is through the formulation of mathematical models that, when analysed analytically or simulated, yield quantitative predictions of the case dynamics and the evolutionary dynamics of the viral population (Holmes & Grenfell 2009). In the case of antigenically variable viruses, these ‘phylodynamic’ models (Grenfell et al. 2004) frequently incorporate multiple antigenically distinct strains and keep track of either the immune status or the infection histories of individuals in the host population.
These multi-strain models range in complexity from the very simple and abstract (e.g. Girvan et al. 2002; Tria et al. 2005) to the more complex and biologically realistic (e.g. Ferguson et al. 2003; Koelle et al. 2006). Many of them yield dynamics that are qualitatively consistent with the dynamics they seek to reproduce. For example, when parametrized with a short duration of infection, the status-based multi-strain model developed by Gog & Grenfell (2002) yields self-organized sets of strains that turn over in time, consistent with empirical patterns of influenza. Other examples are the phylodynamic models developed by Ferguson et al. (2003) and Koelle et al. (2006), both of which yield case dynamics and viral diversity patterns that are qualitatively similar to those observed and phylogenies that resemble the known topology of influenza's haemagglutinin (HA) protein. Although these multi-strain models, among others, have been able to simulate dynamics that are consistent with particular features of the observed data, many of these models embody different mechanistic hypotheses about what factors play dominant roles in shaping the dynamics. For example, the model by Gog & Grenfell (2002) considers only strain-specific immunity, whereas the model by Ferguson et al. (2003) considers the additional role that generalized immunity may play in shaping the evolutionary dynamics of influenza's HA. The model by Koelle et al. (2006) considers a third hypothesis: that the evolutionary dynamics of influenza's HA are shaped by periodic selective sweeps occurring during antigenic cluster transitions.
Given this growing set of phylodynamic models that differ in their mechanistic hypotheses, determining which model performs best when confronted statistically with observed data is now necessary. In the case of phylodynamic models, these data come in two forms: epidemiological (case) data and evolutionary (sequence) data. Interfacing disease models with case data has a long history (e.g. Bobashev et al. 2000; Finkenstädt & Grenfell 2000; Koelle & Pascual 2004; Xia et al. 2005; Ionides et al. 2006; King et al. 2008), with a subset of these analyses focusing on parameter estimation and model selection for antigenically variable RNA viruses (Xia et al. 2005; Fraser et al. 2009). However, phylodynamic models have to date not routinely been tested statistically against observed sequence data.
A quantitative comparison of simulated sequence data against observed sequence data could focus on a number of different sequence-derived patterns. These include divergence and diversity patterns, as well as quantitative comparisons of phylogenies reconstructed from simulated and observed sequences. Although many phylodynamic models have considered at least one of these patterns (Girvan et al. 2002; Ferguson et al. 2003; Tria et al. 2005; Koelle et al. 2006; Minayev & Ferguson 2009a,b), the comparisons against observed data have been only qualitative in nature. The reason for this limitation lies in the current inability of these models to capture these patterns quantitatively. This does not imply that these models are missing the relevant processes at play; rather, the quantitative mismatch between model-simulated sequence data and empirical sequence data results from the way in which sequences have been represented in these models. Specifically, phylodynamic models to date have simplified the representation of viral sequences by considering bitstrings (Girvan et al. 2002; Tria et al. 2005), a subset of codons (Ferguson et al. 2003; Koelle et al. 2006) or a limited number of antigenic loci (Recker et al. 2007; Minayev & Ferguson 2009a,b). These sequence representations have made the models computationally tractable at the cost of simulating sequences that differ in length (or in structure) from the empirical sequences with which they are being compared. In the case of the models that simulate a subset of codons, a quantitative comparison could of course be made between empirical and simulated sequences, if only a subset of the empirical sequences were considered. However, considering a subset of sites introduces several difficulties. First, which subset should be used? Our understanding of which sites are important for antigenic change is still incomplete. Second, if different phylodynamic models represent their sequences differently, a quantitative comparison against sequence data would use different subsets of the data. This would result in models not being compared against the same sequence dataset, making difficult the process of model selection.
To enable a quantitative comparison between simulated and observed sequence data, we here develop a new phylodynamic model that makes explicit the difference between antigenic change and genetic change, and thereby makes it computationally feasible to model sequences in their entirety. Specifically, the model we formulate consists of two tiers. The first tier of the model simulates the ecological dynamics of the virus and its antigenic phenotypes. As such, it builds conceptually on the idea that strain phenotypes (i.e. antigenic variants or clusters), instead of genotypes, can be used as the ‘fundamental particle’ for modelling RNA viruses such as influenza (Plotkin et al. 2002). More recently, Gökaydin et al. (2007) and Ballesteros et al. (2009) have used this phenotype-level approach to consider the invasion dynamics of a new antigenic cluster into a host population with a resident cluster. Most relevant to the research presented here, Koelle et al. (2009) have recently introduced an antigenic tempo model, which, in a modified version, we use here as the first tier of our two-tiered model.
The second tier of the model simulates the molecular evolution of a virus's antigenic protein. It does so by taking as given the epidemiological dynamics simulated in the first tier of the model. Biologically, this separation of ecological dynamics from evolutionary dynamics is of course absurd: the molecular changes of a virus drive the emergence of new antigenic variants, and therewith affect the case dynamics. However, the effect of molecular changes on the epidemiological dynamics is indirect, with the link being the dynamics of the antigenic phenotypes. As such, to simulate case dynamics, a phenomenological model that reproduces the emergence dynamics of antigenic variants can be considered. It is this modular separation of phenotypic dynamics from genotypic dynamics that simplifies the computational complexity of the simulations and thereby allows us to simulate viral sequences that can be statistically compared with empirical sequence data. Below, we first describe the epidemiological submodel and detail (in appendix A) how this model can be mechanistically interpreted in terms of mutations that enable immune escape. We then describe the second tier of the model, the molecular evolution submodel. A schematic overview of the two tiers of the model and the flow of simulated data is shown in the electronic supplementary material, figure S1. The Matlab source code is available for download from the corresponding author's website.
As described here, the two-tiered model assumes that viruses evolve antigenically in a punctuated manner. As such, mutations are assumed to be antigenically neutral or nearly so most of the time, with only the rare mutation resulting in a large antigenic change. This model is, therefore, a simplification of the previously published phylodynamic model of Koelle et al. (2006), which hypothesizes that occasional antigenic innovations and the selective sweeps that accompany their emergence are the two key factors that drive the evolutionary dynamics of influenza A (H3N2)'s HA in humans. The model presented here improves upon this previous model in terms of both its simplicity and its ability to reproduce the quantitative patterns of the observed sequence data.
Following the description of the model, we provide three case studies to illustrate the flexibility of the model and the diversity of dynamics that it can generate. The first application is to influenza A (H3N2) in humans, the second to influenza B in humans and the third to H3N8 in equine hosts. Our first application to influenza A (H3N2) shows that gradual antigenic evolution within antigenic clusters is necessary to reproduce the ecological and evolutionary patterns of the observed data. To our knowledge, it is the first phylodynamic model that can quantitatively reproduce the known patterns of viral diversity and divergence over time. Our second application demonstrates that the model under a different parametrization can generate the emergence and maintenance of two viral lineages, consistent with the evolutionary patterns observed for influenza B. Our third application serves to illustrate the possibility of extending the model. Specifically, we extend the two-tiered model to two patches (representative of North America and Europe) to show that it can reproduce the evolutionary dynamics of influenza A (H3N8) in equine hosts subject to transatlantic quarantine measures.
Although all three of our applications focus on influenza, this two-tiered model is not limited to this virus, as other RNA viruses (e.g. HIV and norovirus) appear to evolve by punctuated immune escape (Cobey & Koelle 2008). The model is also not limited to the assumption of punctuated antigenic evolution, as will be discussed in §4, although it is in this case that the modular advantages of this model are most clearly evident.
2. The two-tiered mathematical model
2.1. Tier 1: the epidemiological submodel
To model the virus's epidemiological dynamics, we improve on the antigenic tempo model recently presented elsewhere (Koelle et al. 2009). This model starts with a given multi-strain model formulation, interpreting strains in terms of major antigenic variants instead of in terms of unique genotypes. As in Koelle et al. (2009), we use a status-based approach to modelling strain interactions (Gog & Grenfell 2002), which assumes polarized immunity. The deterministic dynamics of susceptible and infected individuals belonging to a major antigenic variant i are captured by equations of the form
 |
2.1 |
and
| 2.2 |
where N is the population size, μ is the birth rate and the death rate, γ is the rate of within-variant waning immunity, ν is the recovery rate, β is the transmission rate and n is the total number of antigenic variants that have circulated in the population up to time t. The dynamics of individuals immune to variant i, Ri, are not shown, as they can be easily computed by Ri = N − Si − Ii. As previously described in Koelle et al. (2009), the degree of immunity between variants i and j is given by σij = θlij, where θ is the degree of immunity between a mother–daughter variant pair and lij is the antigenic kinship level between variants i and j. 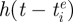 is the per capita antigenic emergence rate, defined as the rate at which individuals infected with variant i give rise to a new antigenic variant. We allow this rate to depend on the age of the variant,
is the per capita antigenic emergence rate, defined as the rate at which individuals infected with variant i give rise to a new antigenic variant. We allow this rate to depend on the age of the variant, 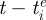 , where
, where 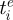 is the time at which variant i emerged in the population. Specifically, using an approach similar to the punctuated model of antigenic change detailed in Koelle et al. (2009), we model the per capita antigenic emergence rate,
is the time at which variant i emerged in the population. Specifically, using an approach similar to the punctuated model of antigenic change detailed in Koelle et al. (2009), we model the per capita antigenic emergence rate, 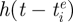 , phenomenologically by using a Weibull hazard function, with scale parameter λ and shape parameter k
, phenomenologically by using a Weibull hazard function, with scale parameter λ and shape parameter k
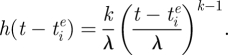 |
2.3 |
When k = 1, this function reduces to a constant rate of antigenic change, which many previous multi-strain models have assumed. However, we consider the case of k > 1, such that the rate of antigenic change increases with the age of the variant. This phenomenological increase in the rate of phenotypic change can be interpreted in several ways. First, it is consistent with ‘rules of thumb’ that have been developed to predict the emergence of antigenically novel variants. These rules of thumb generally specify that a certain number of amino acid changes in epitope regions are required to precipitate a major antigenic change (Wiley et al. 1981; Wilson & Cox 1990). Because it takes time to accumulate these amino acid changes, an endemic viral variant that has been circulating in a population is more likely to give rise to a new variant the longer it persists in the population. Second, an increase in the rate of antigenic change with variant age can be understood in terms of neutral networks in genotype space (Lau & Dill 1990; Koelle et al. 2006; van Nimwegen 2006). As an antigenic variant ages, the sequences that it comprises are accumulating neutral or nearly neutral mutations that are changing the genetic backgrounds of the sequences. Effectively, this exploration of sequence space can therefore lead to a per capita rate of antigenic emergence that increases with variant age. Third, and more formally, we can mechanistically interpret the rate of antigenic change increasing with the age of a variant by considering a Markov model that considers mutation accumulation. We outline this model in appendix A.
The model specified by equations (2.1)–(2.3), and schematically shown as the first tier in the electronic supplementary material, figure S1, differs from the original model formulation described in Koelle et al. (2009) in two ways. First, we allow for the possibility of waning immunity within each antigenic variant (which occurs when γ > 0), such that the dynamics within each antigenic variant are governed by susceptible-infected-recovered-susceptible (SIRS) dynamics instead of susceptible-infected-recovered (SIR) dynamics. Our model is therefore more general, allowing for SIR dynamics when γ = 0. Second, we simplify the model by simulating it stochastically in its entirety instead of using the stochastic hybrid approach described in Koelle et al. (2009). This change to a fully stochastic simulation removes the need to simulate the additional variables that are used to determine the emergence times of new phenotypes. Table 1 maps the deterministic equations constituting the model (equations (2.1)–(2.3)) into Markov chain events and their associated transition rates, which are used to simulate the model stochastically with the Gillespie τ-leap algorithm (Gillespie 2007).
Table 1.
The Markov chain events and their transition rates used to stochastically simulate the epidemiological submodel. Events are shown for a focal variant i. Events and transition rates for all other variants j in the set {1, 2, 3, …, n} are analogous. The population size N is given by Si + Ii + Ri. Several constraints exist in the system and are respected during the simulation. Because Sj + Ij + Rj = N for all variants j, a birth event, occurring at rate μN, increases the number of susceptible hosts for each variant j by 1 (i.e. births do not occur independently for each variant j). Similarly, a death, also occurring at rate μN, decreases the number of hosts S, I or R to each variant j by 1. This decrease is taken from Sj, Ij or Rj with probability Sj/N, Ij/N and Rj/N, respectively. The rate of possible infection with variant j (related to the force of infection) is given by βIj. When i is equal to j, a ‘possible infection’ event results in an infection with probability Si/N. When i is not equal to j, this possible infection event results in a gain of immunity through polarized cross-immunity with probability σij (Si/N). Recovery of an individual infected with variant i and the loss of immunity to variant i occur at rates νIi and γRi, respectively. An antigenic emergence event results in a decrease in the number of individuals infected with variant i and the stochastic appearance of a new antigenic variant, variant n + 1.
| event | change | rate |
|---|---|---|
| birth | (Si,Ii,Ri) → (Si + 1,Ii,Ri) | μN |
| death | (Si,Ii,Ri) → (Si − 1,Ii,Ri) with probability Si/N; (Si,Ii,Ri) → (Si,Ii − 1,Ri) with probability Ii/N; (Si,Ii,Ri) → (Si,Ii,Ri − 1) with probability Ri/N | μN |
| possible infection | for i = j: (Si,Ii,Ri) → (Si − 1,Ii + 1,Ri) with probability Si/N; (Si,Ii,Ri) → (Si,Ii,Ri) with probability (1 − Si/N) for i ≠ j: (Si,Ii,Ri) → (Si − 1,Ii,Ri + 1) with probability σijSi/N; (Si,Ii,Ri) → (Si,Ii,Ri) with probability (1 − σijSi/N) | βIj |
| recovery | (Si,Ii,Ri) → (Si,Ii − 1,Ri + 1) | νIi |
| loss of immunity | (Si,Ii,Ri) → (Si + 1,Ii,Ri − 1) | γRi |
| antigenic emergence | (Si,Ii,Ri) → (Si,Ii − 1,Ri + 1) and (Sn+1, 0,Rn+1) → (Sn+1 − 1, 1,Rn+1) | h(t − tie)Ii |
The majority of the events and their rates shown in table 1 are frequently used in stochastic simulations of disease dynamics. The one exception is the antigenic emergence event, which brings phenotypic novelty into the viral population. When an antigenic emergence event occurs for variant i in the simulation, it results in an individual infected with variant j, where j = n + 1 and n is the number of variants that have been in the population up to time t. The event also results in a decrease in the number of individuals infected with variant i from Ii to Ii − 1 (table 1). The number of hosts susceptible to variant j can be computed at the time of variant j's emergence if the numbers of births, deaths and infections have been tracked over the simulation.
2.2. Tier 2: the molecular evolution submodel
The second tier of the model consists of a molecular evolution submodel, which generates a set of time-stamped viral sequences from which a phylogeny can be inferred and from which diversity and divergence patterns can be constructed. This submodel uses as input the variant-specific disease dynamics from the epidemiological submodel, but does not provide any feedback to it (electronic supplementary material, figure S1). To simulate the molecular evolution submodel, a desired number of sampled sequences s is first specified. Times of isolation are then randomly assigned to each of these s sequences, taking into consideration the number of infected individuals at each time point. This is done by setting the probability of the time of isolation being day k as I(k)/∑iI(i), where I(k) is the total number of individuals infected on day k and index i sums over all days of the simulation. Each time-stamped sequence is then probabilistically assigned an antigenic variant to which it belongs by letting the probability that the sequence belongs to variant j be given by Ij(k)/∑iIi(k) where k is the day of the sequence's isolation and index i sums over all antigenic variants present on day k.
A desired nucleotide length l for the viral sequences is then specified, along with a mutation rate mnuc in units of nucleotide changes per site per year. The per-sequence mutation rate m is given by the product mnucl. Finally, we specify a given model of sequence evolution and provide parameters associated with this model. The model of sequence evolution chosen can be extremely simple and parameter sparse. For example, Kimura's two-parameter model requires only one parameter for the transition rate and one parameter for the transversion rate (Felsenstein 2004). Alternatively, the model chosen can be more complicated. For example, the general time reversible (GTR) model with a proportion of invariant sites (I) and a gamma distribution of rate variation (Γ) would entail specifying 11 parameters: the frequencies of the four nucleotide bases, the transition rates between each pair of bases, the proportion of invariant sites and a parameter α that controls the shape of the gamma distribution (Felsenstein 2004).
Under a given model of sequence evolution and its associated parameter values, site-specific mutation rates are then assigned to each of the l nucleotide locations. Under Kimura's two-parameter model, the mutation rate of each site is mnuc. Under the GTR + I + Γ model, the mutation rate at each site is assigned based on the proportion of invariant sites I and the α parameter of the Γ distribution, such that the per-sequence mutation rate comes out to be m.
To begin the molecular evolution submodel simulation, a single sequence of length l is generated, with each site probabilistically assigned a nucleotide depending on base frequencies specified by the model of sequence evolution. This sequence belongs to antigenic variant i = 1, and all infected individuals on day 0 of the simulation are infected with this strain. Starting with the first antigenic variant, all sampling times of the (genetically yet undetermined) sequences that belong to variant 1 are found, as are all the emergence times of variants that were generated specifically by this variant. Then, starting from day 0, from each day to the next, two processes occur: mutation and transmission of extant sequences. Mutation is simulated by first determining the number of mutations that will occur on a particular day. This number is determined by drawing from a Poisson distribution with mean mI, where I is the number of individuals infected on a particular day. These mutations occur in the viral population (of size I) at sites that are chosen randomly, weighted by their per site mutation rate mnuc. A mutation assigned at a chosen nucleotide site in a chosen individual results in a nucleotide change that reflects the transition rates given by the model of sequence evolution. Transmission is simulated by first calculating, from one day to the next, the number of new infections and the number of recoveries, both only of the first variant. New infections are then drawn from the extant pool of viral sequences belonging to variant 1 and recoveries are chosen from this same pool. Recoveries always occur randomly, while new infections are chosen depending on the selective advantage of each viral sequence.
The selective advantage of a sequence is given by fk, where k is the sequence's nucleotide distance from the founder sequence of the variant and f is a parameter specifying the intensity of the selective advantage a single nucleotide change confers within a single antigenic variant. In the case of purely neutral evolution within antigenic variants (γ = 0 in equation (2.1)), f = 0, such that new infections are chosen randomly from the pool of extant sequences. In the case of gradual antigenic evolution within antigenic variants (γ > 0 in equation (2.1)), f > 0, such that new infections are preferentially chosen from the set of extant sequences that have higher divergence from their founder sequence. (Clearly, there should be a quantitative relationship between the parameters γ and f, with higher values of f associated with higher values of γ. Unfortunately, at this point, we do not have an equation mechanistically linking the value of f to the value of γ as we have for linking λ to m (appendix A).) The implementation of the transmission step thereby captures both demographic stochasticity in disease transmission and the possibility for within-variant selection to act.
Throughout the day-to-day simulation of mutations and transmission events, sampling times of sequences belonging to variant 1 will be reached, as will emergence times of variants that arose from variant 1. When a sampling time is reached, a viral sequence is randomly chosen from the pool of infected individuals; this simulated sequence is now one of the s sequences that can be used in inferring a phylogeny. When an emergence time is reached, a viral sequence is randomly chosen from the pool of infected individuals and mutated at a single nucleotide location; this simulated sequence is now the founder of the antigenic variant born of variant 1 at that time. Alternatively, to be strictly consistent with the Markov chain process detailed above, the viral sequence can be chosen preferentially according to its nucleotide distance k from its founder sequence.
The same iterative procedure of mutation, transmission and sequence sampling is then performed for variant 2 through to the last variant observed in the simulation. The set of sampled sequences that results from this simulation can then be compared against empirical sequence data. Although the simulated sequences will differ from the observed ones genetically, patterns of sequence evolution (e.g. divergence and diversity patterns) can be compared quantitatively across these datasets, and a phylogeny can be inferred from all s sampled sequences and compared against phylogenies inferred from empirical sequences.
3. An application of the model to influenza dynamics in humans
To illustrate its use, we simulate the two-tiered model described above with parameter values that are reasonable for influenza. We use influenza as an application because sequence data are readily available for its dominant antigenic protein, the HA, and because the advantages of the two-tiered model in terms of its modular design are most evident for a system like influenza, where many mutations in the HA protein have been shown to be neutral or nearly so and single mutations have been shown to result in large antigenic changes (Webster & Laver 1980; Nakajima et al. 1983; Berton et al. 1984; Nakagawa et al. 2000, 2001a,b, 2002; Smith et al. 2004).
3.1. Simulating the dynamics of influenza A (H3N2) in humans
Influenza A subtype H3N2 has been circulating in humans since 1968, when a reassortment event between the previously circulating influenza A subtype, H2N2, and a swine influenza virus resulted in its pandemic spread. From 1968 until the present, this subtype has dominated the flu season, with annual H3N2 attack rates estimated to be between 2 and 10 per cent. In temperate regions, H3N2's disease dynamics are largely annual, with occasional years of low activity (figure 1a,b). The evolutionary dynamics of the virus are characterized by the emergence and replacement of antigenic clusters, occurring every 2–8 years (Plotkin et al. 2002; Smith et al. 2004) and, genetically, by the ladder-like phylogeny of its HA protein (figure 1c) (Fitch et al. 1997).
Figure 1.

Observed dynamics of influenza A (H3N2) in humans. (a) The case dynamics of influenza in France over the period 1984–2008 are shown in black (http://www.sentiweb.org/). Starting in 1997, FluNet data were available to estimate variant-specific dynamics (http://gamapserver.who.int/GlobalAtlas/). The subset of cases attributed to influenza A (H3N2) is shown in red after this date. (b) The annual attack rate of influenza in France, computed from the case dynamics shown in (a). Starting in 1997, the attack rate is partitioned into H3N2 cases (red) and H1N1/B cases (grey). Case dynamics are shown for France owing to the availability of data. Similar case dynamics are observed in other temperate regions, with similar estimated attack rates. (c) The phylogeny of influenza A (H3N2)'s HA, inferred from antigenically typed sequences isolated between 1968 and 2003 (Smith et al. 2004). Sequences are coloured by antigenic cluster.
To apply the two-tiered model to influenza A (H3N2) in humans, we first modify the epidemiological submodel shown in equations (2.1) and (2.2) to incorporate a slightly larger degree of realism. Because we will compare the model simulations with the dynamics observed in a temperate region (figure 1a), we include in our simulations seasonal forcing of the transmission rate and a small immigration rate. With these modifications, equations (2.1) and (2.2) become
 |
3.1 |
and
 |
3.2 |
where ɛ is the strength of seasonal forcing, r is the immigration rate and pi is the proportion of cases that belong to antigenic cluster i.
We simulate this submodel under two parameter sets: (i) purely punctuated antigenic evolution and (ii) antigenic evolution that includes components of both punctuated and gradual antigenic change. A third parameter set, with only gradual antigenic evolution, is considered in the electronic supplementary material. Under the first parameter set, there is no waning of immunity within antigenic clusters (γ = 0). In contrast, in simulations with the second parameter set, individuals can become reinfected with a given antigenic cluster after an average duration of immunity 1/γ. All other parameters of the model are chosen to be consistent with values from the literature, where possible (figure 2, legend).
Figure 2.

Epidemiological submodel simulation under a model of purely punctuated antigenic evolution. (a) Simulated case dynamics. Antigenic clusters are colour-coded. (b) Simulated annual attack rates. (c) Proportion of cases belonging to each antigenic cluster. Parameters used: N = 300 million, R0 = 2, μ = 1/70 yr−1, ν = 1/3 d−1, γ = 0 yr−1, ɛ = 0.25, θ = 0.8 (Gill & Murphy 1977), r = 0.01 N person yr−1 and Weibull parameters k = 2, λ = 500. The epidemiological submodel was simulated stochastically, using Gillespie's τ-leap algorithm (Gillespie 2007) with a time interval τ of half a day.
Although the epidemiological submodel parametrized for purely punctuated antigenic evolution reproduces the emergence–replacement dynamics of influenza A (H3N2)'s antigenic clusters (figure 2a,c), it fails to reproduce other features of H3N2's dynamics. Specifically, the simulated annual attack rates are generally too low and the emergence of a new antigenic cluster results in a year having an unrealistically large deviation from other years' attack rates (figure 2a,b). However, the epidemiological submodel parametrized for a combination of punctuated antigenic evolution and gradual antigenic evolution is capable of reproducing the observed patterns of seasonal and interannual variability, as well as the magnitude of H3N2's attack rates (figure 3a,b), while maintaining the ability to reproduce the emergence and replacement of antigenic clusters every 2–8 years (figure 3c). These emergence–replacement dynamics result from the strong selective advantage of novel antigenic variants, arising from the greater number of susceptible hosts available to them (figure 3d). The results shown in figures 2 and 3 are consistent with the recent findings by Ballesteros et al. (2009) that both gradual and punctuated antigenic evolution are necessary to effectively reproduce influenza A (H3N2)'s disease dynamics. A model that considers only gradual antigenic evolution does not reproduce the observed dynamics well, yielding strictly annual cycles with very low incidence rates (electronic supplementary material).
Figure 3.

Epidemiological submodel simulation under a model of punctuated and gradual antigenic evolution. (a) Simulated case dynamics. (b) Simulated annual attack rates. (c) Proportion of cases belonging to each antigenic cluster. (d) The fraction of hosts susceptible to each antigenic cluster, shown over the lifespan of each cluster. Parameters used were as in figure 2, with the exception of γ, which here is γ = 1/8 yr−1.
To simulate the molecular evolution submodel, we specify the length of the HA (l = 987 nucleotides) and the HA mutation rate (mnuc = 5.7 × 10−3 nucleotide substitutions per site per year). We also use a GTR + I + Γ model of sequence evolution, with parameters that were estimated from a maximum-likelihood fit to H3N2 sequences whose phylogeny is shown in figure 1c (figure 4, legend). (Alternatively, we could use a simpler model of sequence evolution; our decision to use the GTR + I + Γ model is based on improving the realism of the model, without requiring additional free parameters.) For the model parametrized for punctuated antigenic evolution, we set f, the degree of within-cluster positive selection, to 0. For the model parametrized for a combination of punctuated and gradual antigenic evolution, we allow for some positive selection within antigenic clusters by setting f = 0.01. Setting f > 0 is qualitatively consistent with antigenic maps for H3N2 that show a scatter of points for each antigenic cluster, indicating imperfect cross-immunity between viral strains belonging to a single cluster (Smith et al. 2004). The phylogenies inferred from the simulated sequences of both models (figure 4a,b) reproduce the cactus-like topology of H3N2's HA phylogeny, consisting of a long trunk and short side branches (figure 1c). Diversity and divergence patterns of the simulated HA sequences can also be plotted and compared against those observed empirically (figure 5a–f). The model with only punctuated antigenic evolution fails to reproduce the rapid divergence that is empirically observed (figure 5c versus 5a). In contrast, the model with a combination of gradual and punctuated evolution captures this pattern quantitatively (figure 5e versus 5a). Although the model with both gradual and punctuated evolution clearly performs better in terms of reproducing divergence patterns, both models are capable of reproducing the sequence diversity patterns of H3N2's HA (figure 5d,f versus 5b). In the electronic supplementary material, we further considered whether the model of only gradual antigenic change could effectively reproduce the evolutionary patterns of influenza's HA. To some extent, it was able to do so. However, we also elaborate on why the molecular evolution submodel, as specified, is not appropriate for this application and review results from an existing model which show that gradual antigenic evolution leads to explosive viral diversity (as previously also shown in Ferguson et al. (2003)), inconsistent with a ladder-like phylogeny.
Figure 4.

Phylogenies inferred from sequences simulated by the molecular evolution submodel. (a) Phylogeny inferred from sequences simulated under a model of purely punctuated antigenic evolution (f = 0). (b) Phylogeny inferred from sequences simulated under a model of punctuated and gradual antigenic evolution (with f = 0.01). Both phylogenetic trees were inferred from 300 simulated HA sequences of length l = 987 nucleotides, spanning 35 years. In both simulations, mnuc = 5.7 × 10−3 mutations per nucleotide site per year (Fitch et al. 1997), and GTR + I + Γ parameters were set to those estimated from the H3N2 phylogeny shown in figure 1c. These were: (πA, πC, πG, πT) = (0.35, 0.22, 0.21, 0.22), transition rates: AC = 1.21, AG = 4.75, AT = 0.62, CG = 0.17, CT = 4.52, GT = 1, gamma distribution parameter α = 1.21 and proportion of invariant sites f0 = 0.28.
Figure 5.

Divergence and diversity patterns of empirical and simulated HA sequences. (a) Divergence of observed HA sequences from influenza A (H3N2) strain BI/16398/68, coloured by antigenic cluster. (b) Diversity of observed HA sequences over time. (c) Divergence of simulated HA sequences from the initial strain, coloured by antigenic cluster, for the purely punctuated model of antigenic evolution. (d) Diversity of simulated HA sequences over time for the purely punctuated model of antigenic evolution. (e) Divergence, as in (c), with sequences simulated from the model of punctuated and gradual antigenic evolution. (f) Diversity, as in (d), with sequences simulated from the model of punctuated and gradual antigenic evolution. For all plots, divergence is defined as the Hamming distance from each sequence to the reference strain, and diversity is defined as the average pairwise nucleotide distance of sequences sampled in a given year.
The results of the simulations shown here and in the appendix indicate, first, that punctuated antigenic change is an important driver of influenza A (H3N2)'s dynamics; second, that gradual antigenic evolution within clusters contributes to influenza A (H3N2)'s ecological and evolutionary dynamics; and, third, that the two-tiered model, when appropriately parametrized, can generate sequence data that quantitatively reproduce patterns in observed sequence data.
3.2. Simulating the dynamics of influenza B in humans
We now consider the dynamics of influenza B in humans, and whether an alternative parametrization of the two-tiered model can yield ecological and evolutionary dynamics consistent with this influenza type. Unlike the largely annual epidemics of influenza A (H3N2), influenza B epidemics occur only every 2–4 years (figure 6a) (Monto & Kioumehr 1975). Also different from H3N2, the evolution of influenza B's HA is characterized by two distinct lineages, the Yamagata lineage and the Victoria lineage (figure 6b) (Rota et al. 1992). Epidemic seasons of influenza B usually have one of these two lineages dominating, although a co-occurrence of strains from both lineages is occasionally observed (Nerome et al. 1998). Within each of these two lineages, genetic and antigenic viral turnover has been documented (Rota et al. 1992; Nerome et al. 1998; Nakagawa et al. 2001a,b).
Figure 6.

Observed versus simulated dynamics of influenza B in humans. (a) As in figure 1a, the case dynamics of influenza in France over the period 1997–2008 are shown in black. Cases attributed to influenza B are shown in red. (b) The phylogeny of influenza B's HA from isolates spanning the years 1973–2008. Sequences were randomly sampled from the influenza virus resource database. (c) Epidemiological submodel simulations of influenza B case dynamics under a model of both punctuated and gradual antigenic change. Antigenic clusters are colour-coded. (d) Proportion of cases shown in (c) belonging to each antigenic cluster. Parameters used: N = 300 million, R0 = 2, μ = 1/70 yr−1, ν = 1/7 d−1, γ = 1/15 yr−1, ɛ = 0.25, θ = 0.80, r = 0.005 and Weibull parameters k = 2, λ = 700. (e) Phylogeny of 600 simulated HA sequences, of length l = 1041 nucleotides and spanning 30 years. The GTR + I + Γ parameters were set to those estimated from the influenza B phylogeny shown in (b). These were: (πA, πC, πG, πT) = (0.35, 0.20, 0.20, 0.25), transition rates: AC = 1.42, AG = 6.15, AT = 0.54, CG = 1.03, CT = 7.19, GT = 1, gamma distribution parameter α = 0.79 and proportion of invariant sites f0 = 0.26. Other parameters for the molecular evolution submodel were mnuc = 2.85 × 10−3 mutations per nucleotide site per year and f = 0.01.
To reflect a lower mutation rate for influenza B (when compared against the mutation rate for influenza A) (Nobusawa & Sato 2006), we reparametrized the epidemiological model with a lower value of γ, a lower value of r and a higher value of λ (figure 6, legend). A lower value of γ reflects slower within-cluster waning of immunity, owing to a lower mutation rate. This lower value of γ also reduces the equilibrium number of individuals infected with influenza B when one cluster is endemic. This in turn would reduce the immigration rate r of infected individuals. Lastly, with a lower mutation rate, we would expect a higher value of the Weibull scale parameter λ (appendix A).
In simulations of the two-tiered model with this alternative parametrization, we find that these changes in parameter values result in lower attack rates and more interannual variability (results not shown), consistent with the observed epidemiological dynamics. However, the model with just these parameter changes did not reproduce the observed pattern of lineage (and cluster) co-circulation.
When the model is simulated with a longer duration of infection, however, two distinct lineages emerge and persist, with antigenic clusters replacing one another periodically within each lineage. Furthermore, the two lineages exhibit asynchronous dynamics, with one lineage usually dominating an influenza B season (figure 6c,d). The phylogeny inferred from the simulated sequences also reproduces the general topology of the phylogeny inferred from the empirical sequences (figure 6e).
We chose to consider a longer duration of infection because influenza B primarily affects children, and children are known to be infectious for longer periods of time than adults. Although these simulations can recover features of the observed evolutionary and ecological dynamics of influenza B, other parameter differences from influenza A (H3N2) not explored here (e.g. in the degree of cross-immunity between antigenic clusters) could result in similar dynamics. Statistical estimation of model parameters will therefore be critical for identifying the parameters that play the greatest role in shaping the dynamical differences between these two types of influenza.
3.3. Simulating the dynamics of influenza H3N8 in equine hosts
To illustrate the ease with which the two-tiered model can be extended, we now consider the evolutionary dynamics of influenza (H3N8) in equine hosts. This subtype of equine influenza A virus (EIV) was first isolated from its hosts in 1963 in Florida (Waddell et al. 1963). Since then, the virus has undergone many genetic and antigenic changes that resulted in EIV outbreaks in horses around the world (Daniels et al. 1985; Kawaoka et al. 1989; Oxburgh et al. 1994). H3N8 is currently the only known strain of EIV circulating in equine hosts; the H7N7 subtype of EIV has not been isolated in horses since 1980 (Webster 1993).
The patterns of genetic and antigenic change in EIV resemble the evolutionary patterns of influenza B virus in humans. Phylogenetic analysis of H3N8 from 1963 to 1986 shows a pattern of relatively linear evolution (Oxburgh et al. 1994) with lineage turnover and low genetic diversity (figure 7a). Some time between 1986 and 1990, H3N8 split into two distinct lineages, referred to as the ‘European’ and ‘American’ lineages, based on their geographical origin (figure 7a) (Lindstrom et al. 1994; Daly et al. 1996; Oxburgh et al. 1998; Oxburgh & Klingeborn 1999; Lai et al. 2004). This split was accompanied by the extinction of the older viral lineages (Daly et al. 1996; Oxburgh et al. 1998; Oxburgh & Klingeborn 1999; Lai et al. 2004). After some time, co-circulation of the two lineages was reported in European countries, such as Sweden and the UK (Daly et al. 1996; Oxburgh & Klingeborn 1999), but, interestingly, only a single isolate of the European lineage was observed in North America (the Canadian isolate A/Eq/Saskatoon/90) (Lai et al. 2004). The American lineage continued to diversify genetically, evolving into several sublineages (Lai et al. 2004; Bryant et al. 2009). In contrast, the European lineage has declined in prevalence and appears to be heading towards extinction, with only a few European isolates having been observed since 1993 (figure 7a) (Oxburgh et al. 1998; Bryant et al. 2009).
Figure 7.

Phylogenies of influenza A H3N8's HA in equine hosts, reconstructed from empirical and simulated viral sequences. (a) Phylogeny inferred from equine influenza virus H3N8 sequences isolated in Europe and North America. Split of the American and European lineages occurred between 1986 and 1990. Sequences isolated from North America are shown in blue. Sequences isolated from Europe are shown in red. Three antigenic variants have been well characterized: the original Mia/63 antigenic variant (yellow), the Ky/76 variant (purple) and an unnamed variant (orange) that appears antigenically similar to Mia/63 (Hinshaw et al. 1983). (b) Phylogeny inferred from 200 simulated H3N8 HA sequences. Prior to quarantine, there is linear evolution of EIV's HA until the lineage split in the late 1980s. Viral isolates belonging to the simulated European lineage were only observed in the European patch, whereas viral isolates belonging to the American lineage were isolated in both the North American patch and the European patch. The GTR + I + Γ parameters used in the molecular sequence evolution model were set to those estimated from the influenza A (H3N8) phylogeny shown in (a). These were: (πA, πC, πG, πT) = (0.35, 0.21, 0.22, 0.22), transition rates: AC = 0.97, AG = 5.65, AT = 0.83, CG = 0.35, CT = 7.74, GT = 1, gamma distribution parameter α = 0.91 and proportion of invariant sites f0 = 0.17. Other parameters for the molecular evolution submodel were mnuc = 5.7 × 10−3 mutations per nucleotide site per year and f = 0.01.
As for flu in humans, the recurrence of H3N8 in the horse population has been attributed to the antigenic drift of the virus's HA protein (Hinshaw et al. 1983; Kawaoka et al. 1989; Berg et al. 1990; Endo et al. 1992). Haemagglutination inhibition assays of H3N8 isolates from 1963 to 1986 have indicated that the antigenic evolution of H3N8 parallels to a large extent the genetic evolution of the virus's HA (figure 7a) (Hinshaw et al. 1983; Kawaoka et al. 1989).
Although similar to influenza B's evolutionary dynamics, the dynamics of H3N8 in equine hosts is complicated by the international movement of race horses. Geographical isolation, through quarantine measures, has been proposed as an explanation for the appearance of the distinct European and North American lineages (Lindstrom et al. 1994; Daly et al. 1996; Lai et al. 2004), and the weakening of quarantine restrictions in Europe could be a potential explanation for the co-circulation of these lineages in Europe. Here, we extend the two-tiered model to allow for the evolutionary dynamics of H3N8 to take place in two geographical locations, representing Europe and North America. Specifically, the epidemiological submodel equations are first modified to consider two ‘patches’
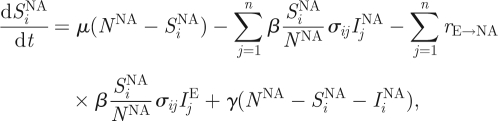 |
3.3a |
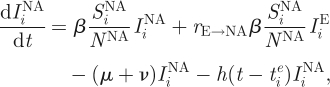 |
3.3b |
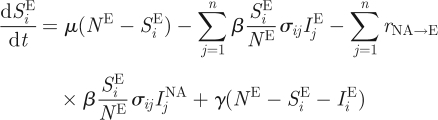 |
3.3c |
and
 |
3.3d |
This extension introduces two more parameters: the degree of transmission reduction from the European patch to the North American patch (rE→NA) and the degree of transmission reduction from the North American patch to the European patch (rNA→E). In the simulated years 1963–1986, we assign both patches the same reduction in the degree of transmission (rE→NA and rNA→E = 1/100). During the simulated years 1986–1999, we assume that transmission between the patches ceases (rE→NA and rNA→E = 0), reflecting effective quarantine between the two continents. After 1999, we let rE→NA remain at 0, but set rNA→E back to 1/100, to reflect the hypothesized breakdown of quarantine measures in Europe.
Simulations of this two-patch epidemiological submodel yield recurrent outbreaks of EIV in North America and in Europe (figure 8a,c), frequently coinciding with the appearance of a new antigenic variant. These patterns are qualitatively similar to the reported dynamics of EIV, with outbreaks occurring every 2–8 years and frequently being associated with new antigenic variants (Waddell et al. 1963; Livesay et al. 1993; Chambers et al. 1994; Daly et al. 1996; Ilobi et al. 1998; Damiani et al. 2008). We do not make an attempt at a quantitative comparison between model-simulated case dynamics and observed case dynamics because H3N8 is not a notifiable disease, and, as such, the data are not sufficient to allow for this comparison.
Figure 8.

Simulated epidemiological dynamics of influenza A (H3N8) in equine host across North America and Europe. (a) Simulated case dynamics in North America. Antigenic clusters are colour-coded. (b) Proportion of cases in North America belonging to each antigenic cluster. (c) Simulated case dynamics in Europe. Antigenic clusters are colour-coded. (d) Proportion of cases in Europe belonging to each antigenic cluster. The black line in 1986 marks the establishment of quarantine. The black line in 1999 marks the breakdown of quarantine for horses transported from North America to Europe. Parameters used: population sizes NNA = 9 500 000 and NE = 4 500 000 (http://www.horsetalk.co.nz/archives/2007/09/105.shtml), R0 = 2, μ = 1/20 yr−1, ν = 1/7 d−1, γ = 8 yr−1, ɛ = 0, θ = 0.6 and Weibull parameters k = 2, λ = 900. Prior to 1986, rE→NA = rNA→E = 0.01. Between 1986 and 1999, rE→NA = rNA→E = 0. After 1999, rNA→E = 0.01 and rE→NA = 0.
Simulations of the epidemiological submodel also show that the model can reproduce antigenic turnover patterns similar to those observed (figure 8b,d). Prior to 1986, both antigenic variants that arose rapidly spread through the two patches and globally replaced the previously endemic variant. After the initiation of the quarantine measures in 1986, a new antigenic variant arose in North America and, later, another in Europe. The emergence of these independent variants corresponds qualitatively to the arrival of the American and European lineages. In 1999, after the quarantine measures were weakened in Europe, the North American variant appeared in Europe, but did not succeed in replacing the European variant. Thus, we observe in our simulations co-circulation of the European and North American lineages in Europe. Owing to the intact quarantine restrictions in North America, only the North American lineage continues to circulate there. As time goes on, we continue to observe antigenic evolution of both the North American and the European lineages.
Following the simulation of these variant specific case dynamics, we simulated the second tier of the model, the molecular evolution submodel, sampling sequences from both continents. The phylogeny reconstructed from the simulated sequences shows both qualitative and quantitative similarities to the tree inferred from observed H3N8 HA sequences (figure 7a,b). Qualitatively, lineage turnover and low genetic diversity are observed in both trees prior to 1986. Both phylogenies then split into two lineages in the late 1980s. In the last decade, both phylogenies show that sequences from the North American lineage were isolated in Europe. In addition to these qualitative similarities, the branch lengths of the tree inferred from simulated sequences are similar to those from the tree inferred from observed sequences.
The application and extension of the two-tiered model to include space in the simulation of equine influenza dynamics testifies to the versatility and adaptability of the model to different host systems and different ecological variables.
4. Discussion
Here, we presented a two-tiered model that can be used to simulate both the ecological and the evolutionary dynamics of rapidly evolving RNA viruses. The model's novelty resides in its modular design: it separates antigenic dynamics from genotypic dynamics, and thereby yields computationally simpler simulations that allow for a more realistic representation of viral sequences. At the heart of the two-tiered model is the antigenic emergence rate, which drives the emergence dynamics of new antigenic variants in the epidemiological submodel. Here, we also showed that this antigenic emergence rate, when parametrized with a shape parameter k of 2, can be mechanistically interpreted in terms of a model that considers neutral mutation accumulation and the probability of immune escape increasing linearly with the number of mutations already accumulated (appendix A). The phenotypic dynamics resulting from this first tier of the model are then used as input for the second tier of the model, the molecular evolution submodel. We showed here that this second submodel simulates sequence data from which quantitative indexes (divergence and diversity metrics) can be computed, which can be compared with empirical sequence data. Furthermore, phylogenies can be inferred from these simulated sequences, with branch lengths that are comparable to those from trees inferred from empirical viral sequences. The two-tiered model in its entirety can, therefore, be used to generate case data and sequence data that can be confronted statistically against empirical datasets of these two types.
The modularity of the two-tiered model is its principal strength and will allow this framework to be adapted to consider alternative hypotheses and to include alternative, and potentially better or faster, submodels. For example, the first tier of the model uses a status-based, reduced infectivity multi-strain model in its implementation (Gog & Grenfell 2002). However, recent work has shown that this model, in contrast to other, more highly dimensional, multi-strain models, overestimates the level of herd immunity to a new antigenic variant (Ballesteros et al. 2009). Owing to its modularity, the two-tiered model can be easily modified to consider alternative epidemiological submodels, for example, the well-known history-based multi-strain model (Andreasen et al. 1997). Furthermore, any of these models can be extended to consider specific questions of interest, such as what climate variables are important drivers of influenza's seasonal dynamics (Shaman et al. 2009), what role population substructure plays in the ecological and evolutionary dynamics of influenza (Truscott et al. 2009) and how cross-immunity may act (e.g. by its separate effects on infectiousness and infectious period; Park et al. 2009). The only requirement of the epidemiological submodel is that it generates variant-specific case dynamics, which are used as input into the second tier of the model (electronic supplementary material, figure S1).
Similarly, the molecular evolution submodel, as described, can be easily replaced with an alternative submodel. One possible alternative submodel that would be computationally faster, but apply to a more limited number of cases, might use approaches based on coalescent theory to yield viral genealogies. A second possibility is to replace the current molecular evolution submodel with a model that has a mechanistic link between the parameter f in the second tier of the model and the parameter γ in the first tier of the model. A third possibility would be to consider not just the process of point mutations, but also to allow for insertions, deletions, recombination and, for segmented viruses, reassortment. Here, the only requirement for the second tier is that it takes in variant-specific case data and generates time-stamped viral sequences.
Following its description, we applied the two-tiered model to influenza A (H3N2) in humans, to influenza B in humans and to influenza A (H3N8) in equine hosts in order to illustrate its use. In the first application, we showed that a model parametrized for a combination of gradual and punctuated antigenic change could quantitatively reproduce the ecological and evolutionary patterns of this subtype in humans. In contrast, and consistent with previous theoretical findings (Ballesteros et al. 2009), a model with purely punctuated antigenic evolution failed to capture these patterns well. In the electronic supplementary material, we also showed that only gradual antigenic evolution was not consistent with all of the observed dynamics of influenza A (H3N2). The ability of only the model with both modes of antigenic change to quantitatively reproduce the dynamic patterns of this variant resolves the seemingly contradictory findings that antigenic change occurs either in a punctuated (Smith et al. 2004; Wolf et al. 2006; Blackburne et al. 2008) or in a gradual (Shih et al. 2007; Suzuki 2008) manner. Both modes of antigenic evolution appear necessary: gradual antigenic evolution is needed to reproduce the observed periodicity of influenza's ecological dynamics and the rapid rate of HA divergence, while punctuated antigenic evolution is needed to reproduce rates of divergence and the overall ladder-like topology of influenza A (H3N2)'s HA protein.
The application of the model to influenza B illustrated the model's ability to generate qualitatively different ecological and evolutionary dynamics under alternative parametrizations. This ability is critical for the model to be effectively interfaced with different empirical datasets, through the development and application of new statistical approaches. The application of the model to influenza A (H3N8) in equine hosts served to illustrate the ease with which the model could be extended to accommodate further hypotheses. Specifically, we considered the hypothesis that the introduction and later weakening of quarantine measures between North America and Europe played a role in shaping the evolutionary dynamics of H3N8. Although the model realizing this hypothesis was able to reproduce features of the ecological and evolutionary dynamics of H3N8, alternative hypotheses could easily be considered within this framework. A statistical comparison between these models' simulated sequences (and possibly case dynamics) could then determine the appropriate level of support for each of the models considered.
In our applications to flu, we parametrized the two-tiered model to consider the effects of humoral immune escape, driven by genetic changes in the virus's dominant antigenic protein. While this parametrization has empirical support in the case of influenza (Smith et al. 2004), we may want to consider alternative mechanisms of immune escape. For example, there is evidence for positive selection of cytotoxic T lymphocyte escape mutants (Gog et al. 2003). Another possible hypothesis is that generalized immunity plays a role in shaping the ecological and evolutionary dynamics of influenza (Ferguson et al. 2003). These hypotheses, as other ones mentioned above, could easily be integrated into this two-tiered modelling framework. This integration would enable us to finally compare these hypotheses in a quantitative way, considering both incidence data and sequence data.
Although our focus here was on the ecological and evolutionary dynamics of RNA viruses at the population level, the two-tiered structure of the model could also be used to consider the dynamics at another level of organization. Specifically, while we modelled the dynamics of susceptible, infected and recovered hosts here, within-host dynamics could instead consider classes of naive cells and cells that are infected with virus of different antigenic phenotypes. In lieu of simulating epidemiological dynamics, the first tier of the model would simulate the viral load dynamics, by antigenic type. The second tier of the model would then be used again to generate viral sequences that could be compared with viral sequences isolated from a single chronically infected host over several time points (e.g. in the case of HIV; Shankarappa et al. 1999).
Regardless of whether the two-tiered framework presented here is applied at the within-host level or the population level, its ability to generate both case data and sequence data that can be statistically confronted with empirical observations will improve our understanding of the key drivers of viral dynamics, and may thereby ultimately help in their control.
Acknowledgements
We thank the three reviewers for their thoughtful comments and suggestions and members of the Koelle research group for detailed feedback. Support for K.K., P.K. and M.K. was provided by grant NSF-EF-08-27416 and by the RAPIDD programme of the Science and Technology Directorate, Department of Homeland Security, and the Fogarty International Center, National Institutes of Health.
Appendix A. A mechanistic interpretation of the Weibull hazard function
We can interpret the rate of antigenic change increasing with the age of a variant (as specified by the Weibull hazard function in equation (2.3)) by considering the following Markov chain on the non-negative integers. Denote by Pn(t) the probability mass function for the compound event that, at time t, a viral sequence belonging to a given antigenic variant i has accrued n mutations from the founder sequence of the variant and that a new antigenic variant j has not yet arisen from the mutations that have accrued in this sequence. Denote by Qn(t) the probability that, at time t, the viral sequence has accrued n mutations and that a new antigenic variant j has already arisen from these accrued mutations. Using these definitions, we can write the following master equation for considering the production of new antigenic variants
| A 1a |
and
| A 1b |
where m is the per-sequence mutation rate and αn is the probability that a mutation to an antigenically preserved sequence that has already accrued n mutations will result in a new antigenic variant. As the simplest possible non-trivial assumption, we let this probability be linearly increasing with the number of mutations n
| A 1c |
where ρ can be thought of as a parameter that captures, inversely, the antigenic protein's robustness to genetic change. Let us consider 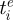 , the time of emergence of antigenic variant i, to be t = 0. At this time, any sequence belonging to cluster i is in an unmutated state
, the time of emergence of antigenic variant i, to be t = 0. At this time, any sequence belonging to cluster i is in an unmutated state
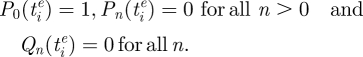 |
To solve this system, we can define the generating function
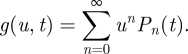 |
A 2 |
Differentiating with respect to t, we have
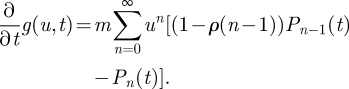 |
A 3 |
Differentiating with respect to u, we have
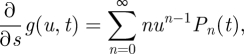 |
A 4 |
so that
| A 5 |
To solve this, we use the method of characteristics. Let u = σ(t,u0) define a family of curves with u0 = υ(0,u0) that covers the plane such that each point (t, u) in the plane lies on one and only one curve in this family and thus can be traced back uniquely to its ‘origin’ (0,u0).
We will produce solutions of equation (A 5) defined on each of these curves and then consider the curves together to form the complete solution. Define the function restricted to the curve with origin at (0,u0)
| A 6 |
We now have
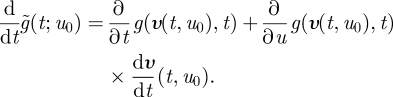 |
A 7 |
Substituting equation (A 5) yields
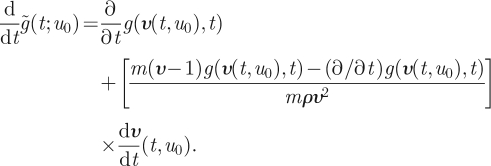 |
A 8 |
We are free to define our curves any way we choose within the constraints given above, so we let
| A 9 |
with solution
| A 10 |
Now, substituting equation (A 9) into equation (A 8) yields
| A 11 |
With the additional substitution of equation (A 10), this yields
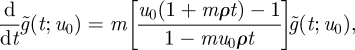 |
A 12 |
which itself has solution
 |
A 13 |
The initial condition at time t = 0 becomes 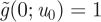 , leaving
, leaving
| A 14 |
Finally, we trace back from u0 to u, solving equation (A 10) for u0
| A 15 |
to get
| A 16 |
The probability of escape by time t is
| A 17 |
To leading order, we have
| A 18 |
or
| A 19 |
The density function for the time of antigenic escape is just the time derivative of PE. To leading order, this is
| A 20 |
which is a Weibull density function with shape parameter k = 2 and  .
.
A mutational process for which the probability of antigenic immune escape increases linearly with the number of mutations already accrued can, therefore, be modelled phenomenologically with a hazard rate with shape parameter k = 2 and a value of λ that depends on the viral mutation rate m and its sensitivity to phenotypic change ρ.
Several modifications to this mechanistic model can be considered. First, instead of assuming that the probability of antigenic immune escape is linearly increasing with the number of accrued mutations (equation (A 1c)), we can consider a second model that is also increasing with the number of accrued mutations, but also has the desired feature of saturating at 1. One such function is αn = ρn/(ρn + 1). However, this function and those of a similar form do not have a closed-form solution for the probability of escape by time t. Although the linear function we use in the derivation of the Weibull hazard function (equation (A 1c)) allows the probability of antigenic immune escape upon the next mutation to exceed 1, we can simulate the epidemiological submodel in a parameter regime where we are ensured that this underlying probability (which we do not explicitly use in the simulations) would never get sufficiently close to 1.
Second, instead of using the hazard rate of the Weibull density function shown in equation (A 20), one could use an exact expression for the rate of generating new antigenic variants that is derived from the Markov model shown in equations (A 1). This rate would be based on the exact expression for the cumulative density function shown in equation (A 17). In our simulations, we instead used the Weibull functional form because it is a simple and well-known distribution that has the ageing properties that we find biologically necessary.
References
- Andreasen V., Lin J., Levin S. A. 1997. The dynamics of co-circulating influenza strains conferring partial cross-immunity. J. Math. Biol. 35, 825–842. ( 10.1007/s002850050079) [DOI] [PubMed] [Google Scholar]
- Ballesteros S., Vergu E., Cazelles B. 2009. Influenza A gradual and epochal evolution: insights from simple models. PLoS ONE 4, e7426 ( 10.1371/journal.pone.0007426) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg M., Desselberger U., Abusugra I. A., Klingeborn B., Linné T. 1990. Genetic drift of equine 2 influenza A virus (H3N8), 1963–1988: analysis by oligonucleotide mapping. Vet. Microbiol. 22, 225–236. ( 10.1016/0378-1135(90)90109-9) [DOI] [PubMed] [Google Scholar]
- Berton M. T., Naeve C. W., Webster R. G. 1984. Antigenic structure of the influenza B virus hemagglutinin: nucleotide sequence analysis of antigenic variants selected with monoclonal antibodies. J. Virol. 52, 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburne B. P., Hay A. J., Goldstein R. A. 2008. Changing selective pressure during antigenic changes in human influenza H3. PLoS Pathog. 4, e1 000 058. ( 10.1371/journal.ppat.1000058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobashev G. V., Ellner S. P., Nychka D. W., Grenfell B. T. 2000. Reconstructing susceptible and recruitment dynamics from measles incidence data. Math. Popul. Stud. 8, 1–29. ( 10.1080/08898480009525471) [DOI] [Google Scholar]
- Bryant N. A., et al. 2009. Antigenic and genetic variations in European and North American equine influenza virus strains (H3N8) isolated from 2006 to 2007. Vet. Microbiol. 138, 41–52. ( 10.1016/j.vetmic.2009.03.004) [DOI] [PubMed] [Google Scholar]
- Chambers T. M., Lai A. C. K., Franklin K. M., Powell D. G. 1994. Recent evolution of the hemagglutinin of equine-2 influenza virus in the USA. In Proc. 7th Int. Conf. Equine Infectious Diseases, Tokyo, 1994, pp. 175–180. Newmarket, UK: R & W Publications (Newmarket). [Google Scholar]
- Cobey S., Koelle K. 2008. Capturing escape in infectious disease dynamics. Trends Ecol. Evol. 23, 572–577. ( 10.1016/j.tree.2008.06.008) [DOI] [PubMed] [Google Scholar]
- Daly J. M., Lai A. C., Binns M. M., Chambers T. M., Barrandeguy M., Mumford J. A. 1996. Antigenic and genetic evolution of equine H3N8 influenza A viruses. J. Gen. Virol. 77, 661–671. ( 10.1099/0022-1317-77-4-661) [DOI] [PubMed] [Google Scholar]
- Damiani A. M., et al. 2008. Genetic characterization of equine influenza viruses isolated in Italy between 1999 and 2005. Virus Res. 131, 100–105. ( 10.1016/j.virusres.2007.08.001) [DOI] [PubMed] [Google Scholar]
- Daniels R. S., Skehel J. J., Wiley D. C. 1985. Amino acid sequences of haemagglutinins of influenza viruses of the H3 subtype isolated from horses. J. Gen. Virol. 66, 457–464. ( 10.1099/0022-1317-66-3-457) [DOI] [PubMed] [Google Scholar]
- Endo A., Pecoraro R., Sugita S., Nerome K. 1992. Evolutionary pattern of the H3 haemagglutinin of equine influenza viruses: multiple evolutionary lineages and frozen replication. Arch. Virol. 123, 73–87. ( 10.1007/BF01317139) [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 2004. Inferring phylogenies. Sunderland, MA: Sinauer Associates, Inc. [Google Scholar]
- Ferguson N. M., Galvani A. P., Bush R. M. 2003. Ecological and immunological determinants of influenza evolution. Nature 422, 428–433. ( 10.1038/nature01509) [DOI] [PubMed] [Google Scholar]
- Finkenstädt B., Grenfell B. 2000. Time series modelling of childhood diseases: a dynamical systems approach. J. R. Stat. Soc. Ser. C 49, 187–205. ( 10.1111/1467-9876.00187) [DOI] [Google Scholar]
- Fitch W. M., Bush R. M., Bender C. A., Cox N. J. 1997. Long term trends in the evolution of H(3) HA1 human influenza type A. Proc. Natl Acad. Sci. USA 94, 7712–7718. ( 10.1073/pnas.94.15.7712) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C., et al. 2009. Pandemic potential of a strain of influenza A (H1N1): early findings. Science 324, 1557–1561. ( 10.1126/science.1176062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill P. W., Murphy A. M. 1977. Naturally acquired immunity to influenza type A: a further prospective study. Med. J. Aust. 2, 761–765. [DOI] [PubMed] [Google Scholar]
- Gillespie D. T. 2007. Stochastic simulation of chemical kinetics. Annu. Rev. Phys. Chem. 58, 35–55. ( 10.1146/annurev.physchem.58.032806.104637) [DOI] [PubMed] [Google Scholar]
- Girvan M., Callaway D. S., Newman M. E. J., Strogatz S. H. 2002. Simple model of epidemics with pathogen mutation. Phys. Rev. E 65, 1–9. [DOI] [PubMed] [Google Scholar]
- Gog J. R., Grenfell B. T. 2002. Dynamics and selection of many-strain pathogens. Proc. Natl Acad. Sci. USA 99, 17 209–17 214. ( 10.1073/pnas.252512799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gog J. R., Rimmelzwaan G. F., Osterhaus A. D. M. E., Grenfell B. T. 2003. Population dynamics of rapid fixation in cytotoxic T lymphocyte escape mutants of influenza A. Proc. Natl Acad. Sci. USA 100, 11 143–11 147. ( 10.1073/pnas.1830296100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gökaydin D., Oliveira-Martins J. B., Gordo I., Gomes M. G. M. 2007. The reinfection threshold regulates pathogen diversity: the case of influenza. J. R. Soc. Interface 4, 137–142. ( 10.1098/rsif.2006.0159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell B. T., Pybus O. G., Gog J. R., Wood J. L. N., Daly J. M., Mumford J. A., Holmes E. C. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303, 327–332. ( 10.1126/science.1090727) [DOI] [PubMed] [Google Scholar]
- Hinshaw V. S., Naeve C. W., Webster R. G., Douglas A., Skehel J. J., Bryans J. 1983. Analysis of antigenic variation in equine 2 influenza A viruses. Bull. World Health Org. 61, 153–158. [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C., Grenfell B. T. 2009. Discovering the phylodynamics of RNA viruses. PLoS Comput. Biol. 5, e1000505 ( 10.1371/journal.pcbi.1000505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilobi C. P., Nicolson C., Taylor J., Mumford J. A., Wood J. M., Robertson J. S. 1998. Direct sequencing of the HA gene of clinical equine H3N8 influenza virus and comparison with laboratory derived viruses. Arch. Virol. 143, 891–901. ( 10.1007/s007050050340) [DOI] [PubMed] [Google Scholar]
- Ionides E. L., Bretó C., King A. A. 2006. Inference for nonlinear dynamical systems. Proc. Natl Acad. Sci. USA 103, 18 438–18 443. ( 10.1073/pnas.0603181103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaoka Y., Bean W. J., Webster R. G. 1989. Evolution of the hemagglutinin of equine H3 influenza viruses. Virology 169, 283–292. ( 10.1016/0042-6822(89)90153-0) [DOI] [PubMed] [Google Scholar]
- King A. A., Ionides E. L., Pascual M., Bouma M. J. 2008. Inapparent infections and cholera dynamics. Nature 454, 877–880. ( 10.1038/nature07084) [DOI] [PubMed] [Google Scholar]
- Koelle K., Pascual M. 2004. Disentangling extrinsic from intrinsic factors in disease dynamics: a nonlinear time series approach with an application to cholera. Am. Nat. 163, 901–913. ( 10.1086/420798) [DOI] [PubMed] [Google Scholar]
- Koelle K., Cobey S., Grenfell B., Pascual M. 2006. Epochal evolution shapes the phylodynamics of influenza A (H3N2) in humans. Science 314, 1898–1903. ( 10.1126/science.1132745) [DOI] [PubMed] [Google Scholar]
- Koelle K., Kamradt M., Pascual M. 2009. Understanding the dynamics of rapidly evolving pathogens through modeling the tempo of antigenic change: influenza as a case study. Epidemics 1, 129–137. ( 10.1016/j.epidem.2009.05.003) [DOI] [PubMed] [Google Scholar]
- Lai A. C., Rogers K. M., Rogers K. M., Chamber T. 2004. Alternate circulation of recent equine-2 influenza viruses (H3N8) from two distinct lineages in the United States. Virus Res. 100, 159–164. ( 10.1016/j.virusres.2003.11.019) [DOI] [PubMed] [Google Scholar]
- Lau K. F., Dill K. A. 1990. Theory for protein mutability and biogenesis. Proc. Natl Acad. Sci. USA 87, 638–642. ( 10.1073/pnas.87.2.638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom S., Endo A., Pecoraro R., Sugita S., Hiromoto Y., Nerome K. 1994. Genetic divergency of equine (H3N8) influenza viruses: cocirculation of earliest and recent strains. In Proc. 7th Int. Conf. Equine Infectious Diseases, Tokyo, 1994. p. 307 Newmarket, UK: R & W Publications (Newmarket). [Google Scholar]
- Livesay G. J., O'Neill T., Hannant D., Yadav M. P., Mumford J. A. 1993. The outbreak of equine influenza (H3N8) in the United Kingdom in 1989: diagnostic use of an antigen capture ELISA. Vet. Rec. 133, 515–519. [DOI] [PubMed] [Google Scholar]
- Minayev P., Ferguson N. 2009a. Improving the realism of deterministic multi-strain models: implications for modelling influenza A. J. R. Soc. Interface 6, 509–518. ( 10.1098/rsif.2008.0333) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minayev P., Ferguson N. 2009b. Incorporating demographic stochasticity into multi-strain epidemic models: application to influenza A. J. R. Soc. Interface 6, 989–996. ( 10.1098/rsif.2008.0467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto A. S., Kioumehr F. 1975. The Tecumseh study of respiratory illness. IX. Occurrence of influenza in the community, 1966–1971. Am. J. Epidemiol. 102, 553–563. [DOI] [PubMed] [Google Scholar]
- Nakagawa N., Kubota R., Maeda A., Nakagawa T., Okuno Y. 2000. Heterogeneity of influenza B virus strains in one epidemic season differentiated by monoclonal antibodies and nucleotide sequences. J. Clin. Microbiol. 38, 3467–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa N., Kubota R., Morikawa S., Nakagawa T., Baba K., Okuno Y. 2001a. Characterization of new epidemic strains of influenza B virus by using neutralizing monoclonal antibodies. J. Med. Virol. 65, 745–750. ( 10.1002/jmv.2099) [DOI] [PubMed] [Google Scholar]
- Nakagawa N., Kubota R., Nakagawa T., Okuno Y. 2001b. Antigenic variants with amino acid deletions clarify a neutralizing epitope specific for influenza B virus Victoria group strains. J. Gen. Virol. 82, 2169–2172. [DOI] [PubMed] [Google Scholar]
- Nakagawa N., Nukuzuma S., Haratome S., Go S., Nakagawa T., Hayashi K. 2002. Emergence of an influenza B virus with antigenic change. J. Clin. Microbiol. 40, 3068–3070. ( 10.1128/JCM.40.8.3068-3070.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S., Nakajima K., Kendal A. P. 1983. Identification of the binding sites to monoclonal antibodies on A/USSR/90/77 (H1N1) hemagglutinin and their involvement in antigenic drift in H1N1 influenza viruses. Virology 131, 116–127. ( 10.1016/0042-6822(83)90538-X) [DOI] [PubMed] [Google Scholar]
- Nerome R., Hiromoto Y., Sugita S., Tanabe N., Ishida M., Matsumoto M., Lindstrom E., Takahashi T., Nerome K. 1998. Evolutionary characteristics of influenza B virus since its first isolation in 1940: dynamic circulation of deletion and insertion mechanism. Arch. Virol. 143, 1569–1583. ( 10.1007/s007050050399) [DOI] [PubMed] [Google Scholar]
- Nobusawa E., Sato K. 2006. Comparison of the mutation rates of human influenza A and B viruses. J. Virol. 80, 3675–3678. ( 10.1128/JVI.80.7.3675-3678.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxburgh L., Klingeborn B. 1999. Cocirculation of two distinct lineages of equine influenza virus subtype H3N8. J. Clin. Microbiol. 37, 3005–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxburgh L., Berg M., Klingeborn B., Emmoth E., Linné T. 1994. Evolution of H3N8 equine influenza virus from 1963 to 1991. Virus Res. 34, 153–165. ( 10.1016/0168-1702(94)90097-3) [DOI] [PubMed] [Google Scholar]
- Oxburgh L., Akerblom L., Fridberger T., Klingeborn B., Linné L. 1998. Identification of two antigenically and genetically distinct lineages of H3N8 equine influenza virus in Sweden. Epidemiol. Infect. 120, 61–70. ( 10.1017/S0950268897008315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A., Daly J. M., Lewis N. S., Smith D. J., Wood J. L. N., Grenfell B. T. 2009. Quantifying the impact of immune escape on transmission dynamics of influenza. Science 326, 726–728. ( 10.1126/science.1175980) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin J. B., Dushoff J., Levin S. A. 2002. Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus. Proc. Natl Acad. Sci. USA 99, 6263–6268. ( 10.1073/pnas.082110799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recker M., Pybus O. G., Nee S., Gupta S. 2007. The generation of influenza outbreaks by a network of host immune responses against a limited set of antigenic types. Proc. Natl Acad. Sci. USA 104, 7711–7716. ( 10.1073/pnas.0702154104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P. A., Hemphill M. L., Whistler T., Regnery H. L., Kendal A. P. 1992. Antigenic and genetic characterization of the haemagglutinins of recent cocirculating strains of influenza B virus. J. Gen. Virol. 73, 2737–2742. ( 10.1099/0022-1317-73-10-2737) [DOI] [PubMed] [Google Scholar]
- Shaman J., Pitzer V., Cecile V., Marc L., Bryan G. 2009. Absolute humidity and the seasonal onset of influenza in the continental US. PLoS Curr. Influenza. See http://knol.google.com/k/plos/plos-currents-influenza/28qm4w0q65e4w/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa R., et al. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73, 10 489–10 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A. C., Hsiao T., Ho M.-S., Li W.-H. 2007. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc. Natl Acad. Sci. USA 104, 6283–6288. ( 10.1073/pnas.0701396104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. J., Lapedes A. S., de Jong J. C., Bestebroer T. M., Rimmelzwaan G. F., Osterhaus A. D. M. E., Fouchier R. A. M. 2004. Mapping the antigenic and genetic evolution of influenza virus. Science 305, 371–376. ( 10.1126/science.1097211) [DOI] [PubMed] [Google Scholar]
- Suzuki Y. 2008. Positive selection operates continuously on hemagglutinin during evolution of H3N2 human influenza A virus. Gene 427, 111–116. ( 10.1016/j.gene.2008.09.012) [DOI] [PubMed] [Google Scholar]
- Tria F., Lässig M., Peliti L., Franz S. 2005. A minimal stochastic model for influenza evolution. J. Stat. Mech. P07008, 1–11. [Google Scholar]
- Truscott J., Fraser C., Hinsley W., Cauchemez S., Donnelly C., Ghani A., Ferguson N. 2009. Quantifying the transmissibility of human influenza and its seasonal variation in temperate regions. PLoS Curr. Influenza. See http://knol.google.com/k/plos/plos-currents-influenza/28qm4w0q65e4w/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nimwegen E. 2006. Influenza escapes immunity along neutral networks. Science 314, 1884–1886. ( 10.1126/science.1137300) [DOI] [PubMed] [Google Scholar]
- Waddell G. H., Teigland M. B., Siegel M. M. 1963. A new influenza virus associated with equine respiratory disease. J. Am. Vet. Med. Assoc. 143, 587–590. [PubMed] [Google Scholar]
- Webster R. G. 1993. Are equine 1 influenza viruses still present in horses? Equine Vet. J. 25, 537–538. [DOI] [PubMed] [Google Scholar]
- Webster R. G., Laver W. G. 1980. Determination of the number of nonoverlapping antigenic areas on Hong Kong (H3N2) influenza virus hemagglutinin with monoclonal antibodies and the selection of variants with potential epidemiological significance. Virology 104, 139–148. ( 10.1016/0042-6822(80)90372-4) [DOI] [PubMed] [Google Scholar]
- Wiley D. C., Wilson I. A., Skehel J. J. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289, 373–378. ( 10.1038/289373a0) [DOI] [PubMed] [Google Scholar]
- Wilson I. A., Cox N. J. 1990. Structural basis of immune recognition of influenza virus hemagglutinin. Annu. Rev. Immunol. 8, 737–787. ( 10.1146/annurev.iy.08.040190.003513) [DOI] [PubMed] [Google Scholar]
- Wolf Y. I., Viboud C., Holmes E. C., Koonin E. V., Lipman D. J. 2006. Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol. Direct 1, 34 ( 10.1186/1745-6150-1-34) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Gog J. R., Grenfell B. T. 2005. Semiparametric estimation of the duration of immunity from infectious disease time-series: influenza as a case study. Appl. Stat. 54, 659–672. ( 10.1111/j.1467-9876.2005.05383.x) [DOI] [Google Scholar]