

Abstract
Background
The clinical use of chromatin-modulating drugs, such as histone deacetylase inhibitors, for the treatment of bone marrow failure and hematopoietic malignancies has increased dramatically over the last few years. Nonetheless, little is currently known concerning their effects on myelopoiesis.
Design and Methods
We utilized an ex vivo differentiation system in which umbilical cord blood-derived CD34+ cells were treated with trichostatin A, sodium butyrate and valproic acid to evaluate the effect of histone deacetylase inhibitor treatment on myeloid lineage development, colony-forming potential, proliferation, and terminal neutrophil differentiation.
Results
Trichostatin A treatment modestly reduced progenitor proliferation, while sodium butyrate and valproic acid resulted in concentration-dependent effects on proliferation and apoptosis. Addition of valproic acid uniquely stimulated CD34+ proliferation. Sodium butyrate treatment inhibited terminal neutrophil differentiation both quantitatively and qualitatively. Addition of 100 μM valproic acid resulted in increased numbers of mature neutrophils with a block in differentiation at increasing concentrations. Sodium butyrate and valproic acid treatment resulted in increased acetylation of histones 3 and 4 while trichostatin A, sodium butyrate and valproic acid had differential effects on the acetylation of non-histone proteins.
Conclusions
Individual histone deacetylase inihibitors had specific effects on cell fate decisions during myeloid development. These data provide novel insights into the effects of histone deacetylase inhibitors on the regulation of normal hematopoiesis, which is of importance when considering utilizing these compounds for the treatment of myeloid malignancies and bone marrow failure syndromes.
Keywords: hematopoiesis, HDAC inhibitors, neutrophil
Introduction
Hematopoiesis is a complex, dynamic and carefully orchestrated series of events involving self-renewal and differentiation of primitive pluripotent stem cells.1 Differentiation of common myeloid progenitors (CMP) generates cells of both the granulocyte/macrophage lineage, leading to the formation of granulocytes, monocytes and macrophages, as well as the megakaryocyte/erythroid lineage, leading to the formation of megakaryocytes, platelets and erythrocytes.2,3 Dysregulation of hematopoietic differentiation can result in the development of a variety of pathological conditions ranging from aplasia of the bone marrow to aberrant differentiation of myeloid progenitors in diseases such as myelodysplastic syndromes and leukemia.4 This process is tightly regulated by signal transduction pathways and transcriptional networks that alter gene expression. In recent years several distinct epigenetic regulatory networks that affect DNA methylation, post-translational histone modifications and alterations in microRNA expression have been identified.5,6 Many malignancies are characterized by such epigenetic modifications, which include alterations in cellular acetylation and methylation profiles.7–9 Abnormal activity of histone acetyltransferases and histone deacetylases (HDAC) leads to transcriptional dysregulation of key genes involved in the control of cell-cycle progression, differentiation and apoptosis.10 For acute promyelocytic leukemia, abnormal recruitment of HDAC by PML-RAR is a key pathogenetic mechanism, and differentiation therapy for this form of leukemia disrupts HDAC-induced repression of retinoic acid target genes.11–13 Moreover, distinct histone modifications, such as the loss of monoacetylated and trimethylated forms of histone 4 (H4) have been found in a large number of tumors.7 These findings have led to a dramatic increase in the number of studies focusing on the use of chromatin-modulating drugs, including HDAC inhibitors, for the treatment of malignancies.14,15 Treatment of cells with HDAC inhibitors can, to a variable extent, induce growth arrest, differentiation and apoptosis both in vitro and in vivo by modulation of both transcription-dependent and transcription-independent mechanisms. cDNA microarray studies have revealed that broad specificity HDAC inhibitors can alter the expression levels of 7–10% of all genes.16–20 However recent studies have suggested that the primary activity of HDAC may also be directed towards non-histone substrates, of which more than 1700 proteins have been identified to date.21,22 These non-histone proteins include transcription factors, such as GATA-1, p53 and STAT-3, but also structural and chaperone proteins such as HSP90.23
Valproic acid (VPA), a short chain fatty acid and potent class I/IIa HDAC inhibitor, has been extensively studied in preclinical and clinical trials involving hematologic malignancies.24,25 Studies in patients with advanced myeloid leukemia and myelodysplastic syndromes have shown that treatment with VPA, as monotherapy or in combination with retinoic acid, results in a reduction of malignant blast cells and hematologic improvement.26–28 In addition, in an ex vivo model of acute leukemia, it has been demonstrated that VPA has the potential to relieve transcriptional repression resulting in cellular differentiation of leukemic blast cells.29 While the clinical use of HDAC inhibitors for the treatment of hematologic malignancies is rapidly increasing, surprisingly little is known about the specific effects of different HDAC inhibitors and their molecular targets in normal hematopoietic cells.
In this study, we investigated the effects of three different HDAC inhibitors on myeloid development.
Design and Methods
Isolation and culture of human CD34+ cells
The procedures used to isolate and culture human CD34+ cells are described in the Online Supplementary Appendix.
Measurement of apoptosis
Cells were harvested at the indicated time points and washed with phosphate-buffered saline (PBS). Samples were subsequently incubated for 15 min with annexin V-fluorescein isothiocyanate (Bender MedSystems, Vienna, Austria) in binding buffer [10 mmol/L HEPES-NaOH (pH 7.4), 150 mmol/L NaCl, 2.5 mmol/L CaCl2] before being washed and resuspended in binding buffer containing 1 μg/mL propidium iodide (Bender MedSystems). Percentages of early apoptotic (annexin V-positive, propidium iodide-negative) and late apoptotic (annexin V- and propidium iodide-positive) cells were determined by FACS analysis (FACS Canto, Becton Dickinson, Alphen a/d Rijn, the Netherlands).
Histochemical staining of hematopoietic cells
May-Grünwald Giemsa staining was used to analyze myeloid differentiation. Cytospins were prepared from 5.0×104 differentiating granulocytes and were fixed in methanol for 3 min. After fixation, cytospins were stained in a 50% eosin methylene blue solution according to May-Grünwald (Sigma Aldrich, Seelze, Germany) for 15 min, rinsed in water for 5 s, and nuclei were counterstained with 10% Giemsa solution (Merck kGaA, Darmstadt, Germany) for 20 min. Neutrophil differentiation occurs through distinct stages from myeloblasts, promyelocytes I, promyelocytes II, myelocytes and metamyelocytes towards neutrophils with banded or segmented nuclei. Mature neutrophils were characterized as cells containing either banded or segmented nuclei. Micrographs were acquired, after staining with May-Grünwald Giemsa solution, with an Axiostar plus microscope (Carl Zeiss, Sliedrecht, the Netherlands) fitted with an 100x/1.3 NA EC Plan Neofluor oil objective using Immersol 518F oil (Carl Zeiss), a Canon Powershot G5 camera (Canon Nederland, Hoofddorp, the Netherlands), and Canon Zoombrowser EX image acquisition software. Photoshop CS3 was used for image processing (Adobe Systems Benelux, Amsterdam, the Netherlands)
Lactoferrin staining
CD34+ cells were cultured in the presence of granulocyte colony-stimulating factor (G-CSF) to induce neutrophil differentiation. After 17 days of differentiation, cells were fixed in 100 μL 0.5% formaldehyde for 15 min at 37°C, after which the cells were permeabilized in 900 μL of ice-cold methanol for 30 min on ice. Cells were subsequently washed with PBS, resuspended with phycoerythrin (PE)-conjugated lactoferrin antibody (Immunotech, Marseille, France) and incubated for 25 min. Cells were then washed again and FACS analysis was performed (FACS Canto, Becton Dickinson).
Western blot analysis
Western blot analysis was performed using standard techniques. In brief, differentiating CD34+ progenitors were lysed in Laemmli buffer [0.12 mol/L Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 0.05 μg/μL bromophenol blue, and 35 mmol/L β-mercaptoethanol], sonicated, and boiled for 5 min. Equal amounts of total lysate were analyzed by 15% sodium dodecylsulfate polyacrylamide gel electrophoresis. Proteins were transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA), incubated with blocking buffer (Tris buffered saline/Tween 20) containing 5% low-fat milk for 1 h at room temperature before incubation with antibodies against either acetyl-histone 4 (lysine 16), acetyl-histone 3 (lysine 9) (Millipore, Billerica, MA, USA), acetyl-lysine (Cell Signaling Technology, Danvers, MA, USA), H3 (Millipore, Billerica, MA, USA) or an antibody against tubulin (Sigma-Aldrich, Zwijndrecht, the Netherlands) overnight at 4°C in a buffer containing Tris buffered saline/Tween 20 with 5% bovine serum albumin (BSA) (Sigma-Aldrich, Zwijndrecht, the Netherlands). Blots were subsequently incubated with peroxidase-conjugated secondary antibodies (Dako, Glostrup, Denmark) for 1 h at room temperature. Chemiluminescence was used as a detection method according to the protocol of the manufacturer (Amersham Pharmacia, Amersham, UK).
Flowcytometric analysis of myeloid progenitor cells
CD34+ cells, isolated from umbilical cord blood, were cultured in the presence of G-CSF to induce neutrophil differentiation as described above. After 3, 7 and 10 days of differentiation, cells were washed and resuspended in PBS/5% FCS (Hyclone) and subsequently incubated for 30 min on ice with a PE-conjugated CD34 antibody (Becton Dickinson). After incubation, cells were again washed and the percentage of CD34+ cells was determined by FACS analysis (FACS Canto, Becton Dickinson).
Hematopoietic progenitor populations were characterized as described by Manz et al.30 and as described in the Online Supplementary Appendix.
Colony-forming unit assay
Five hundred CD34+ cells were plated in Iscove’s modified Dulbecco’s medium (Gibco) supplemented with 35.3% FCS (Hyclone), 44.4% methylcellulose-based medium called Methocult (StemCell Technologies, Vancouver, Canada), 11.1 μmol/L of β-mercaptoethanol, 2.2 u/mL of penicillin, 2.2 μg/mL of streptomycin, and 0.44 mmol/L of glutamine. Colony-forming unit (CFU) assays were performed in the presence of SCF (50 ng/mL), FLT-3 ligand (50 ng/mL), GM-CSF (0.1 nmol/L), IL-3 (0.1 nmol/L), G-CSF (60 ng/mL) and erythropoietin (EPO) (6 IE/mL). Single doses of the HDAC inhibitors trichostatin A (TSA), sodium butyrate (SB) and VPA were added to the medium. CFU-GM (granulocyte/macrophage) and CFU-E (erythrocyte) colonies were scored after 11 days of culture.
Statistics
A one-way ANOVA followed by a Dunnett’s multiple comparison test was performed to compare the differences between the control and HDAC inhibitor-treated cells in all experiments. A P value of 0.05 or less was considered statistically significant.
Results
Histone deacetylase inhibitors have differential effects on CD34+ progenitor proliferation and viability
As previously discussed, little is currently known concerning the effects of HDAC inhibition on the normal hematopoietic compartment. In particular, the effects of HDAC inhibitor treatment on myeloid progenitor cell functionality have not been well characterized. To determine the effect of HDAC inhibition on human CD34+ hematopoietic progenitor functionality, umbilical cord blood-derived CD34+ hematopoietic progenitors were cultured in the presence of G-CSF to induce neutrophil differentiation. Cells were cultured in the absence or presence of increasing concentrations of TSA, SB and VPA, and proliferation and survival were analysed. TSA treatment modestly reduced progenitor proliferation in a concentration-dependent manner (Figure 1A), and this was not accompanied by decreased cell survival (Figure 1B). Moreover these effects were not associated with an up-regulation of p21cip1 expression (Online Supplementary Figure S1). Addition of SB to CD34+ cultures resulted in a significant reduction of progenitor expansion, which was accompanied by increased apoptosis (Figure 1C–D). While 100 μM VPA significantly increased progenitor proliferation, addition of 500 μM VPA resulted in decreased proliferation and a significant increase in the percentage of apoptotic cells (Figure 1E–F). These data demonstrate concentration-dependent, HDAC inhibitor-specific effects on CD34+ progenitor expansion during neutrophil development. To further analyze the effects of HDAC inhibition on CD34+ progenitors specifically, cells were cultured as previously described and both the percentage and absolute number of CD34+ cells were determined. Addition of 25 nM TSA resulted in a small but significant increase in the percentage of CD34+ cells at day 3, which was no longer apparent at day 7 (P=0.063) (Figure 2A), and was not accompanied by an increase in absolute cell numbers (Figure 2B). Addition of SB to cultures resulted in no significant changes in the percentage or absolute numbers of CD34+ progenitors at any time point (Figure 2A, B). Since we previously observed a significant increase in apoptosis after SB treatment of differentiating progenitors (Figure 1D), this suggests that the CD34− cell population is more susceptible to SB-induced apoptosis than the CD34+ one. To investigate this, we analyzed apoptosis at day 7 in CD34+ and CD34− cells treated with SB, and found that, while the basal level of apoptosis is higher in CD34−cells, the fold increase in the percentage of apoptotic cells was similar in both populations (Online Supplementary Figure S2A, B). In contrast, treatment with 200–500 μM VPA resulted in significant increases in the percentage and absolute number of CD34+ progenitors in a concentration-dependent manner (Figure 2A, B). This effect was particularly clear at the later time points suggesting that VPA is capable of inducing expansion of CD34+ progenitors that then retain their undifferentiated phenotype. Taken together, these data demonstrate that TSA, SB and VPA differentially affect CD34+ progenitors, and that VPA, uniquely, can induce CD34+ progenitor proliferation in a concentration-dependent manner.
Figure 1.

HDAC inhibitors differentially modulate myeloid progenitor proliferation and viability. CD34+ cells were cultured in the presence of G-CSF for 17 days to induce neutrophil differentiation. Cells were cultured either in the absence or presence of TSA (5–25 nM) (A–B), SB (100–500 μM) (C–D) or VPA (100–500 μM) (E–F). Proliferation was determined by counting the tryphan blue-negative cell population and data are expressed as fold expansion (A, C, E) (n=4). Apoptosis was determined by annexin-V/propidium iodide staining at days 3, 7 and 10 of differentiation. Data are expressed as percentages of apoptotic cells (B, D, F) (n=3) Error bars represent SEM, (between experiments) *P<0.05, **P<0.01.
Figure 2.

VPA treatment results in increased numbers of CD34+ hematopoietic progenitors. CD34+ cells were cultured in the presence of G-CSF to induce neutrophil differentiation. Cells were cultured in either the absence or presence of TSA (5–25 nM), SB (100–500 μM) or VPA (100–500 μM). At days 3, 7 and 10, FACS using a PE-labeled human-CD34 antibody measured CD34+ expression. Proliferation was determined by counting the tryphan blue-negative cell population. Data are expressed as percentage (A) and absolute numbers (×103) (B) of CD34+ cells. Error bars represent SEM (between experiments, n=3) *P<0.05, **P<0.01.
Sodium butyrate and valproic acid inhibit the transition from common myeloid progenitor to granulocyte/macrophage progenitor
To determine the effect of HDAC inhibition on distinct myeloid progenitor populations within the CD34+ compartment, we analyzed progenitor populations as previously described by Manz et al.30 Based on the expression of CD123 and CD45RA, myeloid progenitors can be divided into CMP (CD123+CD45RA−), GMP (CD123+/CD45RA+) and MEP (CD123−CD45RA−). In addition to the effect on the percentage of CD34+ progenitors early in neutrophil development (Figure 2A), we observed no effects of treatment with TSA on the percentage of CMP (Figure 3A, B). However, at day 3, the percentage of GMP was decreased, and this was accompanied by an increase in the percentage of MEP. This effect was not observed at day 7, suggesting a temporary block of CMP differentiation into GMP (Figure 3C–F). Treatment with SB significantly increased the percentage of CMP at day 7 (Figure 3B), and this was accompanied by a corresponding decrease in the percentage of GMP (Figure 3D). This suggests that SB inhibits the differentiation of CMP towards the GMP lineage. Finally, in addition to its effects on CD34+ progenitor expansion (Figure 2), VPA produced a concentration-dependent increase in the percentage of CMP at day 7 (Figure 3B), accompanied by a significant, concentration-dependent inhibition of the percentage of GMP (Figure 3D). At day 10 we observed no differences in the percentages of CMP and GMP (Online Supplementary Figure S3A) after treatment with any of the three HDAC inhibitors. This suggests that progenitor cells treated with SB or VPA had either differentiated or, alternatively, become apoptotic. In addition we observed a significant increase in the percentage of Lin−CD34− cells in the cultures treated with 500 μM SB (Online Supplementary Figure S3B), suggesting that part of the CD34+ progenitor population differentiated. Unexpectedly, in the absence of EPO and TPO, all HDAC inhibitors increased the percentage of CD123−CD45RA− progenitors at day 3 (Figure 3E). Since we observed no significant differences in the numbers of CD34+ cells at day 3 (Figure 2B), this suggest that HDAC inhibition increases the number of MEP, with this increase being most prominent after addition of 25 nM TSA and 500 μM VPA. This effect was absent at day 7 (Figure 3F), except when 500 μM VPA were added, suggesting a limited effect on immature non-committed progenitors. In addition we observed no effect of HDAC inhibitor treatment on the percentage of MEP within the CD34+ cell population at day 10, indicating that the effect of VPA treatment on CMP differentiation towards MEP was limited (Online Supplementary Figure S3C). Taken together, our data demonstrate that specific HDAC inhibitors modulate differently both the composition and the differentiation mode of the CD34+ progenitor compartment.
Figure 3.

SB and VPA treatment results in inhibition of CMP differentiation. CD34+ cells were cultured in the presence of G-CSF to induce neutrophil differentiation. Cells were cultured in either the absence or presence of TSA (5–25 nM), SB (100–500 μM) or VPA 100–500 μM). At days 3 and 7, a progenitor staining was performed. CD34+CD38+ cells were characterized by FACS based on CD123 and CD45RA expression as CMP (CD123+/CD45RA−), GMP (CD123+/CD45RA−) or MEP (CD123−/CD45RA−). Data are expressed as the percentage of CMP (A–B), percentage of GMP (C–D) and percentage of MEP (E–F) at day 3 (A, C, E) and day 7 (B, D, F). CMP, GMP and MEP represent a percentage of total CD34+ cells. Error bars represent SEM (between experiments, n=3) * P<0.05, ** P<0.01.
Sodium butyrate and valproic acid treatment inhibits granulocyte/macrophage-colony formation
To investigate the effect of HDAC inhibition on colony-forming potential and lineage commitment of CD34+ progenitors, we utilized a semisolid culture system. CD34+ cells were cultured in the presence of a cytokine cocktail including G-CSF and EPO to stimulate production of both granulocyte/macrophage and erythroid colonies. Addition of all HDAC inhibitors resulted in a concentration-dependent reduction in the total number of colonies (Figure 4A). In agreement with the progenitor analysis (Figure 3), this suggests that the differentiation of CD34+ progenitors is inhibited in the presence of HDAC inhibitors. Compared to TSA-and VPA-treated progenitor colonies, SB-treated progenitor colonies appeared less differentiated and smaller (Figure 4B). This is in agreement with our previous data showing inhibition of proliferation in liquid culture (Figure 1). Treatment with all three HDAC inhibitors resulted in an increase in the percentage of GM-colonies (Figure 4C) and concentration-dependent decreases in the percentage of erythroid colonies (Figure 4D), which were primarily responsible for the decrease in the total number of CFU (Figure 4A). Taken together with the data from the progenitor analysis (Figure 3), this suggests that after HDAC inhibition, CMP and GMP still have the potential to differentiate towards the GM-lineage, while further differentiation of MEP towards the erythroid lineage is inhibited.
Figure 4.

HDAC inhibition affects CD34+ progenitor differentiation and lineage commitment. CD34+ cells were cultured in the presence of G-CSF and EPO for 11 days to induce CFU-GM and CFU-E. Cells were cultured in either the absence or presence of TSA (5–25 nM), SB (100–500 μM) or VPA (100–500 μM). Each plate was scored for granulocyte/macrophage colony-forming unit and erythroid burst/colony forming unit growth. Data are expressed as the number of total colonies (A) (n=5), photos were taken to demonstrate GM-colony size (B) and data are expressed as the percentage of granulocyte/macrophage colonies (C) and erythroid colonies (D) (n=4). Error bars represent SEM (between experiments) *P<0.05, **P<0.01.
Histone deacetylase inhibitors have differential effects on terminal neutrophil differentiation
To determine the effect of HDAC inhibition on terminal neutrophil differentiation, CD34+ progenitors were cultured for 17 days in the presence of G-CSF. Neutrophil differentiation was determined based on both cytospin analysis and lactoferrin staining, a protein expressed in secondary granules from the myelocytic stage of neutrophil development. Terminally differentiated neutrophils were characterized as cells containing either banded or segmented nuclei (Figure 5A, see also Design and Methods). Treatment with 5 nM TSA had no effect on either the percentage or the absolute number of mature neutrophils (Figure 5B), but we observed increased segmentation at day 17 (Figure 5A left panel, indicated by arrow). Addition of 25 nM TSA resulted in a modest decrease in the number of mature neutrophils (Figure 5B), which can be explained by reduced progenitor proliferation (Figure 1A). Analysis of lactoferrin expression revealed no effect of TSA treatment (Figure 5C), suggesting that neutrophil differentiation was not inhibited. Treatment with SB impaired neutrophil differentiation in a concentration-dependent manner, resulting in neutrophils with dysplastic features (Figure 5A, indicated by arrows), increased numbers of morphologically apoptotic cells, a relative increase in monocytic cells (data not shown) and a significant decrease in lactoferrin expression in cells treated with 500 μM SB (Figure 5C). Addition of 100 μM SB resulted in a slight decrease in the percentage of mature neutrophils (Figure 5B), which was accompanied by an increased percentage of metamyelocytes (Figure 5D) and no effect on lactoferrin expression (Figure 5C), suggesting that terminal neutrophil differentiation was unaffected. However, mature neutrophils showed delicate dysplastic features such as hypergranulation and ring-shaped nuclei (Figure 5A left panel, indicated by arrows). Taken together, this suggests that treatment with SB impairs neutrophil differentiation both quantitatively and qualitatively. Addition of 100 μM VPA resulted in a modest decrease in the percentage of mature neutrophils (Figure 5B). This effect was accompanied by a slight increase in the absolute number of mature neutrophils, but a significant increase in the absolute number of metamyelocytes (Figure 5D). However, after addition of 500 μM VPA, we observed significant decreases in both the percentage and absolute number of mature neutrophils (Figure 5B), which were accompanied by an increase in immature precursors (data not shown) and a modest decrease in lactoferrin expression (Figure 5C). Since lactoferrin arises from the myelocytic stage of neutrophil differentiation,31 this suggests a differentiation block at the (pro)myelocytic stage. Unlike cells treated with SB, these cells appeared morphologically normal (Figure 5A, left panel).
Figure 5.

Differential effects of HDAC inhibitors on terminal neutrophil differentiation. CD34+ cells were cultured in the presence of G-CSF for 17 days to induce neutrophil differentiation. Cells were cultured in either the absence or presence of TSA (5–25 nM), SB (100–500 μM) or VPA (100–500 μM). Cytospins were made and stained with May-Grünwald Giemsa solution (A). Arrows indicate hypersegmentation (5 nM TSA), hypergranulation and a ring shaped nucleus (100 μM SB). The right panel represents dysplastic neutrophil precursors (500 μM SB). Data are expressed as the percentage and absolute number (×106) of mature neutrophils (banded or segmented nuclei) (n=4) (B), lactoferrin expression (C) (n=2) and percentage and absolute number (×106) of metamyelocytes (D) (n=4). Error bars represent SEM (between experiments) *P<0.05, **P<0.01.
Treatment with sodium butyrate and valproic acid leads to hyperacetylation of histones 3 and 4
To determine whether the observed differential effects of HDAC inhibitors on CD34+ progenitor expansion and neutrophil development were accompanied by differences in histone acetylation, we analyzed protein lysates prepared after 3 and 7 days of neutrophil differentiation. Treatment with TSA resulted in modest changes in total H3-acetylation (Figure 6). In contrast, addition of SB or VPA resulted in a concentration-dependent hyperacetylation of both total H3 and total H4. We further analyzed the effects of HDAC inhibition on histone 3 lysine 9 (H3K9) and histone 4 lysine 16 (H4K16), whose acetylation has been implicated in the regulation of neutrophil development.32,33 While addition of TSA had no substantial effect on H3K9 or H4K16 acetylation, treatment with SB or VPA resulted in concentration-dependent increases in acetylation of H3K9 and H4K16. These data demonstrate that SB and VPA have similar, concentration-dependent effects on histone acetylation in early neutrophil development, while these effects are not observed after addition of TSA. Although SB and VPA have similar effects on histone acetylation, their effects on CD34+ progenitor expansion are distinct (Figures 1 and 2), suggesting that these effects may be due to acetylation of non-histone proteins. To investigate this, we analyzed total protein acetylation of HDAC inhibitor-treated lysates after 7 days of differentiation, finding that HDAC inhibitor treatment does indeed result in differential effects on non-histone protein acetylation (Online Supplementary Figure S4, indicated by arrows)
Figure 6.
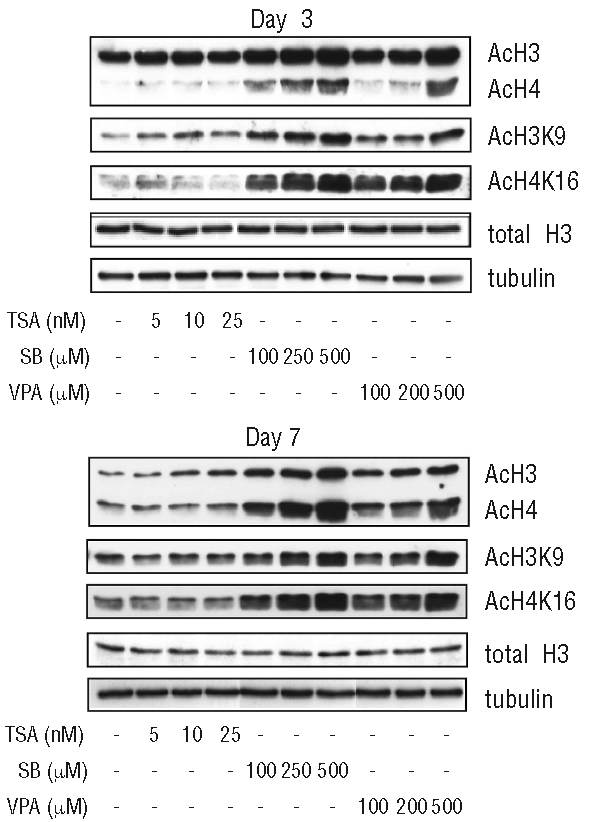
Differential effects of HDAC inhibitor treatment on H3- and H4- acetylation. CD34+ cells were cultured in the presence of G-CSF to induce neutrophil differentiation. Cells were treated overnight with TSA (5–25 nM), SB (100–500 μM) or VPA (100–500 μM) at days 3 and day 7. Lysates were made and western blot analysis was performed with antibodies against acetylated lysines, acetylated H3K9 and acetylated H4K16. As a control for equal loading an antibody against total H3 and an antibody against tubulin was used (n=2)
Discussion
The use of chromatin-modulating drugs, such as HDAC inhibitors, for the treatment of hematologic malignancies has increased dramatically in recent years.15,34 Although these agents have been shown to improve hematologic outcome in patients with myelodysplastic syndromes and acute myeloid leukemia,35–38 little is known about their specific effects and molecular targets. In order to investigate these aspects, we utilized an ex vivo hematopoiesis culture system to study the effects of specific HDAC inhibitors on the proliferation and differentiation of human myeloid progenitors, focusing on neutrophil development. Our data demonstrate that TSA, SB and VPA, despite their large overlap in HDAC specificity, have differential effects on neutrophil progenitor proliferation. Moreover our data show distinct concentration-dependent effects. This was most clearly demonstrated by cells treated with VPA in which 100 μM significantly increased proliferation while 500 μM induced apoptosis of differentiating CD34+ progenitor cells (Figure 1E, F). In agreement with recent studies, we demonstrated that VPA stimulates expansion of CD34+ progenitor cells (Figure 2).39,40 In addition, we showed that HDAC inhibitors have the potential to induce apoptosis in neutrophil progenitors (Figure 1). This is perhaps surprising since it has been previously suggested that untransformed cells are generally more resistant to HDAC inhibitor-induced cell death than transformed cells.41,42 Together, our data suggest specific effects of HDAC inhibition on distinctive progenitor subsets. Supporting this hypothesis, Wada et al. recently demonstrated differential expression of HDAC1-3 in distinct myeloid progenitor populations.43 Importantly, while treatment of leukemic blast cells with HDAC inhibitors generally results in the induction of apoptosis,44–46 Bug et al. demonstrated that numbers of a distinct CD34+CD38− leukemic progenitor population increased after VPA treatment, resulting in stabilization of the leukemic stem cell compartment,47 while Mahlknecht et al. showed that treatment with VPA mobilizes leukemic blast cells from the bone marrow compartment due to reduced VLA-4 expression.48 We further investigated the effect of HDAC inhibition on progenitor differentiation using colony-forming assays and by analyzing progenitor populations during neutrophil development. In agreement with De Felice et al.,40 we demonstrated that VPA and SB have the potential to inhibit myeloid differentiation towards the granulocyte/macrophage lineage, illustrated by an increased percentage of CMP and corresponding decrease in the percentage of GMP (Figure 3). Since we observed no significant effects on the composition of the progenitor compartment after day 7 of neutrophil differentiation (Online Supplementary Figure S3), this suggests that this effect of SB and VPA treatment is limited. In addition to the data on neutrophil progenitor differentiation, we found that specific HDAC inhibitors affect terminal neutrophil differentiation diversely. Interestingly, Skokowa et al. recently reported that nicotinamide, a functionally different, specific class III (sirtuin) HDAC inhibitor, stimulates neutrophil development in patients with congenital neutropenia and in healthy individuals.49 Our data show that, while TSA treatment had no significant effects on neutrophil differentiation, addition of SB impaired differentiation both qualitatively and quantitatively. Treatment with VPA resulted in specific concentration-dependent effects, suggesting that treatment with increasing concentrations finally leads to a differentiation block, in which precursors lose their potential to terminally differentiate (Figure 5). Intriguingly, this inhibitory effect is in contrast to the effect on differentiation of leukemic precursors29,36 and together with other signs of dysmyelopoiesis, has also been described in patients who received long-term treatment with VPA for epilepsy.50,51 In general, the differential effects of HDAC inhibition described in normal hematopoietic progenitors and leukemic precursors may be partially due to distinctive HDAC expression profiles.43,52 The phenotypic effects on proliferation and differentiation observed after HDAC treatment cannot be explained by changes in histone acetylation alone (Figure 6). For example, while both VPA and SB cause almost identical changes in histone acetylation, VPA treatment, unlike SB, results in CD34+ expansion. This suggests that these differential effects may be explained by effects on non-histone protein targets (Online Supplementary Figure S4) of which to date more than 1700 have been defined.21,22 Myeloid development is tightly regulated by key transcription factors including PU.1, GATA-1 and C/EBPα53,54 and in recent years it has become clear that the expression and function of these key transcription factors is regulated by post-translational modifications, including acetylation.5 It is tempting to speculate that the differential effects of HDAC inhibitors observed during CD34+ cultures are the direct effect of the acetylation of key regulators of myeloid differentiation. In summary we describe differential effects of HDAC inhibitors on myeloid progenitors during neutrophil development. Despite the published overlap in HDAC inhibitor specificity, we clearly demonstrate that these inhibitors have specific effects on CD34+ progenitor expansion and neutrophil development. Since the clinical use of HDAC inhibitors is increasing, it is important to define the effects of this family of compounds on the normal hematopoietic compartment. While results of phase I clinical trials with HDAC inhibitors suggest no unfavorable effects on the normal progenitor compartment,24,35,37,38 our data clearly show that both the choice and concentration of HDAC inhibitor can greatly affect the proliferative and differentiation capacity of CD34+ hematopoietic progenitor cells. Furthermore, our results suggest that these effects may be mediated by non-histone targets. Identification of these acetylated proteins may lead to the development of novel, more targeted therapies for the treatment of bone marrow failure and hematologic malignancies.
Footnotes
Funding: this work was supported by a grant from the Dutch Cancer Society (KWF) (UU 2007-3972).
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
MBa: designed and performed experiments, and wrote the paper; CG: performed experiments; MBi, MBu and PJC: designed experiments and wrote the paper.
The authors reported no potential conficts of interest.
References
- 1.Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1(8):661–73. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 2.Metcalf D. Hematopoietic cytokines. Blood. 2008;111(2):485–91. doi: 10.1182/blood-2007-03-079681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weissman IL, Shizuru JA. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood. 2008;112(9):3543–53. doi: 10.1182/blood-2008-08-078220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berliner N. Lessons from congenital neutropenia: 50 years of progress in understanding myelopoiesis. Blood. 2008;111 (12):5427–32. doi: 10.1182/blood-2007-10-077396. [DOI] [PubMed] [Google Scholar]
- 5.Rice KL, Hormaeche I, Licht JD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene. 2007;26(47):6697–714. doi: 10.1038/sj.onc.1210755. [DOI] [PubMed] [Google Scholar]
- 6.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 7.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37(4):391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 8.Fraga MF, Esteller M. Towards the human cancer epigenome: a first draft of histone modifications. Cell Cycle. 2005;4(10):1377–81. doi: 10.4161/cc.4.10.2113. [DOI] [PubMed] [Google Scholar]
- 9.Mahlknecht U, Hoelzer D. Histone acetylation modifiers in the pathogenesis of malignant disease. Mol Med. 2000;6(8):623–44. [PMC free article] [PubMed] [Google Scholar]
- 10.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minucci S, Nervi C, Lo CF, Pelicci PG. Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene. 2001;20(24):3110–5. doi: 10.1038/sj.onc.1204336. [DOI] [PubMed] [Google Scholar]
- 12.Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391 (6669):815–8. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 13.Ferrara FF, Fazi F, Bianchini A, Padula F, Gelmetti V, Minucci S, et al. Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res. 2001;61(1):2–7. [PubMed] [Google Scholar]
- 14.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 15.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 16.Chambers AE, Banerjee S, Chaplin T, Dunne J, Debernardi S, Joel SP, et al. Histone acetylation-mediated regulation of genes in leukaemic cells. Eur J Cancer. 2003;39(8):1165–75. doi: 10.1016/s0959-8049(03)00072-8. [DOI] [PubMed] [Google Scholar]
- 17.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Hideshima T, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci USA. 2004;101(2):540–5. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102(10):3697–702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasakawa Y, Naoe Y, Sogo N, Inoue T, Sasakawa T, Matsuo M, et al. Marker genes to predict sensitivity to FK228, a histone deacetylase inhibitor. Biochem Pharmacol. 2005;69(4):603–16. doi: 10.1016/j.bcp.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Joseph J, Mudduluru G, Antony S, Vashistha S, Ajitkumar P, Somasundaram K. Expression profiling of sodium butyrate (NaB)-treated cells: identification of regulation of genes related to cytokine signaling and cancer metastasis by NaB. Oncogene. 2004;23(37):6304–15. doi: 10.1038/sj.onc.1207852. [DOI] [PubMed] [Google Scholar]
- 21.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23(4):607–18. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 22.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 23.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26(37):5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 24.Kuendgen A, Gattermann N. Valproic acid for the treatment of myeloid malignancies. Cancer. 2007;110(5):943–54. doi: 10.1002/cncr.22891. [DOI] [PubMed] [Google Scholar]
- 25.Bokelmann I, Mahlknecht U. Valproic acid sensitizes chronic lymphocytic leukemia cells to apoptosis and restores the balance between pro- and antiapoptotic proteins. Mol Med. 2008;14(1–2):20–7. doi: 10.2119/2007-00084.Bokelmann. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, et al. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 2006;106(1):112–9. doi: 10.1002/cncr.21552. [DOI] [PubMed] [Google Scholar]
- 27.Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14(3):826–32. doi: 10.1158/1078-0432.CCR-07-0318. [DOI] [PubMed] [Google Scholar]
- 28.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110(7):2302–8. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 29.Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20(24):6969–78. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manz MG, Miyamoto T, Akashi K, Weissman IL. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci USA. 2002;99(18):11872–7. doi: 10.1073/pnas.172384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bainton DF, Miller LJ, Kishimoto TK, Springer TA. Leukocyte adhesion receptors are stored in peroxidase-negative granules of human neutrophils. J Exp Med. 1987;166(6):1641–53. doi: 10.1084/jem.166.6.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121(6):873–85. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 33.Iida S, Watanabe-Fukunaga R, Nagata S, Fukunaga R. Essential role of C/EBPalpha in G-CSF-induced transcriptional activation and chromatin modification of myeloid-specific genes. Genes Cells. 2008;13(4):313–27. doi: 10.1111/j.1365-2443.2008.01173.x. [DOI] [PubMed] [Google Scholar]
- 34.Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol. 2005;23(17):3971–93. doi: 10.1200/JCO.2005.16.600. [DOI] [PubMed] [Google Scholar]
- 35.Kuendgen A, Strupp C, Aivado M, Bernhardt A, Hildebrandt B, Haas R, et al. Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid. Blood. 2004;104(5):1266–9. doi: 10.1182/blood-2003-12-4333. [DOI] [PubMed] [Google Scholar]
- 36.Cimino G, Lo-Coco F, Fenu S, Travaglini L, Finolezzi E, Mancini M, et al. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia. Cancer Res. 2006;66(17):8903–11. doi: 10.1158/0008-5472.CAN-05-2726. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;12(4):981–9. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111(3):1060–6. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 39.Bug G, Gul H, Schwarz K, Pfeifer H, Kampfmann M, Zheng X, et al. Valproic acid stimulates proliferation and self-renewal of hematopoietic stem cells. Cancer Res. 2005;65(7):2537–41. doi: 10.1158/0008-5472.CAN-04-3011. [DOI] [PubMed] [Google Scholar]
- 40.De Felice L, Tatarelli C, Mascolo MG, Gregorj C, Agostini F, Fiorini R, et al. Histone deacetylase inhibitor valproic acid enhances the cytokine-induced expansion of human hematopoietic stem cells. Cancer Res. 2005;65(4):1505–13. doi: 10.1158/0008-5472.CAN-04-3063. [DOI] [PubMed] [Google Scholar]
- 41.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102(3):673–8. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11(1):71–6. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 43.Wada T, Kikuchi J, Nishimura N, Shimizu R, Kitamura T, Furukawa Y. Expression levels of histone deacetylases detremine the cell fate of hematopoietic progenitors. J Biol Chem. 2009;284:30673–83. doi: 10.1074/jbc.M109.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai D, Wang Y, Ottmann OG, Barth PJ, Neubauer A, Burchert A. FLT3-ITD-, but not BCR/ABL-transformed cells require concurrent Akt/mTor blockage to undergo apoptosis after histone deacetylase inhibitor treatment. Blood. 2006;107(5):2094–7. doi: 10.1182/blood-2005-08-3317. [DOI] [PubMed] [Google Scholar]
- 45.Kawagoe R, Kawagoe H, Sano K. Valproic acid induces apoptosis in human leukemia cells by stimulating both caspase-dependent and -independent apoptotic signaling pathways. Leuk Res. 2002;26(5):495–502. doi: 10.1016/s0145-2126(01)00151-5. [DOI] [PubMed] [Google Scholar]
- 46.Tang R, Faussat AM, Majdak P, Perrot JY, Chaoui D, Legrand O, et al. Valproic acid inhibits proliferation and induces apoptosis in acute myeloid leukemia cells expressing P-gp and MRP1. Leukemia. 2004;18(7):1246–51. doi: 10.1038/sj.leu.2403390. [DOI] [PubMed] [Google Scholar]
- 47.Bug G, Schwarz K, Schoch C, Kampfmann M, Henschler R, Hoelzer D, et al. Effect of histone deacetylase inhibitor valproic acid on progenitor cells of acute myeloid leukemia. Haematologica. 2007;92(4):542–5. doi: 10.3324/haematol.10758. [DOI] [PubMed] [Google Scholar]
- 48.Mahlknecht U, Schönlein C. Histone deacetylase inhibitor treatment downregulates VLA-4 adhesion in hematopoietic stem cells and acute myeloid leukemia blast cells. Haematologica. 2008;93(3):443–6. doi: 10.3324/haematol.11796. [DOI] [PubMed] [Google Scholar]
- 49.Skokowa J, Lan D, Thakur BK, Wang F, Gupta K, Cario G, et al. NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med. 2009;15(2):151–8. doi: 10.1038/nm.1913. [DOI] [PubMed] [Google Scholar]
- 50.Bottom KS, Adams DM, Mann KP, Ware RE. Trilineage hematopoietic toxicity associated with valproic acid therapy. J Pediatr Hematol Oncol. 1997;19(1):73–6. doi: 10.1097/00043426-199701000-00011. [DOI] [PubMed] [Google Scholar]
- 51.So CC, Wong KF. Valproate-associated dysmyelopoiesis in elderly patients. Am J Clin Pathol. 2002;118(2):225–8. doi: 10.1309/4TEF-LVGX-WEQ9-R8W8. [DOI] [PubMed] [Google Scholar]
- 52.Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19(10):1751–9. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 53.Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene. 2007;26(47):6816–28. doi: 10.1038/sj.onc.1210764. [DOI] [PubMed] [Google Scholar]
- 54.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132(4):631–44. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]