

Abstract
Sequence comparison and alignment has had an enormous impact on our understanding of evolution, biology and disease. Comparison and alignment of biological networks will probably have a similar impact. Existing network alignments use information external to the networks, such as sequence, because no good algorithm for purely topological alignment has yet been devised. In this paper, we present a novel algorithm based solely on network topology, that can be used to align any two networks. We apply it to biological networks to produce by far the most complete topological alignments of biological networks to date. We demonstrate that both species phylogeny and detailed biological function of individual proteins can be extracted from our alignments. Topology-based alignments have the potential to provide a completely new, independent source of phylogenetic information. Our alignment of the protein–protein interaction networks of two very different species—yeast and human—indicate that even distant species share a surprising amount of network topology, suggesting broad similarities in internal cellular wiring across all life on Earth.
Keywords: network alignment, protein interaction networks, network topology, phylogeny, protein function
1. Introduction and motivation
Advances in high throughput experimental methods have yielded large amounts of biological network data, such as protein–protein interaction (PPI) networks. The two most commonly used high-throughput methods are yeast two-hybrid screening, resulting in binary interaction data (Uetz et al. 2000; Ito et al. 2000, 2001; Rual et al. 2005; Stelzl et al. 2005; Simonis et al. 2008), and protein complex purification methods using mass-spectrometry, resulting in co-complex data (Rigaut et al. 1999; Gavin et al. 2002, 2006; Ho et al. 2002; Krogan et al. 2006; Collins et al. 2008). Just as comparative genomics has led to an explosion of knowledge about evolution, biology and disease, so will comparative proteomics. As more biological network data are becoming available, comparative analyses of these networks across species are proving to be valuable, since such systems biology types of comparisons may lead to transfer of knowledge between species as well as to exciting discoveries in evolutionary biology. The most common methods for such network comparisons are network alignments.
Network alignment is the problem of finding similarities between the structure or topology of two or more networks. In the biological context, comparing networks of different organisms in a meaningful manner is arguably one of the most important problems in evolutionary and systems biology (Sharan et al. 2005). Exactly analogous to sequence alignments between genomes, alignments of biological networks can be useful because we may know a lot about some of the nodes in one network and almost nothing about topologically similar nodes in the other network; then, specialized knowledge about one may tell us something new about the other. Network alignments can also be used to measure the global similarity between complete networks of different species. Given a group of such biological networks, the matrix of pairwise global network similarities can be used to infer phylogenetic relationships.
1.1. Theoretical background
A network (or graph) is a collection of nodes (or vertices), and connections between them called edges. Graphs are used to describe, model and analyse an enormous array of phenomena (Colizza et al. 2006; Guimera et al. 2007), including physical systems such as electrical power grids and communication networks, social systems such as networks of friendships or corporate and political hierarchies, physical relationships such as residue interactions in a folded protein, or software systems such as call graphs or expression and syntax trees.
A graph G(V,E), or G for brevity, is an ordered pair (V,E), where V is a node set and 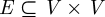 is an edge set. The sheer number and diversity of possible graphs (
is an edge set. The sheer number and diversity of possible graphs (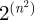 of them exist given n nodes) make graph classification and comparison problems difficult. One particular comparison problem is called subgraph isomorphism, which asks whether a graph G exists as an exact subgraph of another graph H(U,F). This problem is NP-complete, which means that no efficient algorithm is known for solving it (Cook 1971). Network alignment (Sharan & Ideker 2006) is the more general problem of finding the best way to ‘fit’ G into H even if G does not exist as an exact subgraph of H. Some networks, such as the biological ones that we consider below, may also contain noise, i.e. missing edges, false edges or both (Venkatesan et al. 2009). In these cases, and also owing to biological variation, it is not even obvious how to measure the ‘goodness’ of an inexact fit. One measure could be to assess the number of aligned edges—that is, the percentage of edges in E that are aligned to edges in F. We call this the ‘edge correctness’ (EC; Singh et al. 2008; Zaslavskiy et al. 2009). However, it is possible for two alignments to have similar ECs, one of which exposes large, dense, contiguous and topologically complex regions that are similar in G and H, while the other fails to expose such regions of similarity. Additionally, although EC can easily be used to measure the quality of an alignment after the fact, it is not clear how to use it to direct an alignment algorithm; in fact, maximizing EC is an NP-hard problem since it implies solving the subgraph isomorphism problem. Thus, other strategies must be sought to guide the alignment process.
of them exist given n nodes) make graph classification and comparison problems difficult. One particular comparison problem is called subgraph isomorphism, which asks whether a graph G exists as an exact subgraph of another graph H(U,F). This problem is NP-complete, which means that no efficient algorithm is known for solving it (Cook 1971). Network alignment (Sharan & Ideker 2006) is the more general problem of finding the best way to ‘fit’ G into H even if G does not exist as an exact subgraph of H. Some networks, such as the biological ones that we consider below, may also contain noise, i.e. missing edges, false edges or both (Venkatesan et al. 2009). In these cases, and also owing to biological variation, it is not even obvious how to measure the ‘goodness’ of an inexact fit. One measure could be to assess the number of aligned edges—that is, the percentage of edges in E that are aligned to edges in F. We call this the ‘edge correctness’ (EC; Singh et al. 2008; Zaslavskiy et al. 2009). However, it is possible for two alignments to have similar ECs, one of which exposes large, dense, contiguous and topologically complex regions that are similar in G and H, while the other fails to expose such regions of similarity. Additionally, although EC can easily be used to measure the quality of an alignment after the fact, it is not clear how to use it to direct an alignment algorithm; in fact, maximizing EC is an NP-hard problem since it implies solving the subgraph isomorphism problem. Thus, other strategies must be sought to guide the alignment process.
1.2. Previous approaches
Analogous to sequence alignments, there exist local and global network alignments. Thus far, the majority of methods used for alignment of biological networks have focused on local alignments (Berg & Lassig 2004, 2006; Kelley et al. 2004; Flannick et al. 2006; Liang et al. 2006). With local alignments, mappings are chosen independently for each local region of similarity. Many algorithms for local alignment have been developed. PathBLAST searches for high-scoring pathway alignments between two networks, by taking into account both the homology between the aligned proteins and the probabilities that PPIs in the path are true PPIs and not false-positives (Kelley et al. 2004). NetworkBLAST detects conserved protein clusters rather than paths, by deploying a likelihood-based scoring scheme that weighs the denseness of a subnetwork versus the chance of observing such network substructure at random (Sharan et al. 2005). MaWISh defines network alignment as a maximum weight induced subgraph problem and implements an evolution-based scoring scheme to detect conserved clusters; it extends the concepts of evolutionary events in sequence alignments to that of duplication, match and mismatch in network alignments and evaluates the similarity between network structures through a scoring function that accounts for these evolutionary events (Koyuturk et al. 2006). Graemlin, the first method capable of identifying dense conserved subnetworks of arbitrary structure, scores a module by computing the log-ratio of the probability that the module is subject to evolutionary constraints and the probability that the module is under no constraints, while taking into account phylogenetic relationships between species whose networks are being aligned (Flannick et al. 2006).
Local alignments can be ambiguous, with one node having different pairings in different local alignments. By contrast, a global network alignment provides a unique alignment from every node in the smaller network to exactly one node in the larger network, even though this may lead to inoptimal matchings in some local regions. Previous local network alignment algorithms have not generally been able to identify large subgraphs that have been conserved during evolution (Berg & Lassig 2004; Kelley et al. 2004).
Global network alignment has been studied previously in the context of biological networks (Singh et al. 2007; Flannick et al. 2008; Zaslavskiy et al. 2009). Unlike the above algorithms that primarily aim to detect conserved subnetworks, IsoRank (Singh et al. 2007) aims to maximize the overall match between the two networks. It relies on spectral graph theory to compute scores of aligning pairs of nodes from different networks; it does so by using the heuristic that two nodes are a good match if their respective neighbours also match well. Thus, the score of a protein pair depends on the score of their neighbours, that, in turn, depend on the neighbours of their neighbours, and so on. Once these ‘topological’ scores are computed for all node pairs, sequence-based BLAST scores are included in the pairwise alignment scores. IsoRank then constructs the node alignment with the repetitive greedy strategy of identifying among all protein pairs the highest scoring pair, outputting that pair, and removing all scores involving any of the two identified nodes (Singh et al. 2007). The more recent IsoRankN relies on the notion of node-specific rankings and uses a method similar to PageRank-Nibble algorithm (Liao et al. 2009). Graemlin has been extended to allow global network alignment by relying on a learning algorithm that uses a training set of known network alignments and their phylogenetic relationships to learn parameters for its scoring function, and by automatically adapting the learned objective function to any set of networks (Flannick et al. 2008).
1.3. Our contribution
At best, all previous algorithms depend only implicitly or indirectly on the topology of the network, as they heavily rely on some a priori information about nodes, such as sequence similarities of proteins in PPI networks (§3.2). Sequences are a very valuable source of biological information. Since proteins aggregate to perform a function instead of acting in isolation, system-level analyses of complex wiring around a protein in a PPI network could also provide valuable biological information and give deep insights into the inner working of cells. Network topology and protein sequences might give insights into complementary slices of biological information. For example, there exist identical protein sequences that can fold in different ways in different environments, leading to different functions and PPI network topologies (Whisstock & Lesk 2003; Watson et al. 2005; Komili et al. 2007; Kosloff & Kolodny 2008). In such cases, homology information is more correctly encoded in network topology than in sequence similarity (Memišević et al. in press). Thus, one could lose this information by focusing on sequence alone. We propose a network alignment algorithm with the cost function based solely and explicitly on a strong, theoretically grounded, direct measure of network topological similarity. Since our method does not use protein sequence information, it can align any two networks, not just biological ones. For example, our algorithm can be applied to road maps or social networks, which obviously have no genetic or protein sequence associated with them. Note that if we are trying to understand complex biological phenomena, we should try to use all biological data that are available, including sequences and topology. This is because integration of different data sources could provide deeper understanding. Thus, we design our method in a way that allows for inclusion of sequence information into the alignment cost function (see §2). However, it is of importance to uncover how much biological information can be extracted from network topology alone. Thus, our study addresses the problem of finding a good network alignment algorithm that relies solely on network topology.
We apply our method to align two PPI networks and demonstrate that our alignment exposes far more topologically complex regions of similarity than existing methods. Also, we use our method to compute a pairwise all-to-all network similarity matrix between a group of species, and then build a phylogenetic tree that bears a striking resemblance to the one based on sequence comparison. The importance of these results is that they extract biological knowledge from a new source of biological information, pure network topology, independently of any other source of biological information. We believe that the results in this paper just barely scratch the surface of the information that can be extracted from network topology.
2. Material and Methods
A graph G(V,E), or G for brevity, has node set V and edge set E. Given  nodes, the maximum number of undirected edges is M = n(n − 1)/2, and the number of possible undirected graphs on n nodes is thus 2M. The sheer number and diversity of possible graphs make graph classification and comparison problems difficult. One of those problems is called subgraph isomorphism: given two arbitrary graphs G(V,E) and H(U,F) such that
nodes, the maximum number of undirected edges is M = n(n − 1)/2, and the number of possible undirected graphs on n nodes is thus 2M. The sheer number and diversity of possible graphs make graph classification and comparison problems difficult. One of those problems is called subgraph isomorphism: given two arbitrary graphs G(V,E) and H(U,F) such that 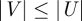 , does G exist as a subgraph of H? That is, is there a discrete map σ :V → U defined
, does G exist as a subgraph of H? That is, is there a discrete map σ :V → U defined  such that
such that  ? This problem is NP-complete, which means that no efficient algorithm is known for finding the mapping σ—the only known generally applicable way is to search through all possible mappings from V to U (Cook 1971). Since the number of such mappings is exponential in both
? This problem is NP-complete, which means that no efficient algorithm is known for finding the mapping σ—the only known generally applicable way is to search through all possible mappings from V to U (Cook 1971). Since the number of such mappings is exponential in both 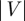 and
and  , this is considered an intractable problem.
, this is considered an intractable problem.
2.1. Graphlet degree signatures and signature similarities
GRAAL aligns a pair of nodes originating in different networks based on a similarity measure of their local neighbourhoods (Milenković & Pržulj 2008). This measure generalizes the degree of a node, which counts the number of edges that the node touches, into the vector of graphlet degrees, counting the number of graphlets that the node touches, for all 2–5-node graphlets (see figure 1). Note that the degree of a node is the first coordinate in this vector, since an edge (graphlet G0 in figure 1) is the only 2-node graphlet. Since it is topologically relevant to distinguish between, for example, nodes touching graphlet G1 at an end or at the middle, the notion of automorphism orbits (or just orbits, for brevity) is used. By taking into account the ‘symmetries’ between nodes of a graphlet, there are 73 different orbits across all 2- to 5-node graphlets. We number the orbits from 0 to 72 (Pržulj 2007). The full vector of 73 coordinates is the signature of a node (figure 2).
Figure 1.

All the connected graphs on up to five nodes. When appearing as an induced subgraph of a larger graph, we call them graphlets. They contain 73 topologically unique node types, called ‘automorphism orbits’. In a particular graphlet, nodes belonging to the same orbit are of the same shade. Graphlet G0 is just an edge, and the degree of a node historically defines how many edges it touches. We generalize the degree to a 73-component ‘graphlet degree’ vector that counts how many times a node is touched by each particular automorphism orbit. This figure is adapted from Pržulj (2007).
Figure 2.

An illustration of how the degree of node v in the leftmost panel is generalized into its ‘graphlet degree vector’, or ‘signature’, that counts the number of different graphlets that the node touches, such as triangles (middle panel) or squares (rightmost panel). Values of the 73 coordinates of the graphlet degree vector of node v, GDV (v), are presented in the table.
The signature of a node provides a novel and highly constraining measure of local topology in its vicinity and comparing the signatures of two nodes provides a highly constraining measure of local topological similarity between them. The signature similarity (Milenković & Pržulj 2008) is computed as follows. For a node 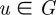 , ui denotes the ith coordinate of its signature vector, i.e. ui is the number of times node u is touched by an orbit i in G. The distance Di(u,v) between the ith orbits of nodes u and v is defined as Di(u,v) =
wi × |log(ui + 1) − log(vi + 1)|/log(max{ui, vi} + 2), where wi is a weight of orbit i that accounts for dependencies between orbits; for example, differences in counts of orbit 3 will imply differences in counts of all orbits that contain a triangle, such as orbits 10–14, 25, 26, etc., and thus, a higher weight is assigned to orbit 3, w3, than to the orbits that contain it (Milenković & Pržulj 2008). The total distance D(u,v) between nodes u and v is defined as: D(u,v) =
, ui denotes the ith coordinate of its signature vector, i.e. ui is the number of times node u is touched by an orbit i in G. The distance Di(u,v) between the ith orbits of nodes u and v is defined as Di(u,v) =
wi × |log(ui + 1) − log(vi + 1)|/log(max{ui, vi} + 2), where wi is a weight of orbit i that accounts for dependencies between orbits; for example, differences in counts of orbit 3 will imply differences in counts of all orbits that contain a triangle, such as orbits 10–14, 25, 26, etc., and thus, a higher weight is assigned to orbit 3, w3, than to the orbits that contain it (Milenković & Pržulj 2008). The total distance D(u,v) between nodes u and v is defined as: D(u,v) =  . The distance D(u,v) is in [0, 1], where distance 0 means that signatures of nodes u and v are identical. Finally, the signature similarity, S(u,v), between nodes u and v is S(u,v) = 1 − D(u,v).
. The distance D(u,v) is in [0, 1], where distance 0 means that signatures of nodes u and v are identical. Finally, the signature similarity, S(u,v), between nodes u and v is S(u,v) = 1 − D(u,v).
2.2. GRAAL (GRAph ALigner) algorithm
When aligning two graphs G(V,E) and H(U,F), GRAAL first computes costs of aligning each node v in G with each node u in H. The cost of aligning two nodes takes into account the signature similarity between them, modified to reduce the cost as the degrees of both nodes increase, since higher degree nodes with similar signatures provide a tighter constraint than correspondingly similar low degree nodes (see the electronic supplementary material1). In this way, we align the densest parts of the networks first. If we denote by deg(v) the degree of a node v in network G, by max_deg(G), the maximum degree of nodes in G, by S(v,u), the signature similarity of nodes v and u, and by α a parameter in [0, 1] that controls the contribution of the node signature similarity to the cost function (that is, 1 − α is the parameter that controls the contribution of node degrees to the cost function), then the cost of aligning nodes v and u is computed as
 |
A cost of 0 corresponds to a pair of topologically identical nodes v and u, while a cost close to 2 corresponds to a pair of topologically different nodes.
It is also possible to add protein sequence component to the cost function, to balance between topological and sequence similarity of aligned nodes. This can be done trivially by adding another parameter β to the cost function that would control the contribution of the current topologically derived costs, while 1 − β would control the contribution of node sequence similarities to the total cost function; a similar method has been used in other relevant studies (Singh et al. 2007; Liao et al. 2009; Zaslavskiy et al. 2009). Thus, if S′(v,u) is the sequence similarity between nodes v and u, the new cost of aligning nodes v and u obtained by including sequence similarity can be computed as
However, since we aim to extract only biological information encoded in network topology, analysing how balancing between the topological and sequence similarity affects the resulting alignments is out of the scope of our manuscript and is the subject of future work.
GRAAL chooses as the initial seed a pair of nodes (v,u), 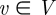 and
and 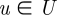 , which have the smallest cost. Ties are broken randomly, which could result in different alignments across different runs, although we empirically show that about 60 per cent of the entire alignment is consistent across different runs (§3.2). Once the seed is found, GRAAL builds ‘spheres’ of all possible radii around nodes v and u. A sphere of radius r around node v is the set of nodes SG (v,r) =
, which have the smallest cost. Ties are broken randomly, which could result in different alignments across different runs, although we empirically show that about 60 per cent of the entire alignment is consistent across different runs (§3.2). Once the seed is found, GRAAL builds ‘spheres’ of all possible radii around nodes v and u. A sphere of radius r around node v is the set of nodes SG (v,r) = 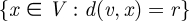 that are distance r from v where the distance d(v,x) is the length of the shortest path from v to x. Spheres of the same radius in two networks are then greedily aligned together by searching for the pairs
that are distance r from v where the distance d(v,x) is the length of the shortest path from v to x. Spheres of the same radius in two networks are then greedily aligned together by searching for the pairs 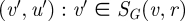 and
and 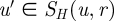 that are not already aligned and that can be aligned with the minimal cost. When all spheres around the seed (v,u) have been aligned, some nodes in both networks may remain unaligned. For this reason, GRAAL repeats the same algorithm on a pair of networks (Gp, Hp) for p = 1, 2 and 3, and searches for the new seed again, if necessary. We define a network Gp as a new network Gp = (V, Ep) with the same set of nodes as G and with
that are not already aligned and that can be aligned with the minimal cost. When all spheres around the seed (v,u) have been aligned, some nodes in both networks may remain unaligned. For this reason, GRAAL repeats the same algorithm on a pair of networks (Gp, Hp) for p = 1, 2 and 3, and searches for the new seed again, if necessary. We define a network Gp as a new network Gp = (V, Ep) with the same set of nodes as G and with  if, and only if, the distance between nodes v and x in G is less than or equal to p, i.e.
if, and only if, the distance between nodes v and x in G is less than or equal to p, i.e. 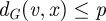 . Note that G1 = G. Using Gp, p > 1 allows us to align a path of length p in one network to a single edge in another network, which is analogous to allowing ‘insertions’ or ‘deletions’ in a sequence alignment. GRAAL stops when each node from G is aligned to exactly one node in H.
. Note that G1 = G. Using Gp, p > 1 allows us to align a path of length p in one network to a single edge in another network, which is analogous to allowing ‘insertions’ or ‘deletions’ in a sequence alignment. GRAAL stops when each node from G is aligned to exactly one node in H.
GRAAL produces global alignments. We note that optimal global alignments are not necessarily unique. Given any particular cost function, there may be many distinct alignments that all share the optimal cost. In this paper, we analyse just one specific alignment that we believe is a good one, although it may not be optimal even according to our measure. Enumerating all optimal (or at least good) alignments requires extending our algorithm to allow many-to-many mappings between the nodes in the two networks, and is the subject of the future work. Thus, many more predictions of equal validity to those in this paper are likely to be possible. However, we empirically demonstrate that a large portion (about 60%) of the entire alignment is conserved across different runs of the algorithm; thus, this core alignment is independent of the randomness in the algorithm.
The algorithm's pseudo code and details about the complexity analysis are presented in the electronic supplementary material. The software and data used in this paper are available upon request.
2.3. Statistical significance of our yeast–human alignment
Given a GRAAL alignment of two networks G(V,E) and H(U,F), we compute the probability of obtaining a given or better edge correctness score at random. For this purpose, an appropriate null model of random alignment is required. A random alignment is a random mapping f between nodes in two networks G(V,E) and H(U,F), f : V → U. GRAAL produces global alignments, so that all nodes in the smaller network (smaller in terms of the number of nodes) are aligned with nodes in the larger network. In other words, f is defined as  . This is equivalent to aligning each edge from G(V,E) with a pair of nodes (not necessarily an edge) in H(U,F). Thus, we define our null model of random alignment as a random mapping g : E → U × U. We define
. This is equivalent to aligning each edge from G(V,E) with a pair of nodes (not necessarily an edge) in H(U,F). Thus, we define our null model of random alignment as a random mapping g : E → U × U. We define 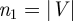 ,
,  ,
,  and
and 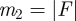 . We also define the number of node pairs in H as p = n2(n2 − 1)/2, and let EC = x% be the EC of the given alignment. We let k = [m1 × EC] = [m1 × x] be the number of edges from G that are aligned to edges in H. Then, the probability P of successfully aligning k or more edges by chance is the tail of the hypergeometric distribution:
. We also define the number of node pairs in H as p = n2(n2 − 1)/2, and let EC = x% be the EC of the given alignment. We let k = [m1 × EC] = [m1 × x] be the number of edges from G that are aligned to edges in H. Then, the probability P of successfully aligning k or more edges by chance is the tail of the hypergeometric distribution:
 |
For our yeast2–human1 alignment, we find 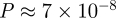 .
.
Now, we describe how to estimate the statistical significance of the amount of similarity we find between yeast2 and human1 in our alignment. To do that, we need to estimate how much similarity one would expect to find between two random networks and doing that, in turn, requires us to specify how we generate model random networks. Given two models that purport to fit a set of observations, we generally consider as superior the one that has fewer tunable parameters. For example, the STICKY and ER-DD models are constructed to preserve the degree distribution of the data. These and other data-driven models of random networks (Snijders 2002; Thorne & Stumpf 2007; Kuchaiev & Pržulj 2009) are thus expected to model particular PPI networks better than theoretical network models. However, they are not an appropriate choice to judge whether the yeast2 and human1 networks share a significant amount of structural similarity; this is because these models are strongly conditioned on these particular networks and thus they might transfer onto the model networks the similarities between yeast2 and human1 that we aim to detect in the first place. Thus, we search for a well-fitting theoretical null model. Arguably the best currently known theoretical model for PPI networks, requiring the fewest tunable parameters, is the geometric random graph model (‘GEO’; Pržulj et al. 2004; Pržulj 2007; Higham et al. 2008), in which proteins are modelled as existing in a metric space and are connected by an edge if they are within a fixed, specified distance of each other.
Although early, incomplete PPI datasets were modelled well by scale-free networks because of their power-law degree distributions (Barabási & Albert 1999; Jeong et al. 2001), it has been argued that such degree distributions were an artefact of noise (Agrafioti et al. 2005; Han et al. 2005; de Silva et al. 2006). In the light of new PPI network data, several studies (Pržulj et al. 2004; Pržulj 2007; Higham et al. 2008) have presented compelling evidence that the structure of PPI networks is closer to geometric than to scale-free networks. This was done by comparing frequencies of graphlets in real-world and model networks (Pržulj et al. 2004) and by measuring a highly constraining agreement between ‘graphlet degree distributions’ (Pržulj 2007). Finally, it has been shown that PPI networks can be successfully embedded into a low-dimensional Euclidean space, thus directly confirming that they have a geometric structure (Higham et al. 2008). The superior fit of the GEO model to PPI networks over other models may not be surprising, since it can be biologically motivated. Our intuition is based on the observation that genes (and proteins as gene products) exist in some biochemical space. The currently accepted paradigm is that genomes have evolved through series of gene duplication and mutation events (Ohno 1970). This can be modelled in the above-mentioned biochemical space as follows. When a gene gets duplicated, its child is in the same position in the biochemical space. Then natural selection acts on the duplicated genes so that one of them gets moved in the biochemical space (via mutations) from the other. The larger the mutation level, the larger the distance in the biochemical space. Consequently, the closer in space the duplicated genes, the larger the number of common interacting partners. These processes can naturally be modelled by GEO (see Pržulj et al. 2010 for details). In addition to PPI networks, GEO is a well-fitting theoretical null model for other biological networks, e.g. brain function networks (Kuchaiev et al. 2009) and protein structure networks (Milenković et al. 2009).
Accepting GEO as the optimal null model for PPI networks, we compute the probability of obtaining the EC of 11.72 per cent in our alignment of yeast2 and human1 to be 8.4 × 10−3. We do so by aligning with GRAAL pairs of GEO networks of the same size as yeast2 and human1 and by applying the following form of the Vysochanskij–Petunin inequality: P(|X − μ| ≥ λσ) ≤ (4/9λ2). Since GEO networks that are aligned have the same number of nodes and edges as the data, it is reasonable to assume that the distribution of their alignment scores is unimodal. Thus, we use the Vysochanskij–Petunin inequality, since it is more precise than Chebyshev's inequality for unimodal distributions. More details are supplied in the electronic supplementary material.
3. Results and discussion
Obviously, if one is to build meaningful alignments based solely upon network topology, one must first have a highly constraining measure of topological similarity. The simplest description of the topology of a node is its degree, which is the number of edges that touch it. Our much more highly constraining measure is a generalization of the degree of a node. We define a graphlet as a small, connected, induced subgraph of a larger network (Pržulj et al.
2004, 2006; Pržulj 2007). An induced subgraph on a node set 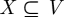 of G is obtained by taking X and all edges of G having both end-nodes in X. Figure 1 shows all the graphlets on 2, 3, 4 and 5 nodes. For a particular node v in a large network, we define a vector of ‘graphlet degrees’ (Milenković & Pržulj 2008) that counts the number of each kind of graphlet that touch v (figure 2). This vector, or signature, of v describes the topology of its neighbourhood and captures its interconnectivities out to a distance of 4 (see §4.1 and figure 2; Milenković & Pržulj 2008). Since this measure is based on all up to 5-node graphlets, it is very effective in quantifying local topological similarities between nodes in many real-world networks, owing to their small-world nature (Watts & Strogatz 1998).
of G is obtained by taking X and all edges of G having both end-nodes in X. Figure 1 shows all the graphlets on 2, 3, 4 and 5 nodes. For a particular node v in a large network, we define a vector of ‘graphlet degrees’ (Milenković & Pržulj 2008) that counts the number of each kind of graphlet that touch v (figure 2). This vector, or signature, of v describes the topology of its neighbourhood and captures its interconnectivities out to a distance of 4 (see §4.1 and figure 2; Milenković & Pržulj 2008). Since this measure is based on all up to 5-node graphlets, it is very effective in quantifying local topological similarities between nodes in many real-world networks, owing to their small-world nature (Watts & Strogatz 1998).
For our purposes, an alignment of two networks G and H consists of a set of ordered pairs (x,y), where x is a node in G and y is a node in H. Our algorithm, called GRAAL (GRAph ALigner), incorporates facets of both local and global alignments. We match pairs of nodes originating in different networks based on their signature similarity (Milenković & Pržulj 2008), where a higher signature similarity between two nodes corresponds to a higher topological similarity between their extended neighbourhoods (out to distance 4). The cost of aligning two nodes is modified to align the densest parts of the networks first; the cost is reduced as the degrees of both nodes increase, since higher degree nodes with similar signatures provide a tighter constraint than correspondingly similar low degree nodes (see §2 and the electronic supplementary material); α is a parameter in [0,1] that controls the contribution of the node signature similarity to the cost function, the other contribution being simply the degree of the node (see §2). In the case of two node alignments comparing equally, the tie is broken randomly. Thus, different runs of the alignment algorithm can produce different results. However, we find that for PPI networks that we analyse below, a deterministic ‘core’ alignment containing 60 per cent of all aligned pairs remains across all runs (see §3.2).
We align each node in the smaller network to exactly one node in the larger network. The matching proceeds using a technique analogous to the ‘seed and extend’ approach of the popular BLAST (Altschul et al. 1990) algorithm for sequence alignment: we first choose a single ‘seed’ pair of nodes (one node from each network) with high signature similarity. We then expand the alignment radially outward around the seed as far as practical using a greedy algorithm (see §2). Although local in nature, our algorithm produces large and dense global alignments. By ‘dense’ we mean that the aligned subgraphs share many edges, which would not be the case in a low-quality or random alignment.
3.1. Evaluation of GRAAL algorithm
To measure the performance of GRAAL, we align the largest connected component of the high-confidence yeast Saccharomyces cerevisiae PPI network (Collins et al. 2008), consisting of 8323 interactions amongst 1004 proteins, with its synthetic (or ‘noisy’) counterparts obtained by: (1) random removal of nodes from the network; (2) random removal of edges from the network; (3) random addition of edges to the network; and (4) addition of low confidence PPIs to the network (thus, this is the addition of real data to the network, but the data is of low confidence). For each of these four ‘noise types,’ we run experiments with different percentages of added noise: 5, 10, 15, 20 and 25 per cent. Owing to randomness, we run each experiment 30 times and average results over the 30 runs. Example for experiment of type (1) above and the 5 per cent noise level, we randomize the data 30 times by random removal of 5 per cent of nodes from the data network; this results in 30 randomized networks and we align each of them with the original data network to obtain 30 EC scores that we average. The only exception is noise type (4) above, the additions of low-confidence PPIs: no randomness exists for this noise type, since top k per cent most confident PPIs (k = 5, 10, 15, 20, 25) from the lower-confidence data set are added. The results are presented in the electronic supplementary material, figures S7 and S8. With these tests, depending on the noise type and level, we demonstrate that our algorithm is capable of producing high-quality alignments with EC of about 90 per cent (see section ‘Evaluation of GRAAL algorithm’ of electronic supplementary material, figures S7 and S8). This indicates that, given high topological similarity of the aligned networks, our algorithm is capable of discovering alignments with high EC.
3.2. Pairwise alignment of yeast and human PPI networks
Using GRAAL, we align the human PPI network of Radivojac et al. (2008) to the Collins et al. (2008) yeast PPI network which we call ‘human1’ and ‘yeast2,’ respectively. We chose yeast as our second species because currently it has a high quality PPI network, with 16 127 interactions (edges) among 2390 proteins (nodes). The ‘best’ alignment (defined below) found by GRAAL aligns 1890 of the edges in yeast2 to edges in human1. Thus, the EC of our alignment is 11.72 per cent. There are 970 nodes involved in these ‘correct’ edge alignments, representing 40 per cent of all yeast2 nodes. We obtained similar EC for aligning other yeast (Stark et al. 2006; Collins et al. 2008) and human (Peri et al. 2004; Rual et al. 2005; Stark et al. 2006) networks (see the electronic supplementary material, figure S1). The best alignment is defined as follows. Owing to the existence of the α parameter in the cost function (as explained above) and some randomness in the GRAAL algorithm (see §2.2 and the electronic supplementary material for details), the actual alignments and ECs vary across different values of α, and across different runs of the algorithm for the same α. With this in mind, the best alignment is the alignment with the highest EC over all values of α, and over all runs for the given α. The highest EC is obtained for α of 0.8; the minimum EC over all runs for this α is higher than the maximum EC over all runs for any other α between 0 and 1 in increments of 0.1. Thus, we focus on alignments produced for α of 0.8. Variation of EC over different runs for this α is small, with minimum and maximum EC of 11.5 and 11.72 per cent, respectively. Moreover, intersection of alignments from up to 40 different runs at α of 0.8 contains 1433 pairs, i.e. about 60 per cent of the entire alignment. We call this intersection the core alignment.
In addition to counting aligned edges, it is important that the aligned edges cluster together to form large and dense connected subgraphs, in order to uncover such regions of similar topology. We define a common connected subgraph (CCS) as a connected subgraph (not necessarily induced) that appears in both networks. The largest CCS in our best alignment (figure 3a) has 900 interactions amongst 267 proteins, which comprises 11.2 per cent of the proteins in the yeast2 network. Our second largest CCS has 286 interactions amongst 52 nodes, depicted in figure 3b. The entire common subgraph is presented in the electronic supplementary material, figure S2.
Figure 3.

The alignment of yeast2 and human1 PPI networks. An edge between two nodes means that an interaction exists in both species between the corresponding protein pairs. Thus, the displayed networks appear, in their entirety, in the PPI networks of both species. (a) The largest common connected subgraph (CCS) consisting of 900 interactions amongst 267 proteins. (b) The second largest CCS consisting of 286 interactions amongst 52 proteins; each node contains a label denoting a pair of yeast and human proteins that are aligned.
3.3. Quality of GRAAL's yeast–human alignment
Optimizing EC is an NP-hard problem and therefore we cannot say with certainty whether EC of 11.72 per cent is the maximum EC for the yeast and human PPI networks that we analyse. However, since we demonstrate on synthetic data that GRAAL can correctly align similar networks (§3.1), we believe that GRAAL's alignment with EC of 11.72 per cent between PPI networks of distant species such as yeast and human is good. We further support this belief by demonstrating the statistical significance and biological validity of this alignment (see below). Also, we show that both the size and the quality of our alignment is superior to the alignments produced by competing methods (see §3.4).
We look at several distinct ways in which to judge the quality of GRAAL's yeast–human alignment. First, we judge its topological quality compared with a random alignment of these two particular networks. We find that the probability of obtaining EC of 11.72 per cent or better (p-value) in a random alignment of yeast and human PPI networks is lower than 7 × 10−8 (for details about the null model of random alignment that we use, see §2.3 and the electronic supplementary material). The probability of obtaining a large CCS would be significantly smaller, so this represents a weak upper bound on our p-value.
Second, we comment on the amount of topological similarity found between yeast and human in GRAAL's alignment. We compare GRAAL's yeast–human alignment with GRAAL's alignment of random networks (from a particular model) of the same size as the data. If we align with GRAAL networks drawn from several different random graph models (Milenković et al. 2008) that have the same number of nodes and edges as yeast2 and human1, we find that EC between random networks of at most 8.8 ± 0.39 per cent is significantly lower than EC of GRAAL's yeast2–human1 alignment of 11.72 per cent, with a p-value of less than 8.4 × 10−3 (as explained below). Specifically, aligning two Erdös–Rényi random graphs with the same degree distribution as the data (‘ER-DD’) gives an EC of only about 0.31 ± 0.22 per cent. Similar alignments of Barabási-Albert type scale-free networks (‘SF-BA’; Barabási & Albert 1999), stickiness model networks (‘STICKY’; Pržulj & Higham 2006), or three-dimensional geometric random graphs (‘GEO-3D’; Pržulj et al. 2004), give EC scores of only 2.86 ± 0.57, 5.89 ± 0.39 and 8.8 ± 0.39 per cent, respectively. Even though we use five different network models as null models, we only report the p-value of GRAAL's alignment of yeast and human when evaluated against GEO as the null model. This is not only because GEO has already been shown to be the best-fitting model for PPI networks (see §3), but also because when GRAAL aligns GEO to GEO networks (of the size of the data), and when we use these alignments to compute statistical significance of GRAAL's yeast–human alignment, we get a higher p-value than when we use any of the other four network models. So we choose the null model that is the worst-case scenario for evaluating the statistical significance of GRAAL's yeast–human alignment and we still get the p-value of 8.4 × 10−3. This tells us that yeast and humans, two very different species, enjoy more network similarity than chance would allow.
Third, we analyse the biological quality of our alignment by examining whether: (i) aligned protein pairs perform the same biological function; (ii) GRAAL is capable of identifying evolutionary conserved functional modules; and (iii) GRAAL's alignment of yeast and human is supported by sequence alignments.
We count how many of our aligned pairs share common gene ontology (GO) terms (The Gene Ontology Consortium 2000). GO terms succinctly describe the many biological properties that a given protein may have. For this analysis, we consider the ‘complete’ GO annotation data set, containing all GO annotations, independent of GO evidence code. GO annotation data were downloaded in September 2009. Across our entire best yeast2–human1 alignment, 45.1, 15.6, 5.1 and 2.0 per cent of aligned protein pairs share at least one, two, three, and four GO terms, respectively. Compared with random alignments, the p-values for these percentages are all in the 10−6 to 10−8 range. Furthermore, the results improve across GRAAL's core yeast2–human1 alignment: 50.9, 19.3, 7.3 and 3.0 per cent of aligned protein pairs share at least one, two, three and four GO terms, respectively; the p-values for these percentages are all in the 10−8 to 10−9 range.
We find that GRAAL aligns network regions of yeast and human in which a large percentage of proteins perform the same biological function in both species. In the ‘best’ alignment (defined in §2.2), GRAAL aligns a 52-node subnetwork between yeast and human in which 98 per cent of yeast and 67 per cent of human proteins are involved in splicing. This result is encouraging, since splicing is known to be conserved even between distant eucaryotes (Wentz-Hunter & Potashkin 1995; Collins & Penny 2005; Lorkovic et al. 2005). Additionally, it aligns a 24-node subnetwork in which 96 per cent of yeast and 39 per cent of human proteins are involved in transcription. Furthermore, it aligns another 12-node subnetwork in which 25 per cent of yeast and 40 per cent of human proteins are involved in transcription. Finally, it aligns a 10-node subnetwork in which 100 per cent of yeast and 80 per cent of human proteins are involved in translation. Similar results are obtained for the ‘core’ alignment (defined in §2.2): GRAAL aligns a 48-node subnetwork in which 98 per cent of yeast and 69 per cent of human proteins are involved in splicing; also, it aligns a 12-node subnetwork in which 25 per cent of yeast and 40 per cent of human proteins are involved in transcription.
We examine GRAAL's core alignment of yeast and human PPI networks and we find that 19 per cent of aligned yeast–human protein pairs have sequence identities above the ‘twilight zone’ threshold of 30 per cent for evolutionary relatedness (Doolittle 1981; Rost 1999). Rost (1999) analysed more than a million alignments of protein sequences from the Protein Data Bank and found that 90 per cent of aligned protein pairs with sequence identities above this threshold were homologous. Moreover, about 70 per cent of protein pairs in GRAAL's core alignment have sequence identities in the ‘twilight zone,’ i.e. between 20 and 30 per cent. Although evolutionary closeness cannot be claimed with certainty for sequences with identities in the ‘twilight zone’ (Doolittle 1981; Rost 1999) the vast majority of homologues had been shown to have less than 30 per cent sequence identity. Hence, GRAAL's yeast–human alignment appears to be partially supported by the sequence alignment.
Section 2 and the electronic supplementary material provide more details on all of the above.
3.4. Comparison with other methods
GRAAL produces by far the most complete topological alignments of biological networks to date and uncovers CCSs that are substantially larger and denser than those produced by currently published algorithms, as demonstrated below. The best currently published global alignment of similar networks is the alignment of yeast and fly by IsoRank (Singh et al. 2007), which uses sequence information in addition to topological information. It aligns 1420 edges, but its largest CCS contains just 35 nodes and 35 edges. We applied IsoRank to our yeast2–human1 data using only topological information. We found that it aligns 628 interactions, giving an edge correctness of only 3.89 per cent, compared with GRAAL's EC of 11.72 per cent. Hence, we align three times more edges than IsoRank does. IsoRank's largest CCS has just 261 interactions among 116 proteins, compared with GRAAL's largest CCS with 900 interactions amongst 267 proteins. Thus, GRAAL's largest CCS is 2.3 and 3.5 times larger than IsoRank's largest CCS in terms of the number of nodes and edges, respectively. Note that we do not include sequence information in IsoRank's alignment cost function, since Singh et al. (2007) have shown that the highest EC is obtained when topology alone is used.
Additionally, our results are better than those achieved by IsoRank with respect to the number of shared GO terms even though GRAAL does not use any protein sequence information. In the global alignment produced by IsoRank, 44.2, 14.1, 4.1 and 1.5 per cent of aligned protein pairs have at least one, two, three, and four GO terms in common, respectively, compared with GRAAL's percentages of 45.1, 15.6, 5.1 and 2.0 per cent, respectively. Furthermore, if we restrict our analysis only to the largest CCS, in IsoRank's CCS, the percentages are 60.6, 11.9 and 0 per cent for sharing at least 1, 2 and 3 common GO terms, respectively, while in GRAAL's CCS, these percentages are 67.2, 22.0 and 5.2 per cent, respectively.
Recently, IsoRankN, an algorithm for global alignment of multiple networks, has been introduced (Liao et al. 2009). However, a comparison with GRAAL is not feasible, since the output of the two algorithms is different. While GRAAL outputs a list of one-to-one node mappings between the networks being aligned, IsoRankN's alignment contains sets of aligned proteins, where no two sets overlap, but each set can contain more than one node (i.e. many-to-many node mappings) from each of the networks being aligned. Thus, IsoRankN's output cannot be quantified topologically with EC, since one many-to-many node alignment can produce exponentially many one-to-one node alignments and enumerating all of them is computationally infeasible.
Another popular global network alignment method is Graemlin (Flannick et al. 2008). We do not compare our alignment to the one produced by Graemlin because Graemlin requires a variety of other input information, including phylogenetic relationships between the species being aligned. By contrast, GRAAL's output can be used to infer phylogenetic relationships.
Finally, other methods potentially better than IsoRank exist (Zaslavskiy et al. 2009). However, their current implementations failed to process networks of the size of yeast2 and human1 (M. Zaslavskiy & J.-P. Vert 2009, personal communication with the authors). Moreover, we do not benchmark these methods on the yeast and fly data analysed by Zaslavskiy et al. (2009) because they did not try to align the entire yeast and fly networks but they focused only on their smaller induced subgraphs defined on proteins covered by Inparanoid clusters. Thus, although their ‘global’ yeast-fly alignment aligns each node in the smaller subnetwork (defined above) to a node in the larger subnetwork, it is not truly global, as it aligned only parts of the original yeast and fly networks. Therefore, we found it inappropriate to evaluate GRAAL's global alignment of the entire yeast and fly networks with their ‘global’ alignments of partial yeast and fly networks. Moreover, we believe that a good network alignment algorithm should both produce high-quality alignments and be capable of dealing with large datasets; this is especially true for biological networks, since their sizes will only continue to grow. Thus, the methods by Zaslavskiy et al. (2009) that failed to process any larger dataset are not relevant to the large networks we consider.
3.5. Application to protein function prediction
With the above validations in hand (§3.3), we believe that GRAAL's alignments can be used to predict biological characteristics (i.e. GO molecular function (MF), biological process (BP) and cellular component (CC)) of un-annotated proteins based on their alignments with annotated ones.
Here, we distinguish between two different sets of GO annotation data: the complete set described above, containing all GO annotations, independent of GO evidence codes, and biologically based set, containing GO annotations obtained by experimental evidence codes only (see The Gene Ontology Consortium (2000) for details). Since in the complete GO annotation dataset, many GO terms were assigned to proteins computationally (e.g. from sequence alignments), that set is biologically less confident than the biologically based one. We make predictions with respect to both GO annotation datasets, as described below.
First, we analyse GRAAL's best yeast2–human1 alignment (i.e. the alignment with the highest EC over all runs for alpha of 0.8, as explained in §2) to identify aligned protein pairs where one of the proteins is annotated with a ‘root’ GO term only: GO:0003674 for MF, GO:0008150 for BP, or GO:0005575 for CC; this means that one of the proteins in the pair has no known functional information (The Gene Ontology Consortium 2000). Next, we check whether aligned partners of such proteins with unknown function are annotated with a known MF, BP or CC GO terms, with respect to both the complete and biologically based GO annotation datasets. If so, we assign all known MF, BP or CC GO terms to the unannotated protein.
With respect to the complete GO dataset, we predict MF for 44 human and 435 yeast proteins, BP for 53 human and 157 yeast proteins and CC for 52 human and 54 yeast proteins. Since the GO database offers a list with an explicit note that a protein is not associated with a given GO term, we were able to examine directly whether our predictions contradicted this list. We found no contradictions in the GO database for any of the yeast or human proteins with respect to MF or BP; and we found contradiction only for one of our human predictions with respect to CC. We also attempted to validate all of our predictions using the literature search and text mining tool CiteXplorer (Labarga et al. 2007). For 34.1, 43.4 and 46.2 per cent of our MF, BP, and CC human predictions, respectively, this tool found at least one article mentioning the protein of interest in the context of at least one of our predictions for that protein. For yeast, these percentages are 42.07, 3.18 and 12.96 per cent, respectively. Our human and yeast predictions made with respect to the complete GO dataset are presented in electronic supplementary material, tables S1 and S2, respectively.
With respect to the biologically based GO dataset, we predict MF for 30 human and 214 yeast proteins, BP for 42 human and 41 yeast proteins and CC for 45 human and 17 yeast proteins. None of these predictions were contradicted in the GO database. We validated with CiteXplorer 10, 4.76 and 20 per cent of our biologically based MF, BP, and CC human predictions, respectively. We also validated 48.1 per cent of our biologically based MF yeast predictions. Our human and yeast predictions made with respect to the biologically based GO dataset are presented in electronic supplementary material, tables S3 and S4, respectively.
3.6. Reconstruction of phylogenetic trees by aligning metabolic pathways across species
Finally, we describe a completely different application: how purely topological alignment of metabolic networks obtained by GRAAL can be used to recover phylogenetic relationships.
Several studies analysing metabolic pathways in different species have aimed to find an evolutionary relationship between the species and construct their phylogenetic trees (Forst & Schulten 2001; Heymans & Singh 2003; Suthram et al. 2005; Zhang et al. 2006). Different distance metrics have been used for constructing phylogenetic trees. For example, similarities between pathways have been computed from sequence similarities between corresponding substrates and enzymes from individual pathways (Forst & Schulten 2001) or as a combination of similarities of enzymes from individual metabolic networks and topologies of these networks (Heymans & Singh 2003; Suthram et al. 2005). The similarity of enzymes is based on the similarity of their sequences, structures or Enzyme Commission numbers (Webb 1990). The topological similarity of two pathways has been based on the similarity between nodes (corresponding to enzymes) and the similarity of their neighbourhoods, measuring whether a node influences similar nodes and whether it is influenced by similar nodes itself (Heymans & Singh 2003). In addition, topological similarity of metabolic pathways combining global network properties, such as the diameter and clustering coefficient, and similarities of shared node (i.e. enzyme) neighbourhoods has been used (Zhang et al. 2006).
Therefore, although related attempts exist (Suthram et al. 2005), they all still use some biological or functional information external to network topology, such as sequence similarities, to define node similarities and derive phylogenetic trees from pathways. Since we use only network topology to define protein similarity, our information source is fundamentally different. Thus, our algorithm recovers phylogenetic relationships (but not the evolutionary timescale of species divergence at this point) in a completely novel and independent way from all existing methods for phylogenetic recovery.
It has been shown that PPI network structure has subtle effects on the evolution of proteins and that reasonable phylogenetic inference can only be done between closely related species (Agrafioti et al. 2005). In the KEGG pathway database, there are 17 eucaryotic organisms with fully sequenced genomes (Kanehisa & Goto 2000), of which seven are protists, six are fungi, two are plants and two are animals. Here we focus on protists (see the electronic supplementary material for fungi). For each organism, we extract the union of all metabolic pathways from KEGG, and then find all-to-all pairwise network alignments between species using GRAAL. The EC scores between pairs of protist networks range from 29.6 to 76.7 per cent. We create phylogenetic trees using the average distance algorithm,2 with pairwise EC as the distance measure. We compare our phylogenetic trees to the published ones3 obtained from genetic or amino acid sequence alignments (Keeling et al. 2000; Pennisi 2003). Figure 4 presents our phylogenetic tree for protists and shows that it is very similar to that found by sequence comparison (Pennisi 2003). We can estimate the statistical significance of our tree by measuring how it compares to trees built from random networks of the same size as the metabolic networks (see the electronic supplementary material); we find that the p-value of our tree is less than 1.3 × 10−3. Phylogenetic trees based on alignments made by IsoRank do not differ significantly from random ones (see the electronic supplementary material). We also find that the topologies of the entire metabolic networks of Cryptosporidium parvum and Cryptosporidium hominis are very similar, having an EC of 75.72 per cent. This result is encouraging since these organisms are two morphologically identical species of Apicomplexan protozoa with 97 per cent genetic sequence identity, but with strikingly different hosts (Tanriverdi & Widmer 2006) that contribute to their divergence (Xu et al. 2004).
Figure 4.

Comparison of the phylogenetic trees for protists obtained by genetic sequence alignments and by GRAAL's metabolic network alignments. Left: the tree obtained from genetic sequence comparison. Right: the tree obtained from GRAAL. The following abbreviations are used for species: CHO, Cryptosporidium hominis; DDI, Dictyostelium discoideum; CPV, Cryptosporidium parvum; PFA, Plasmodium falciparum; EHI, Entamoeba histolytica; TAN, Theileria annulata; TPV, Theileria parva. The species are grouped into the following classes: ‘Alveolates,’ ‘Entamoeba,’ and ‘cellular slime mould’.
Note that all of the metabolic networks that we align are derived from a mix of experimentally obtained data and network reconstructions based on orthology relationships between species. Hence, the fact that we largely recover the phylogenetic trees obtained from sequence alignments is a strong validation of our method. Moreover, the phylogenetic tree in the literature is obtained from sequence alignments of mitochondrial proteins or ribosomal RNA, whereas metabolic networks in KEGG are partially obtained by sequence alignments of protein sequences. Therefore, since a different source of sequence data is used for reconstructing phylogenetic trees in the literature and for reconstructing metabolic networks, the phylogenetic trees obtained from our network alignments might already be viewed as new and independent sources of phylogenetic information. This will gain in biological importance when purely experimentally obtained networks become available, further providing validation of sequence-based phylogeny.
Given that our phylogenetic tree is slightly different from that produced by sequence, there is no reason to believe that the sequence-based one should a priori be considered the correct one. Sequence-based phylogenetic trees are built based on multiple alignment of gene sequences and whole genome alignments. Multiple alignments can be misleading owing to gene rearrangements, inversions, transpositions and translocations that occur at the substring level. Furthermore, different species might have an unequal number of genes or genomes of vastly different lengths. Whole genome phylogenetic analyses can also be misleading owing to non-contiguous copies of a gene or non-decisive gene order (Out & Sayood 2003). Finally, the trees are built incrementally from smaller pieces that are ‘patched’ together probabilistically (Pennisi 2003), so probabilistic errors in the tree are expected. Our tree suffers from none of these problems, but it may suffer from other problems, such as noise and incompleteness of PPI networks.
4. Conclusions
In summary, we present evidence that it is possible to extract biological knowledge from network topology only. We introduce a new global network alignment algorithm that is based solely on network topology. As such, it can be applied to any network type, not just biological ones. We apply our method to align PPI networks of yeast and human and demonstrate that it produces topologically statistically significant alignments in which many aligned proteins perform the same biological function. Given the high quality of our yeast–human alignment, we predict biological function of unannotated proteins based on the function of their annotated aligned partners, validating a large number of our predictions in the literature. Additionally, we successfully reconstruct phylogenetic trees from topological alignments of metabolic networks, demonstrating that network topology can be used as a novel and independent source of phylogenetic information.
Network alignment has applications across an enormous span of domains, from social networks to software call graphs. In the biological domain, the mass of currently available network data will only continue to increase and we believe that high-quality topological alignments can yield new and pivotal insights into function, evolution and disease.
Acknowledgements
We thank M. Rašajski for computational assistance. This project was supported by the NSF CAREER IIS-0644424 grant.
Footnotes
The supplementary information is available at http://www.ics.uci.edu/~bio-nets/GRAAL_suppl_inf/.
References
- Agrafioti I., Swire J., Abbott J., Huntley D., Butcher S., Stunpf M. P. 2005. Comparative analysis of the Saccharomyces cerevisiae and Caenorhabdits elegans protein interaction networks. BMC Evol. Biol. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Lipman D. J. 1990. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Barabási A., Albert R. 1999. Emergence of scaling in random networks. Science 286, 509–512. ( 10.1126/science.286.5439.509) [DOI] [PubMed] [Google Scholar]
- Berg J., Lassig M. 2004. Local graph alignment and motif search in biological networks. Proc. Natl Acad. Sci. USA 101, 14 689–14 694. ( 10.1073/pnas.0305199101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J., Lassig M. 2006. Cross-species analysis of biological networks by Bayesian alignment. Proc. Natl Acad. Sci. USA 103, 10 967–10 972. ( 10.1073/pnas.0602294103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colizza V., Flammini A., Serrano M. A., Vespignani A. 2006. Detecting rich-club ordering in complex networks. Nat. Phys. 2, 110–115. ( 10.1038/nphys209) [DOI] [Google Scholar]
- Collins S., Kemmeren P., Zhao X., Greenblatt J., Spencer F., Holstege F., Weissman J., Krogan N. 2008. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol. Cell. Proteom. 6, 439–450. ( 10.1074/mcp.M600381-MCP200) [DOI] [PubMed] [Google Scholar]
- Collins L., Penny D. 2005. Complex spliceosomal organization ancestral to extant eukaryotes. Mol. Biol. Evol. 22, 1053–1066. ( 10.1093/molbev/msi091) [DOI] [PubMed] [Google Scholar]
- Cook S. 1971. The complexity of theorem-proving procedures. Proc. 3rd Annu. ACM Symp. Theory of Computing, pp. 151–158. New York, NY: Association for Computing Machinery. [Google Scholar]
- de Silva E., Thorne T., Ingram P., Agrafioti I., Swire J., Wiuf C., Stumpf M. P. 2006. The effects of incomplete protein interaction data on structural and evolutionary inferences. BMC Biol. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle R. F. 1981. Similar amino-acid sequences: chance or common ancestry? Science 214, 149–159. ( 10.1126/science.7280687) [DOI] [PubMed] [Google Scholar]
- Flannick J., Novak A., Balaji S., Harley H., Batzglou S. 2006. Graemlin general and robust alignment of multiple large interaction networks. Genome Res. 16, 1169–1181. ( 10.1101/gr.5235706) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannick J., Novak A. F., Do C. B., Srinivasan B. S., Batzoglou S. 2008. Automatic parameter learning for multiple network alignment. In Proc. Int. Conf. Research in Computational Molecular Biology—RECOMB, pp. 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forst C., Schulten K. 2001. Phylogenetic analysis of metabolic pathways. J. Mol. Evol. 52, 471–489. [DOI] [PubMed] [Google Scholar]
- Gavin A. C., et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147. ( 10.1038/415141a) [DOI] [PubMed] [Google Scholar]
- Gavin A. C., et al. 2006. Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636. ( 10.1038/nature04532) [DOI] [PubMed] [Google Scholar]
- Guimera R., Sales-Pardo M., Amaral L. A. N. 2007. Classes of complex networks defined by role-to-role connectivity profiles. Nat. Phys. 3, 63–69. ( 10.1038/nphys489) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. D. H., Dupuy D., Bertin N., Cusick M. E., Vidal M. 2005. Effect of sampling on topology predictions of protein–protein interaction networks. Nat. Biotechnol. 23, 839–844. ( 10.1038/nbt1116) [DOI] [PubMed] [Google Scholar]
- Heymans M., Singh A. 2003. Deriving phylogenetic trees from the similarity analysis of metabolic pathways. Bioinformatics 19, i138–i146. ( 10.1093/bioinformatics/btg1018) [DOI] [PubMed] [Google Scholar]
- Higham D., Rašajski M., Pržulj N. 2008. Fitting a geometric graph to a protein–protein interaction network. Bioinformatics 24, 1093–1099. ( 10.1093/bioinformatics/btn079) [DOI] [PubMed] [Google Scholar]
- Ho Y., et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183. ( 10.1038/415180a) [DOI] [PubMed] [Google Scholar]
- Ito T., Tashiro K., Muta S., Ozawa R., Chiba T., Nishizawa M., Yamamoto K., Kuhara S., Sakaki Y. 2000. Toward a protein-protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl Acad. Sci. USA 97, 1143–1147. ( 10.1073/pnas.97.3.1143) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl Acad. Sci. USA 98, 4569–4574. ( 10.1073/pnas.061034498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H., Mason S. P., Barabási A. L., Oltvai Z. N. 2001. Lethality and centrality in protein networks. Nature 411, 41–42. ( 10.1038/35075138) [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. ( 10.1093/nar/28.1.27) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling P., Luker M., Palmer J. 2000. Evidence from β-tubulin phylogeny that microsporidia evolved from within the fungi. Mol. Biol. Evol. 17, 23–31. [DOI] [PubMed] [Google Scholar]
- Kelley B. P., Bingbing Y., Lewitter F., Sharan R., Stockwell B. R., Ideker T. 2004. PathBLAST: a tool for alignment of protein interaction networks. Nucleic Acids Res. 32, W83–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komili S., Farny N. G., Roth F. P., Silver P. A. 2007. Functional specificity among ribosomal proteins regulates gene expression. Cell 131, 557–571. ( 10.1016/j.cell.2007.08.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosloff M., Kolodny R. 2008. Sequence-similar, structure-dissimilar protein pairs in the PDB. Proteins 71, 891–902. ( 10.1002/prot.21770) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuturk M., Kim Y., Topkara U., Subramaniam S., Szpankowski W., Grama A. 2006. Pairwise alignment of protein interaction networks. J. Comput. Biol. 13, 182–199. ( 10.1089/cmb.2006.13.182) [DOI] [PubMed] [Google Scholar]
- Krogan N. J., et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643. ( 10.1038/nature04670) [DOI] [PubMed] [Google Scholar]
- Kuchaiev O., Pržulj N. 2009. Learning the structure of protein–protein interaction networks. In Proc. 2009 Pacific Symp. Biocomputing (PSB), Big Island, Hawaii, 4–8 January 2010, pp. 39–50. [PubMed] [Google Scholar]
- Kuchaiev O., Wang P. T., Nenadić Z., Pržulj N. 2009. Structure of brain functional networks. In 31st Annu. Int. Conf. IEEE Engineering in Medicine and Biology Society (EMBC '09), Minneapolis, Minnesota, USA, 2–6 September 2009. [DOI] [PubMed] [Google Scholar]
- Labarga A., Valentin F., Andersson M., Lopez R. 2007. Web services at the European bioinformatics institute. Nucleic Acids Res. 35, W6–W11. ( 10.1093/nar/gkm291) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z., Xu M., Teng M., Niu L. 2006. NetAlign: a web-based tool for comparison of protein interaction networks. Bioinformatics 22, 2175–2177. ( 10.1093/bioinformatics/btl287) [DOI] [PubMed] [Google Scholar]
- Liao C. S., Lu K., Baym M., Singh R., Berger B. 2009. IsoRankN: spectral methods for global alignment of multiple protein networks. Bioinformatics 25, i253–i258. ( 10.1093/bioinformatics/btp203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorkovic Z. J., Lehner R., Forstner C., Barta A. 2005. Evolutionary conservation of minor u12-type spliceosome between plants and humans. RNA 11, 1095–1107. ( 10.1261/rna.2440305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memišević V., Milenković T., Pržulj N. In press Complementarity of network and sequence structure in homologous proteins. J. Integr. Bioinformatics. [DOI] [PubMed] [Google Scholar]
- Milenković T., Filippis I., Lappe M., Pržulj N. 2009. Optimized null model for protein structure networks. PLoS ONE 4, e5967 ( 10.1371/journal.pone.0005967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenković T., Pržulj N. 2008. Uncovering biological network function via graphlet degree signatures. Cancer Inform. 6, 257–273. [PMC free article] [PubMed] [Google Scholar]
- Milenković T., Lai J., Pržulj N. 2008. Graphcrunch: a tool for large network analyses. BMC Bioinformatics 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. Berlin, Germany: Springer. [Google Scholar]
- Out H., Sayood K. 2003. A new sequence distance measure for phylogenetic tree construction. Bioinformatics 19, 2122–2130. ( 10.1093/bioinformatics/btg295) [DOI] [PubMed] [Google Scholar]
- Pennisi E. 2003. Modernizing the tree of life. Science 300, 1692–1697. ( 10.1126/science.300.5626.1692) [DOI] [PubMed] [Google Scholar]
- Peri S. 2004. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 32, D497–D501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pržulj N. 2007. Biological network comparison using graphlet degree distribution. Bioinformatics 23, e177–e183. [DOI] [PubMed] [Google Scholar]
- Pržulj N., Higham D. 2006. Modelling protein–protein interaction networks via a stickiness index. J. R. Soc. Interface 3, 711–716. ( 10.1098/rsif.2006.0147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pržulj N., Corneil D. G., Jurisica I. 2004. Modeling interactome: scale-free or geometric? Bioinformatics 20, 3508–3515. ( 10.1093/bioinformatics/bth436) [DOI] [PubMed] [Google Scholar]
- Pržulj N., Corneil D. G., Jurisica I. 2006. Efficient estimation of graphlet frequency distributions in protein–protein interaction networks. Bioinformatics 22, 974–980. [DOI] [PubMed] [Google Scholar]
- Pržulj N., Kuchaiev O., Stevanović A., Hayes W. 2010. Geometric evolutionary dynamics of protein interaction networks. In Proc. 2009 Pacific Symp. Biocomputing (PSB), Big Island, Hawaii, 4–8 January 2010, pp. 178–189. [DOI] [PubMed] [Google Scholar]
- Radivojac P., Peng K., Clark W. T., Peters B. J., Mohan A., Boyle S. M., Mooney S. D. 2008. An integrated approach to inferring gene-disease associations in humans. Proteins 72, 1030–1037. ( 10.1002/prot.21989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Seraphin B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnol. 17, 1030–1032. ( 10.1038/13732) [DOI] [PubMed] [Google Scholar]
- Rost B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12, 85–94. ( 10.1093/protein/12.2.85) [DOI] [PubMed] [Google Scholar]
- Rual J., et al. 2005. Towards a proteome-scale map of the human protein–protein interaction network. Nature 437, 1173–1178. ( 10.1038/nature04209) [DOI] [PubMed] [Google Scholar]
- Sharan R., Ideker T. 2006. Modeling cellular machinery through biological network comparison. Nat. Biotechnol. 24, 427–433. ( 10.1038/nbt1196) [DOI] [PubMed] [Google Scholar]
- Sharan R., Suthram S., Kelley R. M., Kuhn T., McCuine S., Uetz P., Sittler T., Karp R. M., Ideker T. 2005. Conserved patterns of protein interaction in multiple species. Proc. Natl Acad. Sci. USA 102, 1974–1979. ( 10.1073/pnas.0409522102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis N., et al. 2008. Empirically controlled mapping of the Caenorhabditis elegans protein–protein interactome network. Nat. Meth. 6, 47–54. ( 10.1038/nmeth.1279) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Xu J., Berger B. 2007. Pairwise global alignment of protein interaction networks by matching neighborhood topology. In Research in computational molecular biology, pp. 16–31. Berlin, Germany: Springer. [Google Scholar]
- Singh R., Xu J., Berger B. 2008. Global alignment of multiple protein interaction networks. Proc. Pacific Symp. Biocomputing 13, 303–314. ( 10.1142/9789812776136_0030) [DOI] [PubMed] [Google Scholar]
- Snijders T. A. 2002. Markov chain Monte Carlo estimation of exponential random graph models. J. Soc. Struct. 3, 2–40. [Google Scholar]
- Stark C., Breitkreutz B., Reguly T., Boucher L., Breitkreutz A., Tyers M. 2006. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U., et al. 2005. A human protein–protein interaction network: a resource for annotating the proteome. Cell 122, 957–968. ( 10.1016/j.cell.2005.08.029) [DOI] [PubMed] [Google Scholar]
- Suthram S., Sittler T., Ideker T. 2005. The plasmodium protein network diverges from those of other eukaryotes. Nature 438, 108–112. ( 10.1038/nature04135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanriverdi S., Widmer G. 2006. Differential evolution of repetitive sequences in Cryptosporidium parvum and Cryptosporidium hominis. Infect. Genet. Evol. 6, 113–122. ( 10.1016/j.meegid.2005.02.002) [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium 2000. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne T., Stumpf M. 2007. Generating confidence intervals on biological networks. BMC Bioinformatics 8, 467 ( 10.1186/1471-2105-8-467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P., et al. 2000. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627. ( 10.1038/35001009) [DOI] [PubMed] [Google Scholar]
- Venkatesan K., et al. 2009. An empirical framework for binary interactome mapping. Nat. Meth. 6, 83–90. ( 10.1038/nmeth.1280) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. D., Laskowski R. A., Thornton J. M. 2005. Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol. 15, 275–284. ( 10.1016/j.sbi.2005.04.003) [DOI] [PubMed] [Google Scholar]
- Watts D. J., Strogatz S. H. 1998. Collective dynamics of ‘small-world’ networks. Nature 393, 440–442. ( 10.1038/30918) [DOI] [PubMed] [Google Scholar]
- Webb E. C. 1990. Enzyme nomenclature 1992: recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the nomenclature and classification of enzymes, the University of Michigan. [Google Scholar]
- Wentz-Hunter K., Potashkin J. 1995. The evolutionary conservation of the splicing apparatus between fission yeast and man. Nucleic Acids Symp. 33, 226–228. [PubMed] [Google Scholar]
- Whisstock J. C., Lesk A. M. 2003. Prediction of protein function from protein sequence and structure. Q. Rev. Biophys. 36, 307–340. ( 10.1017/S0033583503003901) [DOI] [PubMed] [Google Scholar]
- Xu P., et al. 2004. The genome of Cryptosporidium hominis. Nature 431, 1107–1112. ( 10.1038/nature02977) [DOI] [PubMed] [Google Scholar]
- Zhang Y., et al. 2006. Phylophenetic properties of metabolic pathway topologies as revealed by global analysis. BMC Bioinformatics 7 ( 10.1186/1471-2105-7-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslavskiy M., Bach F., Vert J. P. 2009. Global alignment of protein–protein interaction networks by graph matching methods. Bioinformatics 25, i259–i267. [DOI] [PMC free article] [PubMed] [Google Scholar]