

Abstract
Background
Clones of glycosylphosphatidylinositol-anchor protein-deficient cells are characteristic in paroxysmal nocturnal hemoglobinuria and are present in about 40–50% of patients with severe aplastic anemia. Flow cytometry has allowed for sensitive and precise measurement of glycosylphosphatidylinositol-anchor protein-deficient red blood cells and neutrophils in severe aplastic anemia.
Design and Methods
We conducted a retrospective analysis of paroxysmal nocturnal hemoglobinuria clones measured by flow cytometry in 207 consecutive severe aplastic anemia patients who received immunosuppressive therapy with a horse anti-thymocyte globulin plus cyclosporine regimen from 2000 to 2008.
Results
The presence of a glycosylphosphatidylinositol-anchor protein-deficient clone was detected in 83 (40%) patients pre-treatment, and the median clone size was 9.7% (interquartile range 3.5–29). In patients without a detectable clone pre-treatment, the appearance of a clone after immunosuppressive therapy was infrequent, and in most with a clone pre-treatment, clone size often decreased after immunosuppressive therapy. However, in 30 patients, an increase in clone size was observed after immunosuppressive therapy. The majority of patients with a paroxysmal nocturnal hemoglobinuria clone detected after immunosuppressive therapy did not have an elevated lactate dehydrogenase, nor did they experience hemolysis or thrombosis, and they did not require specific interventions with anticoagulation and/or eculizumab. Of the 7 patients who did require therapy for clinical paroxysmal nocturnal hemoglobinuria symptoms and signs, all had an elevated lactate dehydrogenase and a clone size greater than 50%. In all, 18 (8.6%) patients had a clone greater than 50% at any given time of sampling.
Conclusions
The presence of a paroxysmal nocturnal hemoglobinuria clone in severe aplastic anemia is associated with low morbidity and mortality, and specific measures to address clinical paroxysmal nocturnal hemoglobinuria are seldom required.
Keywords: paroxysmal nocturnal hemoglobinuria, severe aplastic anemia
Introduction
Prior to the introduction of hematopoietic stem cell transplantation (HSCT) and immunosuppressive therapy (IST), management of severe aplastic anemia (SAA) was limited to androgens and transfusion support,1,2 and the majority of patients died from infectious or bleeding complications 1–2 years after diagnosis.2 HSCT is curative in SAA but graft rejection, infections and graft-versus-host disease (GVHD) have limited the success of this treatment modality. IST is the most common treatment modality employed for SAA worldwide and is used in patients who are not suitable candidates for HSCT due to age, comorbidities, lack of a histocompatible donor, inaccessibility to transplant or personal preference. The long-term outcome with IST is comparable to that of HSCT, but unresponsiveness to immunosuppression, relapse and clonal evolution to myelodysplasia limit the success of IST.3
Clones of paroxysmal nocturnal hemoglobinuria (PNH) cells are characterized by deficiency of glycosylphosphatidylinositol-anchored proteins (GPI-AP) on the cell surface due to an acquired mutation of the PIG-A gene in one or more hematopoietic stem cells.4 For many years, testing for PNH was performed with the Ham test, now substituted by a more sensitive and less cumbersome flow cytometric assay.5,6 The emergence of GPI-anchored protein-deficient cells primarily occurs in the setting of bone marrow failure, and about 40–50% of AA patients have a PNH clone detected at the time of diagnosis.7 The mechanism by which the expansion of PNH cells occurs in AA remains unknown; one hypothesis is that the PNH cells have a proliferative advantage over non-PNH cells by an immune mechanism of selection.7
Since 2000, all SAA treatment protocols at our institution included the measurement of PNH clones by flow cytometry in erythrocytes and granulocytes before and after IST. In order to determine the evolution of PNH clones after IST, we conducted a retrospective analysis in 207 patients who were treated with horse anti-thymocyte globulin (h-ATG) plus cyclosporine (CsA) from 2000 to 2008 at the Clinical Center of the National Institutes of Health.
Design and Methods
Patient details
Patients who fulfilled entry criteria for SAA were enrolled in three different treatment protocols from February 2000 to April 2008 at the Warren Grant Magnuson Clinical Center and the Mark O. Hatfield Clinical Research Center at the National Institutes of Health in Bethesda, Maryland. Consecutive patients treated with h-ATG/CsA in these sequential immunosuppression protocols were analyzed. All adult patients or parents (legal guardians) of children (< 18 years of age) signed informed consent documents approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute. For protocol entry purposes, SAA was defined as severe pancytopenia with at least two of the following peripheral blood count criteria: 1) absolute neutrophil count (ANC) < 0.5×109/L; 2) absolute reticulocyte count (ARC) < 60×109/L; 3) platelet count < 20×109/L;8,9 and 4) bone marrow cellularity of less than 30%. Response was defined as no longer meeting these blood count criteria for SAA and was determined at three and six month landmark visits following ATG.8–10
Bone marrow biopsy and aspiration, for morphology and cytogenetics, were performed before enrolment. Children and young adults (< 40 years of age) had chromosomes assayed after in vitro exposure of peripheral blood lymphocytes to diepoxybutane and in some cases also to mitomycin C to exclude the diagnosis of Fanconi anemia. Testing for PNH by a flow cytometric assay5 was introduced into all treatment protocols from 2000 onwards and was performed pre-treatment and then at three, six, and 12 months after ATG and then yearly thereafter. The presence of a clone was defined as the absence of GPI-anchored surface proteins (CD55 and 59) on greater than 1% of neutrophils or red cells, and the size of the clone was defined by the highest level of red blood cells or neutrophils lacking GPI-AP.
Study design and treatment regimens
Three h-ATG-based regimens previously described were included in the analysis: standard h-ATG/CsA,9 h-ATG/CsA/mycophenolate mofetil10 and h-ATG/CsA/sirolimus.11 H-ATG (ATGAM, Pfizer), CsA, and prednisone (for serum sickness prophylaxis) administration was the same for all 3 regimens. CsA was discontinued after six months in all but 50 patients who received h-ATG/CsA followed by a slow CsA taper in the subsequent 18 months. Mycophenolate mofetil (MMF) was dosed on the first day of ATG at 600 mg/m2 twice daily for children aged 11 years or younger and at 1 g twice daily for children aged 12 or over and adults, for a total of 18 months.10 Sirolimus was initiated on day 1 at 2 mg/d in adults and 1 mg/m2/d in children (< 40 kg) and administered for six months to a trough between 5–15 ng/ml.11
Statistical methods
The continuous variables without a normal distribution were expressed as medians and 25–75% interquartile range (IQR). The two-tailed Kruskall-Wallis test was used to compare the medians between groups and Spearman’s rank correlation coefficient was used to measure correlations. Comparisons were performed using GraphPad Prism version 5.00 (San Diego, CA) and considered significant when P values were less than 0.05. Since the GPI- neutrophil population was greater than the GPI- red blood cells in over 95% of cases when a PNH clone was detected (and considered more representative of the clone size), the GPI- neutrophil measurement was used for the purpose of comparison between the pre- and post-treatment time points.
Results
A total of 207 patients received an h-ATG-based regimen for SAA at our institution from March 2000 to April 2008 on three different treatment protocols: h-ATG/CsA (73 patients), h-ATG/CsA plus mycophenolate mofetil (99 patients),10 and h-ATG/CsA plus sirolimus (35 patients).11 All regimens yielded virtually identical outcomes and were therefore combined for this analysis. The response rate for all patients in the cohort was 62% (129/207) which is in accordance with our previous published experience with h-ATG/CsA. There was no difference in response rate between patients with or without a pre-treatment PNH clone. At six months following IST, 200 patients were evaluable for response; 4 patients died from infectious complications and 3 patients received a second course of IST prior to their six month assessment after initial h-ATG due to worsening pancytopenia and clinical deterioration. Of the 71 non-responders to IST at six months, 25 underwent HSCT and 40 received a second course of IST. A total of 34 (16%) patients died at a median of 24 (IQR, 12–37) months after IST. The median follow-up was 48 (IQR, 24–64) months for surviving patients and testing for PNH was performed a median of 5 (IQR, 3–7) times per patient after IST. Patients’ characteristics are shown in Table 1.
Table 1.
Patients’ characteristics.
Evolution of GPI-AP-deficient cells in patients without a PNH clone pre-treatment (< 1%)
A total of 124 (60%) patients did not have a PNH clone prior to treatment. After IST, a PNH clone was present at least once in 26 of these patients (21%) but persisted (identified more than twice) in only 12 (10%). The median peak of PNH clone size after IST was 3% (IQR, 1.5–8.9) and occurred at a median of six (IQR, 3–24) months after treatment. A large PNH clone (>50%) developed in only one patient, who also had an elevated lactate dehydrogenase (LDH) but no episodes of dark urine, esophageal spasms, abdominal pain or thrombotic events. All other patients with smaller clones did not have clinical evidence of hemolysis or thrombosis. The appearance of a PNH clone after IST was equally observed in responders to treatment (13 patients) and non-responders (13 patients).
Evolution of GPI-AP-deficient cells in patients with a PNH clone pre-treatment (≥1%)
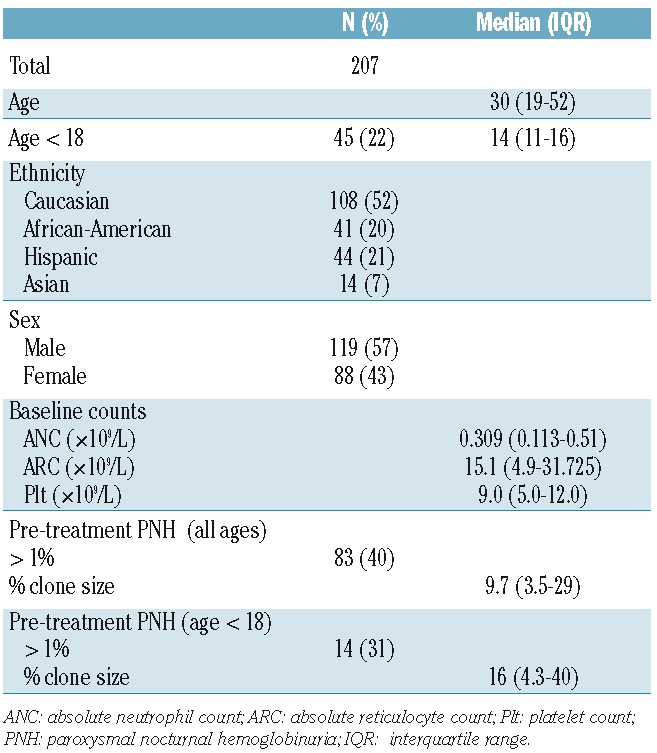
A total of 83 (40%) patients had a PNH clone detected prior to treatment, with a median clone size of 9.7% (IQR, 3.5–29). After IST, a PNH clone was not detected in 10 patients; the median clone size pre-treatment for these patients was 2.3% (IQR, 1.3–6). In the remaining 73 patients who had PNH testing at least once after IST, a gradual decrease in the PNH clone size was noted in the months following IST (P=0.0191; Figure 1, top panel). When only patients who had a PNH clone measured at all the landmark visits within two years of follow-up (44 patients) were analyzed (thus excluding those who had died, underwent HSCT or who had been further treated within two years from analysis), a similar decrease of the PNH clone size with time was observed (P=0.0286; Figure 1, bottom panel). The depiction of the PNH clone size for each patient over time is shown in Online Supplementary Figure S1. This pattern of clone size decline was similar for both responders and non-responders to IST.
Figure 1.
Evolution of GPI- neutrophils after immunosuppressive therapy (IST). In patients with a pre-treatment PNH clone detected prior to IST, a gradual decrease in the clone size occurred in the months after treatment. This decrease was observed when all patients who were tested at least once after IST (73 patients) were analyzed (P=0.0191; top panel) as well as in patients with PNH clone measured in all landmark visits (3, 6, 12, and 24 months; 44 patients) in the 2-year period after IST (P=0.0286; bottom panel). The median bar is shown. *P<0.05; Kruskall-Wallis test.
Of the patients with a pre-existing PNH clone who had at least 2 measurements in the two years after IST, an increase in clone size was observed in 30 patients. The increase was modest and occurred about 3–6 months after IST (Figure 2, top left). Of these 30 patients who had a PNH clone measured at all the landmark visits in the two years of follow-up (three, six, 12 and 24 months), a similar pattern of increase in clone size was observed (Figure 2, top right). In all 30 patients, the highest PNH clone size (peak) after IST was about twice that of the pre-treatment levels and follow-up values after the peak were usually lower (Figure 2, bottom left). A similar pattern of decreasing clone size was noted when only patients who had at least one (post peak 1) and two measurements (post peaks 1 and 2) after the highest level of a clone for each patient were analyzed (Figure 2, bottom right). Of the 41 patients who had a pre-treatment PNH clone less than 10% in size, only one developed a clone that was greater than 50% after IST. Of the patients who had a PNH clone greater than 50% after IST, the median pre-treatment PNH clone size was 39% (IQR, 20.4–50.6).
Figure 2.

Kinetics of PNH clone expansion. In 30 patients an expansion of the pre-treatment PNH clone was observed at least once at any time point after immunosuppressive therapy (IST) (top left). In the 18 patients for whom a measurement was obtained for every landmark time point after IST, a similar pattern of clonal expansion was noted (top right). The highest PNH clone size (peak) after IST on all patients was about twice that of the pre-treatment PNH clone size (bottom left). The clone measurement at subsequent landmark visits following the peak is depicted as post peak 1 and in the subsequent landmark visit as post peak 2. When only patients who had at least one measurement after the peak are analyzed, a similar pattern of decreasing clone size was observed (bottom right, first 3 time points on the left side) and the same occurred when only those who had at least 2 measurements after the peak were analyzed (bottom right, last 4 time points on the right side).
In the majority of cases, the presence of a PNH clone was not associated with an elevated LDH or evidence of hemolysis. Of the 423 time points where a PNH clone greater than 1% was measured, a corresponding elevated LDH greater than 250 U/L (normal range 113–226) was identified in 114 and a LDH greater than 300 U/L was identified in 77 patients. A stronger positive correlation was observed between the GPI- red blood cells and LDH compared to the GPI- neutrophils (Figure 3). Patients with a PNH clone between 25–50% and a LDH greater than 250 U/L did not experience bouts of hemolysis, abdominal pain, esophageal spasm, or thrombosis. Of the 14 patients with a PNH clone greater than 50% and an elevated LDH, 4 have experienced significant hemolysis and 3 have had thrombotic complications (2 in the central nervous system and one in the upper extremities). Of these 7 symptomatic patients, 2 are receiving eculizumab and 6 are receiving oral anticoagulation (one patient was on both eculizumab and oral anticoagulation). Of the remaining 7 patients, a large clone (>50%) was detected only transiently, no significant clinical complications from PNH were observed and/or thrombocytopenia precluded anticoagulation therapy. In all 18 (8.6%) patients had a clone greater than 50% at any given time of sampling.
Figure 3.
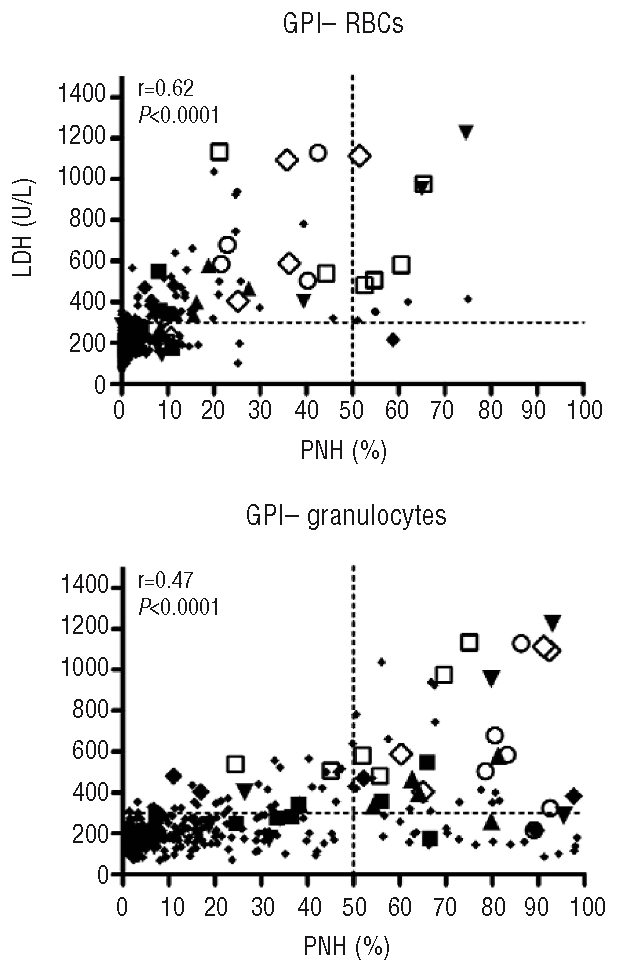
Correlation between GPI- cells and lactate dehydrogenase (LDH). A positive correlation was noted for both GPI- RBCs and GPI-neutrophils to LDH. Patients with clinical evidence of hemolysis and/or thrombosis all developed a GPI- neutrophil population greater than 50%. Larger symbols depict different patients who experienced clinically significant hemolysis and/or thrombosis. Each symbol depicts the different clone measurements over time for a patient. Patients depicted by symbols had clones greater than 50% at diagnosis or after IST and experienced symptoms related to PNH. The horizontal dotted line is drawn at y=300 and the vertical line at x=50.
Pediatric patients
Of the 207 patients in the cohort, 47 were less than 18 years of age, of which 14 had a PNH clone greater than 1% at baseline. The median size of the PNH clone in these 14 patients pre- and post-IST was 16% (IQR, 4.3–40) pre-treatment; 12% (IQR, 2.2–42.2) at three months; 9.3% (IQR, 0–40) at six months; and 5.5% (IQR, 1.4–34.3) at 12 months. Two patients with a baseline PNH clone underwent HSCT for refractory disease or relapse. Thus, the trend of PNH clone size after IST in pediatric patients was similar to that observed in patients of all ages. In none of the pediatric patients was there clinically significant hemolysis or thrombotic complications.
Discussion
By using a flow cytometric assay since 2000, our data allow quantitation of PNH clone size and evolution in SAA patients who received standard h-ATG/CsA. Our analysis shows that the development of a PNH clone in patients who do not have a pre-existent clone prior to IST is uncommon, and, in those with a pre-existing clone, a decrease in clone size is likely to occur in the years following IST. In only about 25% of those with a pre-treatment clone did the clone expand and in only 15% was a large clone, greater than 50%, detected after IST. Although an elevated LDH could accompany an expanded clone, hemolysis was mild and subclinical in most patients. Symptoms occurred primarily when the clone size was large (>50%) and very few patients in our cohort (<5%) required specific interventions for PNH. Thus, in our experience, clinical PNH becomes problematic in only a minority of SAA patients who receive IST, and specific interventions are seldom required. Thrombotic complications in our cohort were particularly infrequent.
Until recently, the management of patients with symptomatic PNH consisted of red cell transfusion support and anticoagulation in selected cases.12 A short course of corticosteroids has been suggested to be of benefit13,14 but is not routine practice. Eculizumab, a monoclonal antibody directed against the complement protein C5, is very effective in blocking intravascular hemolysis in patients with PNH15 and, in a retrospective analysis, to reduce the incidence of thromboembolic events in patients with a prior history of clotting.16 However, eculizumab is expensive, with an estimated yearly cost of $400,000; in addition, as it has no effect on the underlying stem cell abnormality in PNH, and indefinite therapy may be required in most patients.17
That PNH clones can be present in SAA patients is well known but their significance and consequence for patients are not fully understood. For many years GPI-AP-deficient clones were detected in red blood cells only by testing these cells ability to tolerate an acid environment (Ham test). In the last decade, the detection of CD55-/CD59-cells by flow cytometry greatly increased the sensitivity of detecting a PNH clone in both red blood cells and leuko-cytes, minimizing the effects of red cell transfusions on estimating clone size. Since the introduction of flow cytometry, quantitative and kinetic differences of GPI-AP-deficient clones have been reported between patients with PNH with and without marrow failure, in which patients with less pronounced cytopenias appear more likely to have a larger PNH clone and clinical hemolysis.18 The presence of a clone has been reported to be predictive of response to IST in SAA,19 but others have not observed this finding.20 One of the problems with comparison between studies is the lack of consensus for defining PNH clonal expansion, with thresholds ranging from 0.003% to 1%.
In clinical practice, patients with SAA are treated with IST regardless of the presence of a PNH clone as resumption of hematopoiesis should be the primary goal once eculizumab does not have a role in improving marrow function and averting the clinical consequences of cytopenias in these patients. Our experience indicates that a PNH clone post-IST in the majority of patients will remain subclinical, and in only a few patients is specific intervention required with anticoagulation and/or eculizumab. This information may help relieve the considerable anxiety patients often express on the introduction of a second diagnosis of a rare disease with potentially dire consequences in its classic form. Patients who are likely to benefit from eculizumab are those who are symptomatic or at risk for thrombosis due to a large clone (>50%). It is our practice to offer oral anticoagulation in those with a clone greater than 50% (if the platelet count is over 50,000/uL) and eculizumab in those with significant symptoms associated with PNH and/or if there is a history of thrombosis at unusual sites. Since further prospective studies are unlikely to be undertaken to define the role of eculizumab for PNH clones in SAA patients, our retrospective analysis of the evolution and clinical consequence of a PNH clone in this setting should help physicians determine those at a high risk of complications and the need for specific therapy.
Footnotes
Funding: this research was supported by the Intramural Research Program of the NIH, National Heart, Lung and Blood Institute.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
PS participated in the primary conception, data collection and analysis, and drafted the manuscript; MM participated in data collection, analysis and manuscript preparation; ON participated in the data collection; NSY participated in the primary conception, data analysis, interpretation, interim discussions, and writing of the manuscript.
The authors have no conflict of interest to declare.
References
- 1.Sanchez-Medal L, Gomez-Leal A, Duarte L, Guadalupe Rico M. Anabolic androgenic steroids in the treatment of acquired aplastic anemia. Blood. 1969;34(3):283–300. [PubMed] [Google Scholar]
- 2.Williams DM, Lynch RE, Cartwright GE. Drug-induced aplastic anemia. Semin Hematol. 1973;10(3):195–223. [PubMed] [Google Scholar]
- 3.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509–19. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyata T, Takeda J, Iida Y, Yamada N, Inoue N, Takahashi M, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. 1993;259(5099):1318–20. doi: 10.1126/science.7680492. [DOI] [PubMed] [Google Scholar]
- 5.Dunn DE, Tannawattanacharoen P, Boccuni P, Nagakura S, Green SW, Kirby MR, et al. Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes. Annals of Internal Medicine. 1999;131(6):401–8. doi: 10.7326/0003-4819-131-6-199909210-00002. [DOI] [PubMed] [Google Scholar]
- 6.Piedras J, López-Karpovitch X. Flow cytometric analysis of glycosylphosphatidylinositol-anchored proteins to assess paroxysmal nocturnal hemoglobinuria clone size. Cytometry. 2000;42(4):234–8. doi: 10.1002/1097-0320(20000815)42:4<234::aid-cyto3>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 7.Young NS. Paroxysmal nocturnal hemoglobinuria and myelodysplastic syndromes: clonal expansion of PIG-A-mutant hematopoietic cells in bone marrow failure. Haematologica. 2009;94(1):3–7. doi: 10.3324/haematol.2008.001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289(9):1130–5. doi: 10.1001/jama.289.9.1130. [DOI] [PubMed] [Google Scholar]
- 9.Rosenfeld SJ, Kimball J, Vining D, Young NS. Intensive immunosuppression with antithymocyte globulin and cyclosporine as treatment for severe acquired aplastic anemia. Blood. 1995;85(11):3058–65. [PubMed] [Google Scholar]
- 10.Scheinberg P, Nunez O, Wu C, Young NS. Treatment of severe aplastic anaemia with combined immunosuppression: anti-thymocyte globulin, ciclosporin and mycophenolate mofetil. Br J Haematol. 2006;133(6):606–11. doi: 10.1111/j.1365-2141.2006.06085.x. [DOI] [PubMed] [Google Scholar]
- 11.Scheinberg P, Wu CO, Nunez O, Boss C, Sloand EM, Young NS. Treatment of severe aplastic anemia with a combination of horse antithymocyte globulin and cyclosporine, with or without sirolimus: a prospective randomized study. Haematologica. 2009;94(3):348–54. doi: 10.3324/haematol.13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2003;102(10):3587–91. doi: 10.1182/blood-2003-01-0009. [DOI] [PubMed] [Google Scholar]
- 13.Bourantas K. High-dose recombinant human erythropoietin and low-dose corticosteroids for treatment of anemia in paroxysmal nocturnal hemoglobinuria. Acta Haematol. 1994;91(2):62–5. doi: 10.1159/000204254. [DOI] [PubMed] [Google Scholar]
- 14.Issaragrisil S, Piankijagum A, Tang-naitrisorana Y. Corticosteroids therapy in paroxysmal nocturnal hemoglobinuria. Am J Hematol. 1987;25(1):77–83. doi: 10.1002/ajh.2830250108. [DOI] [PubMed] [Google Scholar]
- 15.Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–43. doi: 10.1056/NEJMoa061648. [DOI] [PubMed] [Google Scholar]
- 16.Hillmen P, Muus P, Duhrsen U, Risitano AM, Schubert J, Luzzatto L, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–8. doi: 10.1182/blood-2007-06-095646. [DOI] [PubMed] [Google Scholar]
- 17.Parker C. Eculizumab for paroxysmal nocturnal haemoglobinuria. The Lancet. 2009;373(9665):759–67. doi: 10.1016/S0140-6736(09)60001-5. [DOI] [PubMed] [Google Scholar]
- 18.Maciejewski JP, Rivera C, Kook H, Dunn D, Young NS. Relationship between bone marrow failure syndromes and the presence of glycophosphatidyl inositol-anchored protein-deficient clones. Br J Haematol. 2001;115(4):1015–22. doi: 10.1046/j.1365-2141.2001.03191.x. [DOI] [PubMed] [Google Scholar]
- 19.Sugimori C, Chuhjo T, Feng X, Yamazaki H, Takami A, Teramura M, et al. Minor population of CD55-CD59- blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107(4):1308–14. doi: 10.1182/blood-2005-06-2485. [DOI] [PubMed] [Google Scholar]
- 20.Scheinberg P, Wu CO, Nunez O, Young NS. Predicting response to immunosuppressive therapy and survival in severe aplastic anaemia. Br J Haematol. 2009;144(2):206–16. doi: 10.1111/j.1365-2141.2008.07450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]