

Abstract
Background
Chronic lymphocytic leukemia is a neoplastic disorder that arises largely as a result of defective apoptosis leading to chemoresistance. Stromal cell-derived factor-1 and its receptor, CXCR4, have been shown to play an important role in chronic lymphocytic leukemia cell trafficking and survival.
Design and Methods
Since histone acetylation is involved in the modulation of gene expression, we evaluated the effects of suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, on chronic lymphocytic leukemia cells and in particular on cell survival, CXCR4 expression, migration, and drug sensitization.
Results
Here, we showed that treatment with suberoylanilide hydroxamic acid (20 μM) for 48 hours induced a decrease in chronic lymphocytic leukemia cell viability via apoptosis (n=20, P=0.0032). Using specific caspase inhibitors, we demonstrated the participation of caspases-3, -6 and -8, suggesting an activation of the extrinsic pathway. Additionally, suberoylanilide hydroxamic acid significantly decreased CXCR4 mRNA (n=10, P=0.0010) and protein expression (n=40, P<0.0001). As a result, chronic lymphocytic leukemia cell migration in response to stromal cell-derived factor-1 (n=23, P<0.0001) or through bone marrow stromal cells was dramatically impaired. Consequently, suberoylanilide hydroxamic acid reduced the protective effect of the microenvironment and thus sensitized chronic lymphocytic leukemia cells to chemotherapy such as fludarabine.
Conclusions
In conclusion, suberoylanilide hydroxamic acid induces apoptosis in chronic lymphocytic leukemia cells via the extrinsic pathway and down-regulates CXCR4 expression leading to decreased cell migration. Suberoylanilide hydroxamic acid in combination with other drugs represents a promising therapeutic approach to inhibiting migration, chronic lymphocytic leukemia cell survival and potentially overcoming drug resistance.
Keywords: suberoylanilide hydroxamic acid, chronic lymphocytic leukemia, CXCR4, migration
Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the western world and is characterized by a gradual accumulation of leukemic cells. It is now well accepted that this accumulation could largely be explained by a defect in apoptosis.1 Different factors contribute to the resistance of CLL cells to apoptosis, such as intrinsic defects in apoptotic pathways and interactions with non-leukemic cells in the microenvironment of lymph nodes, bone marrow, and other tissues.2 However, recent studies suggested that CLL is not exclusively a disease of accumulation, but also one of proliferation.3
CLL has a heterogeneous clinical course which can be predicted by many classical or molecular prognostic factors.4 Of these the absence of hypersomatic mutations of the immunoglobulin heavy chain region (IgVH),5 zeta-associated protein 70 (ZAP70)6 and CD387 expression have been abundantly described and are associated with poor outcome. Moreover, despite the introduction of novel therapeutic agents, drug resistance remains a great obstacle to the treatment of CLL and this leukemia is still considered an incurable disease justifying the need to develop alternate therapeutic approaches.
The chemokine (C-X-C motif) receptor 4 (CXCR4) is a G-protein 7-transmembrane domain receptor that is specific for the chemokine stromal cell-derived factor-1 (SDF-1), also known as CXCL12 (C-X-C motif ligand 12).8 SDF-1 plays an important role in B-lymphocyte development and trafficking.9 B-cell precursors deficient in the expression of the SDF-1 receptor, CXCR4, are not retained within the bone marrow microenvironment, thus demonstrating the importance of this pathway.10 SDF-1 is constitutively produced by bone marrow stromal cells and acts as a chemoattractant supporting the homing of stem cells and contributing to the tropism of malignant cells for the bone marrow.11
CLL cells depend upon the supportive interaction within their microenvironment.12 Several studies have demonstrated that CLL cells can interact with their microenvironment through the CXCR4/SDF-1 axis.11,13 Furthermore CLL cells express a high level of CXCR4 surface receptors.11 Lagneaux et al. showed that stromal cells could rescue CLL cells from apoptosis after they had migrated beneath a bone marrow stromal microenvironment,12 while Burger et al. highlighted the protective function of nurse-like cells.14 The CXCR4/SDF-1 axis has, therefore, been considered as a potential target for new therapeutic strategies.
Histone deacetylases (HDAC) play an important role in transcriptional regulation and the pathogenesis of cancer. HDAC inhibitors are members of a new class of agents that are capable of regulating gene expression by changing chromatin structure. By modifying the epigenetic code, this novel class of therapeutic agents may suppress aberrant gene expression or activate gene transcription to inhibit tumor growth.15 Suberoylanilide hydroxamic acid (SAHA), also known as vorinostat (Zolinza, Merck, Whitehouse Station, NJ, USA), is a small molecule inhibitor of the HDAC class I and II enzymes16 which can induce cell cycle arrest and apoptosis via gene expression modulation.17 This drug has demonstrated activity against hematologic malignancies when used alone18,19 or in combination with other chemotherapy.20 Vorinostat has already been tested in clinical trials, has a known safety profile, and is effective in the treatment of cutaneous T-cell lymphoma.21,22 Since the chemokine receptor CXCR4 plays a crucial role in the survival and migration of CLL cells, we evaluated the effects of SAHA on CLL cells and, in particular, on cell survival and migration.
Design and Methods
Patients
This study was approved by the Bordet Institute Ethics Committee and was based on peripheral blood samples obtained with informed consent from 40 CLL patients who presented with a typical CD19+CD5+CD23+ phenotype. Patients were either untreated or had received no treatment for at least 6 months before the study. A summary of the patients’ characteristics is presented in Online Supplementary Table S1. Cytoplasmic ZAP70 expression was determined by three-color flow cytometry (CD3/ZAP70/CD19) and confirmed by quantitative real-time polymerase chain reaction (PCR) analysis.23 CD38 expression, standard karyotype analysis and interphase fluorescence in situ hybridization screening for most common aberrations and IgVH gene mutational analysis were performed as previously described.24
Cell culture, suberoylanilide hydroxamic acid treatment, and establishment of the bone marrow stromal layer
SAHA was obtained from Alexis Biochemicals (Lausanne, Switzerland). Mononuclear cells were isolated from peripheral blood using density gradient centrifugation (Linfosep, Biomedics, Spain) and stromal layers of mesenchymal stromal cells were prepared as previously described.25 SAHA was added at the beginning of the culture period. Culture conditions are detailed in the Online Supplementary Appendix.
Viability assay, and apoptotic assays
Cell viability was determined by the trypan blue dye exclusion assay and all results were confirmed on a higher number of cells using 7AAD/CD19 staining counted by flow cytometry as previously described.26 Phosphatidylserine exposure was measured by the binding of annexin V and allophycocyanin-conjugated CD19 monoclonal antibody (Miltenyi Biotec) using the protocol outlined in the ApoTarget Kit (Biosource International, Camarillo, CA, USA). DNA fragmentation was evaluated by standard propidium iodide staining as previously described.27
Caspase inhibition, caspase-3 activity assessment and western blot
To evaluate the involvement of caspases in SAHA-induced apoptosis, patients’ cells (with a purity >90% of CD19/CD5) were preincubated with the pan-caspase inhibitor ZVAD-FMK (N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone) (20 μM) (Eurobiochem, Bierges, Belgium) for 1 h prior to SAHA exposure. To examine which caspases are critical for inducing apoptosis, cells were preincubated with selective inhibitors of caspase-3, -6, -8 and -9 (Z-DEVD-FMK, Z-VEID-FMK, Z-IETD-FMK, Z-LEHD-FMK, respectively; Biosource, Nivelles, Belgium) 1 h prior to SAHA treatment. The enzymatic activity of caspases-3, -6, -8 and -9 was also determined using the ApoTarget Apoptosis colorimetric detection kit (Biosource). Caspase-3 activation was indirectly evaluated by PARP cleavage using a fluorescein isothiocyanate (FITC)-conjugated rabbit anti-PARP cleavage site antibody (Biosource). Western blot analysis was performed as previously described:24 control or SAHA-treated (20 μM) cultured CLL cells were harvested after 48 h of treatment and lysed in RIPA buffer. Anti-actin (C2 – sc-8432), anti-PARP-1 (F-2 – sc-8007) and anti-FLIPL/S (G-11 – sc-5276) were obtained from Santa Cruz (Heidelberg, Germany).
Measurement of CXCR4 mRNA expression by semi-quantitative reverse transcriptase polymerase chain reaction analysis
CLL cell total RNA was extracted with classical TriPure isolation reagent (Roche Applied Science, Indianapolis, IN, USA ) in a single step extraction method.28 Three CLL samples were purified using a CD19+ magnetic-bead system (MidiMACS, Miltenyi Biotec, Bergish Gladbash, Germany) and their RNA was extracted to verify the influence of CD19− cells. Mean B-cell purity, as measured by flow cytometry, was greater than 99%. cDNA synthesis was performed by a standard reverse transcription reaction and subjected to PCR analysis for quantification of CXCR4 (610 bp) and β-actin (610 bp) mRNA. Primer sequences and additional details are provided in the Online Supplementary Appendix.
Quantification of CXCR4 expression on leukemic cells
Untreated or SAHA-treated CLL cells were incubated for 30 min with FITC-conjugated anti-CD19 monoclonal antibody (Immunotech, Marseille, France) and phycoerythrin-conjugated anti-CXCR4 (BD Biosciences Pharmigen, San Diego, CA, USA). The viable cells were gated based on their forward and side scatter features in order to dissociate apoptotic and viable cells as described previously.29 CXCR4 expression was evaluated in comparison with that of cells incubated with an isotype control antibody and measured on the CD19+ population. CLL samples (n=4) were also purified using a CD19+ magnetic-bead system (MidiMACS, Miltenyi Biotec) in order to assess the influence of other cell types on the expression of these antigens.
Chemotaxis assays and in vitro migration assay of chronic lymphocytic leukemia cells into stromal layers
Chemotaxis assays in response to SDF-1 were performed as previously described.30 Cells were pre-incubated alone or with SAHA for 24 h before performing the migration assay. Additional details are provided in the Online Supplementary Appendix. For migration into a stromal layer, 5×106 untreated or SAHA-treated cells from six different patients were suspended in 1 mL of RPMI and added to stromal layers that were established from normal subjects. After incubation for 3 h, cells in suspension in the medium were removed. The stromal layer containing cells that had migrated was carefully washed twice with phosphate-buffered saline. Adherent CLL cells were harvested by Triple Select treatment (Gibco, Invitrogen, Merelbeke, Belgium) and labeled with allophycocyanin-conjugated CD19 monoclonal antibody (Miltenyi Biotec) and 100 μL of cell suspension were counted with the MACSQUANT® flow cytometer using the absolute volumetric cell counting function. The absolute number of CD19+ cells was then determined. In other experiments, 2×106 CLL cells/mL were plated with or without SAHA (20 μM) in the presence or absence of a stromal microenvironment. Viability and apoptosis were evaluated after 48 h.
Drug treatment and viability assay in a chronic lymphocytic leukemia/mesenchymal stromal cell co-culture model
Fludarabine (FLUDARA®; Schering AG, Berlin, Germany) was dissolved in stock solution with water at 1 mM. Stock solutions were stored at − 20°C. CLL cells (2×106/mL) from six patients were pre-incubated for 4 h with or without a low dose of SAHA (1 μM), and were then plated on a stromal layer with or without fludarabine (10 μM). Viability and apoptosis were evaluated after 48 h. To evaluate the synergism between SAHA and fludarabine, a Cooperative Index (CI) was calculated as previously described.31 This index is the sum of specific apoptosis obtained by treatment with a single agent divided by the specific apoptosis obtained by combined treatment. CI values less than 1, equal to 1 and more than 1 mean that the effect is synergistic, additive or less than additive, respectively.
Statistical analysis
The Wilcoxon signed ranks test was used to analyze the statistical significance of experimental results. All tests were two-sided. An effect was considered to be statistically significant when the P value was less than 0.05, and all analyses were performed with GraphPad Prism 5.0 software. For normalized data, the P values stated in this paper were obtained on primary data (before normalization) since a P value could not be calculated on data with the same rank.
Results
Suberoylanilide hydroxamic acid reduces viability and induces apoptosis in chronic lymphocytic leukemia cells after 48 hours of treatment
Mononuclear cells from ten patients were cultured in the presence or absence of different concentrations of SAHA (1, 10 and 20 μM). After a 24-h incubation, cell viability (assessed by the trypan blue dye exclusion assay and confirmed by flow cytometry counting after 7AAD/CD19 staining) was unchanged in all patients evaluated (97±14% of viability after treatment with SAHA 20 μM, P=ns). However, viability was reduced after 48 h of exposure to SAHA 10 and 20 μM (P<0.05) (Figure 1A). The level of apoptosis, assessed by annexin binding, increased after 48 h of exposure to SAHA and was significantly higher in SAHA-treated cells: 45.14±5.47% (at 10 μM, P<0.001) and 51.63±4.08% (20 μM, P<0.001) of apoptotic cells versus 29.51±4.35% for untreated cells) (Figure 1B). Furthermore, we corroborated these results measuring apoptosis by propidium iodide staining to analyze DNA fragmentation, and by morphological analysis. A representative experiment is presented in Online Supplementary Figure S1.
Figure 1.
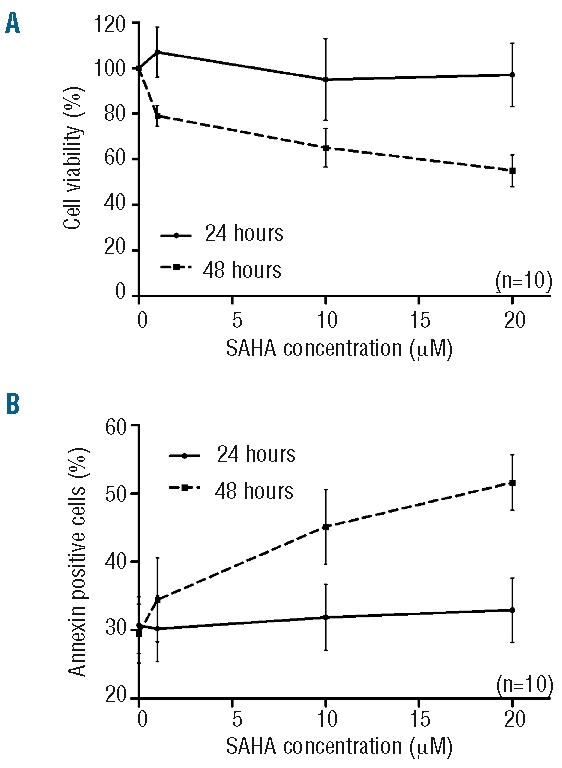
Viability and apoptotic effect of SAHA on CLL cells. (A) Mononuclear cells were incubated for 24 and 48 h with SAHA (1, 10 and 20 μM). Cell viability rates stated in the figure were normalized to untreated cells. (B) Phosphatidylserine exposure after SAHA treatment. Rates stated in the figure include early and late apoptosis. Each bar represents the mean ± SEM of 10 experiments.
Suberoylanilide hydroxamic acid induces caspase-dependent apoptosis
Apoptosis can occur through both caspase-dependent and -independent pathways. To investigate the requirement of caspases for SAHA cytotoxicity, CLL cells were incubated in the presence or absence of SAHA and the pan-caspase inhibitor Z-VAD-FMK. Apoptosis was measured using two different methods: annexin-V assay and propidium iodide staining. As shown in Figure 2A, pre-treatment of cells with Z-VAD before SAHA treatment had no effect on phosphatidylserine externalization. In contrast, addition of Z-VAD inhibited SAHA-induced DNA degradation. These data indicate that phosphatidylserine externalization is independent of caspases, although caspases are critical for the induction DNA fragmentation. We next examined whether the processing of DNA fragmentation could be inhibited by selective caspase inhibitors. Treatment with DEVD, VEID and IETD decreased the percentage of cells with degraded DNA demonstrating that caspases-3, -6 and -8 are critical for inducing cell apoptosis (Figure 2B). In addition, the inhibition of caspase-9, the initiator caspase in the intrinsic/mitochondrial pathway, did not influence cell apoptosis. Thus, the extrinsic pathway appears to be activated during SAHA-induced apoptosis. The enzymatic activities of caspases-3, -6 and -8 were also confirmed by a colorimetric reaction assay (data not shown). Flow cytometric analysis of caspase-3 activity demonstrated that SAHA activates caspase-3. In addition, cleavage of the caspase-3 substrate, PARP, was detected in all treated samples. This cleavage was confirmed by western blot analysis (Figure 2C) and flow cytometry. A representative CLL sample showing increased caspase-3 activity and PARP cleavage detected by flow cytometry is provided in Online Supplementary Figure S2. Finally, it was demonstrated that FLIPL decreases after SAHA treatment (Figure 2C).
Figure 2.
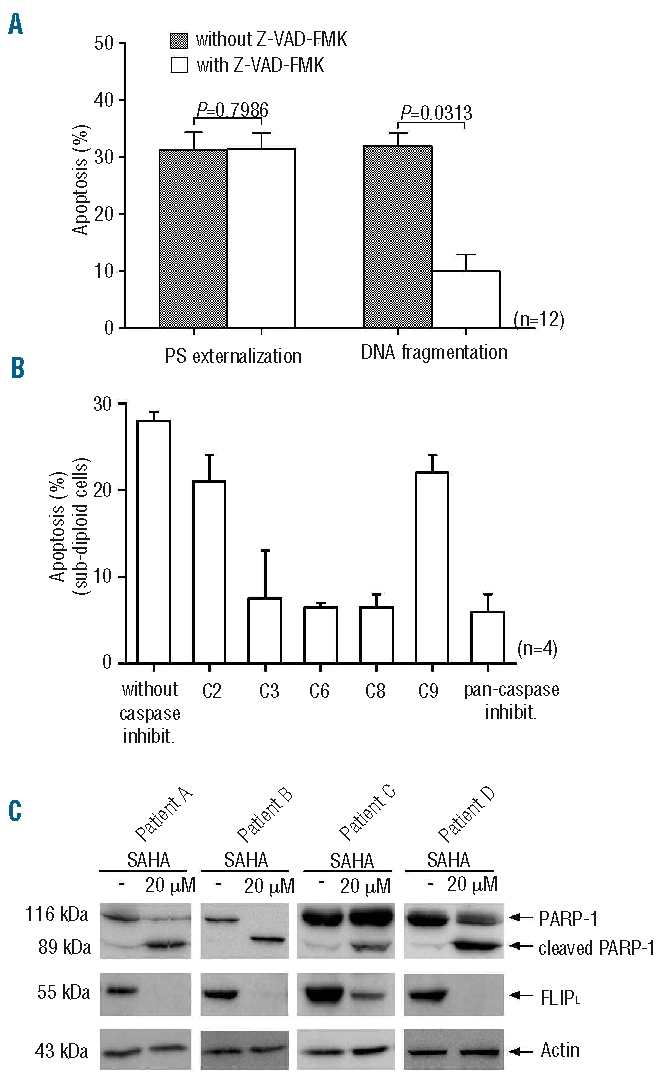
The effect of SAHA on caspase activity, phosphatidylserine (PS) externalization, DNA fragmentation and PARP and FLIPL. (A) CLL cells were preincubated for 1 h in the absence or presence of Z-VAD-FMK and then treated with 20 μM SAHA. (B) One hour prior to exposure to SAHA (20 μM), cells were preincubated with selective inhibitors of caspase-3 (C3), -6 (C6), -8 (C8) and -9 (C9), and the pan-caspase inhibitor as a control. (C) Protein extract of CLL cells from four patients plated with or without 20 μM SAHA for 48 h were analyzed by western blotting for PAPRP-1, FLIPL and actin expression.
Suberoylanilide hydroxamic acid down-regulates CXCR4 mRNA and protein
CXCR4 mRNA expression was examined by semi-quantitative reverse transcriptase PCR analysis. Since tumor necrosis factor-α (TNF-α) has been shown to induce down-regulation of CXCR4 mRNA in various cell types, TNF-α was used as a positive control. RNA was isolated from cells incubated in the presence or absence of SAHA and TNF-α. Amplified products were electrophoresed and analyzed by densitometry using the β-actin band as an internal control. As shown in Figure 3A, SAHA at 10 and 20 μM induced significant changes in CXCR4 mRNA [32.29±12.63% (P<0.01) and 53.84±6.11% (P<0.01) inhibition of control ratio, respectively], suggesting an effect at the transcriptional level. The results for one representative sample are shown in Figure 3B. The results were similar for unpurified and purified CLL samples indicating that CD19− cells did not influence CXCR4 mRNA quantification (data not shown). To investigate the effect of SAHA on CXCR4 protein level, CLL cells were cultured in the presence or absence of SAHA at various concentrations for 24 h, and subsequently washed and then analyzed by flow cytometry. We observed that SAHA, even at a low concentration, significantly reduced CXCR4 levels in all patients tested (n=40): inhibition of control ratios were 32.53±4.71%, 38.98±3.49% and 42.39±3.31% (P<0.0001) after treatment with SAHA 1 μM, 10 μM and 20 μM, respectively (Figure 3C). The results for one representative sample are shown in Figure 3D. Similar results were observed on purified CLL samples (n=4) indicating that other cell types did not alter the results obtained with the unpurified population.
Figure 3.

SAHA-induced down-regulation of CXCR4 mRNA and protein. (A) After treatment for 24 h with various concentrations of SAHA, RNA was isolated and subjected to reverse transcriptase PCR analysis. The results are expressed as mean ratios of CXCR4/β-actin for the ten CLL patients evaluated. (B) Electrophoresis of one representative sample. (C) Cells were cultured in medium alone or with SAHA and stained with anti-CD19 and anti-CXCR4-phycoer ythrin. Results are expressed a mean of fluorescence intensity ratio (MFIR). (D) Representative flow cytometric analysis of surface CXCR4 protein on untreated or SAHA-treated CD19 positive cells.
Suberoylanilide hydroxamic acid reduces migration of chronic lymphocytic leukemia cells in response to stromal-derived factor-1 and into stromal layer
We evaluated the effect of SAHA on CLL cell chemotaxis in response to SDF-1 (n=23). As shown in Figure 4A, 10 and 20 μM SAHA significantly decreased the migration index in response to SDF-1 [with 10 μM of SAHA (P=0.0005); with 20 μM of SAHA (P<0.0001)]. These results correspond to an inhibition of 65.35±5.32% (10 μM) and 61.23±5.84% (20 μM) of control migration. Actin polymerization after SDF-1 stimulation was also clearly inhibited by preincubation of CLL cells with SAHA (Online Supplementary Figure S3). We also compared the migration of untreated or SAHA-treated (20 μM) CLL cells (n=6) into a bone marrow stromal layer established from normal subjects: of the input CLL cells (5×106), an average of 11.50±2.76% of cells migrated into the stromal layer. Following 24 h of 20 μM SAHA treatment, only 57.30±7.35% (n=6) of cells compared with untreated control samples (100%) had migrated after 3 h. This value was 65.27±4.77% (n=6) after pertussis toxin treatment, although this was not significantly different. In addition, viability and apoptosis were evaluated after 48 h of co-culture with a stromal layer with or without SAHA (20 μM). After normalization to the cells plated alone without SAHA (100% of viable cells), we observed that the decrease of viability induced by SAHA was greater in the presence of a stromal layer (Figure 4B). Indeed, SAHA induced only 33.97±4.32% of cell death in the absence of a stromal layer while cell death reached 75.86±5.75% in the presence of a stromal layer (P<0.05) indicating that SAHA would be more efficient in an in vivo context. Similar results were observed with annexin staining after SAHA treatment with higher, albeit not statistically significantly so, apoptosis observed in the presence of stromal cells (Figure 4C).
Figure 4.

SAHA reduces cell migration and synergizes with fludarabine in a mesenchymal stromal cell/CLL co-culture model. (A) CLL cells were cultured in the presence or absence of SAHA for 24 h before plating onto 5-μm Transwell microporous membranes for the migration assay. Results are expressed as a mean (± SEM) of the migration index. Cell viability normalized to untreated (B) and percentage of apoptotic cells (C) were evaluated after 48 h of incubation with SAHA (20 μM) in the presence or absence of a stromal layer. Cell viability normalized to untreated (D) and percentage of apoptotic cells (E) were evaluated on mononuclear cells pre-incubated for 4 h with SAHA (1 μM) and then incubated with SAHA and fludarabine for 48 h on a stromal layer.
Suberoylanilide hydroxamic acid synergizes with fludarabine to induce apoptosis in a chronic lymphocytic leukemia/mesenchymal stromal cell co-culture model
As suggested previously by Kurtova et al., a co-culture model with a protective microenvironment seems to be very important to study drug-induced apoptosis of CLL cells.32 Indeed, such a model is essential since SAHA decreases CXCR4 expression on CLL cells. We, therefore, measured viability and apoptosis of CLL cells co-cultured with a stromal layer (composed of mesenchymal stromal cells) without drug, with a low concentration of SAHA (1 μM), with fludarabine (10 μM) or with both drugs (n=6). After normalization to cells plated alone without SAHA (100% of viable cells), we observed that the decrease of viability induced by SAHA or fludarabine alone was relatively low but that the combination of both drugs induced a dramatic decrease of live cells (39.25±7.03) (P<0.05) (Figure 4D). Similar results were observed for apoptosis (Figure 4E). The CI were less than 1 (mean of 0.65±0.13; range, 0.20–0.97), indicating that the observed effects were synergistic.
Discussion
In the present study, we have demonstrated that SAHA, the prototypic member of a series of hydroxamic acid-based HDAC inhibitors, induces CLL cell death in a dose-dependent manner after 48 h of treatment. This induction of apoptosis was demonstrated by several criteria: cell shrinkage, externalization of phosphatidylserine and DNA fragmentation. These results are consistent with previous reports demonstrating SAHA activity in various pathologies such as prostate cancer,33 breast cancer,34 acute T-cell leukemia, myeloma and in malignant B cells.35 However, the mechanism underlying the apoptotic effect induced by SAHA has not been completely elucidated. Our results demonstrated that SAHA-mediated apoptosis in CLL cells was caspase-dependent since the pan-caspase inhibitor Z-VAD protected cells from SAHA. However, Z-VAD inhibited DNA fragmentation but not phosphatidylserine externalization suggesting that this last step is caspases-independent. Previous studies demonstrated that two different mechanisms (caspase-dependent and -independent) could exist in phosphatidylserine externalization and are dependent upon the cell type.36 The existence of both mechanisms is also supported by the fact that Z-VAD inhibits phosphatidylserine externalization induced by anti-CD3 but not by glucocorticoid in T cells.37
In the caspase-dependent pathway, caspase-3 serves as an effector molecule by cleaving cellular proteins, including PARP. Here, we detected caspase-3 activation in SAHA-treated cells and demonstrated PARP cleavage by western blotting and flow cytometry. Caspase-dependent apoptosis can be initiated through a variety of signaling cascades including the TNF receptor (extrinsic pathways via caspase-8) and the mitochondria (intrinsic pathways via caspase-9) pathways of apoptosis.38 Current treatments for CLL (such as fludarabine) induce apoptosis via a mitochondria-dependent pathway that involves the activation of the protease caspase-9.39 Using specific caspase inhibitors, we demonstrated the role of caspases-8, -6 and -3 in the apoptotic effect of SAHA suggesting that this drug uses the cell death receptor pathway of apoptosis to activate the initiator caspase-8 leading to caspase-3 activation and finally to PARP cleavage. In the case of adult T-cell leukemia and myeloma cells, SAHA-induced cell death was caspase-independent but appeared to be mediated by transcriptional events resulting in Bid cleavage, disruption of the mitochondrial membrane and production of reactive oxygen species.17,40 In contrast, in breast cancer cells and U937 leukemia cells, caspase-3 activation and PARP degradation have been described.35 Similar results have been observed for other HDAC inhibitors such as valproic acid: our group previously showed that caspase-8 activation and caspase-9 mRNA increase after valproic acid treatment,41 while Bokelmann et al. reported that caspase-8 was the first caspase activated by valproic acid but a mitochondrial amplification loop seemed to be required.42
In CLL cells, SAHA induces caspase-dependent apoptosis via the extrinsic pathway. This alternative pathway involves caspase-8 and could be triggered by different death ligands such as FAS-L, TNF or TRAIL. However, studies have shown that this pathway is generally not functional in CLL. In our study, while we observed an increase in CD95 expression after SAHA treatment (Online Supplementary Appendix and Online Supplementary Figure S4) (probably caused by a stress-induced response activating the JNK/SAPK pathways), activation of the caspase-8 pathway did not appear to require CD95. This phenomenon has been previously described: data from experiments using FAS-blocking antibody confirmed that drug-induced apoptosis via the extrinsic pathway can indeed occur via a FAS-independent mechanism.43–45 Furthermore, extrinsic pathways could be activated via TRAIL receptors (such as DR4 or DR5), as was previously reported.46,47 However, in this study we did not observe a modulation of these receptors after SAHA treatment suggesting that SAHA induces activation of extrinsic pathways via an independent mechanism. Another way to induce extrinsic pathways could be down-regulation of the expression of multiple inhibitors. Indeed, we found a dramatic down-regulation of c-FLIP after SAHA treatment. This down-regulation may thus induce indirect activation of receptor aggregation.
Our data also demonstrated that SAHA down-regulated CXCR4 mRNA and protein levels in CLL cells. Our findings are in line with those of Crazzolara et al. who reported CXCR4 down-regulation after SAHA or butyrate treatment in acute lymphoblastic leukemia cells.48 Interactions between CLL cells and accessory cells such as bone marrow stromal cells confer a growth advantage, extended cell survival and drug resistance to the leukemic cells.49 In this context, the SDF-1/CXCR4 system mediates an important physical interaction between leukemic cells and the microenvironment regulating apoptosis. The SDF-1/CXCR4 pathway may, therefore, be an attractive target for the development of novel therapeutic approaches. It has been demonstrated that SDF-1-induced migration of leukemic cells is correlated with surface CXCR4 levels and can be specifically blocked by antibodies against CXCR4 or CXCR4 antagonists.50 Here, we showed that SAHA treatment induced CXCR4 down-regulation, and a reduction of actin polymerization in response to SDF-1, which is likely responsible for reduced migration of leukemic cells towards bone marrow stromal cells. Since stromal cells could protect against fludarabine-induced apoptosis, and regarding the fact that SAHA could reduce CLL/microenvironment cell interactions, we tested the combination of these two drugs and observed a clear synergistic effect in the presence of a stromal layer. These results strongly suggest that SAHA exposure could disrupt the cross-talk between CLL cells and their protective microenvironment and thus sensitize CLL cells to chemotherapy. Compared to other disrupting agents (such as CXCR4 antagonist peptide),50 SAHA has an additional intrinsic pro-apoptotic activity, thus acting on different pathways to induce cell death. A small concentration of SAHA (1 μM) in combination with other standard treatments could, therefore, be a new therapeutic strategy for the management of CLL.
In conclusion, we showed that SAHA induces apoptosis via both caspase-dependent and caspase-independent pathways in CLL cells. The mechanism of cytotoxicity involves the extrinsic pathway leading to caspase-8 activation. SAHA potently represses CXCR4 receptor expression and significantly impairs cell migration into stromal cell layers. Our results suggest that SAHA in combination with other drugs represents a promising approach to overcome drug resistance and to improve the outcome of patients with CLL.
Footnotes
Funding: this research was supported by the “Fonds National de la recherche scientifique – FNRS” (F.R.S-F.N.R.S.) and the Belgian Foundation against Cancer. This work was also financed by an F.R.I.A. grant (Fonds de Recherche pour l’Industrie et l’Agriculture) and the Télévie fund, both of which are affiliated with the F.R.S-F.N.R.S. (Fonds de la Recherche Scientifique – FNRS). BS (Postdoctoral Researcher), CDB. (Scientific Research Worker), LL (Senior Research Associate) are members of the F.N.R.S.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
BS performed research and the statistical analysis, analyzed data, made the figures and tables, and wrote the manuscript. AD contributed to the morphological analyses. CDB contributed to the flow cytometry experiments. NM and DB contributed patients’ samples and data. LL performed, supervised and designed the research, and revised the manuscript.
The authors declare no competing financial interests.
References
- 1.Dighiero G, Travade P, Chevret S, Fenaux P, Chastang C, Binet JL. B-cell chronic lymphocytic leukemia: present status and future directions. French Cooperative Group on CLL. Blood. 1991;78(8):1901–14. [PubMed] [Google Scholar]
- 2.Bannerji R, Byrd JC. Update on the biology of chronic lymphocytic leukemia. Curr Opin Oncol. 2000;12(1):22–9. doi: 10.1097/00001622-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest. 2005;115(3):755–64. doi: 10.1172/JCI23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Bockstaele F, Verhasselt B, Philippe J. Prognostic markers in chronic lymphocytic leukemia: a comprehensive review. Blood Rev. 2009;23(1):25–47. doi: 10.1016/j.blre.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–54. [PubMed] [Google Scholar]
- 6.Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med. 2003;348(18):1764–75. doi: 10.1056/NEJMoa023143. [DOI] [PubMed] [Google Scholar]
- 7.Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–7. [PubMed] [Google Scholar]
- 8.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382(6594):829–33. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 9.Egawa T, Kawabata K, Kawamoto H, Amada K, Okamoto R, Fujii N, et al. The earliest stages of B cell development require a chemokine stromal cell-derived factor/pre-B cell growth-stimulating factor. Immunity. 2001;15(2):323–34. doi: 10.1016/s1074-7613(01)00185-6. [DOI] [PubMed] [Google Scholar]
- 10.Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, et al. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA. 1998;95(16):9448–53. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohle R, Failenschmid C, Bautz F, Kanz L. Overexpression of the chemokine receptor CXCR4 in B cell chronic lymphocytic leukemia is associated with increased functional response to stromal cell-derived factor-1 (SDF-1) Leukemia. 1999;13(12):1954–9. doi: 10.1038/sj.leu.2401602. [DOI] [PubMed] [Google Scholar]
- 12.Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. 1998;91(7):2387–96. [PubMed] [Google Scholar]
- 13.Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94(11):3658–67. [PubMed] [Google Scholar]
- 14.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–63. [PubMed] [Google Scholar]
- 15.Richon VM, O’Brien JP. Histone deacetylase inhibitors: a new class of potential therapeutic agents for cancer treatment. Clin Cancer Res. 2002;8(3):662–4. [PubMed] [Google Scholar]
- 16.Glick RD, Swendeman SL, Coffey DC, Rifkind RA, Marks PA, Richon VM, et al. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer Res. 1999;59(17):4392–9. [PubMed] [Google Scholar]
- 17.Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci USA. 2001;98(19):10833–8. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He LZ, Tolentino T, Grayson P, Zhong S, Warrell RP, Jr, Rifkind RA, et al. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J Clin Invest. 2001;108(9):1321–30. doi: 10.1172/JCI11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111(3):1060–6. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Gonzalez B, Yang H, Bueso-Ramos C, Hoshino K, Quintas-Cardama A, Richon VM, et al. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood. 2006;108(4):1174–82. doi: 10.1182/blood-2005-09-008086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24(1):166–73. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- 22.Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 23.Stamatopoulos B, Meuleman N, Haibe-Kains B, Duvillier H, Massy M, Martiat P, et al. Quantification of ZAP70 mRNA in B cells by real-time PCR is a powerful prognostic factor in chronic lymphocytic leukemia. Clin Chem. 2007;53(10):1757–66. doi: 10.1373/clinchem.2007.089326. [DOI] [PubMed] [Google Scholar]
- 24.Stamatopoulos B, Meuleman N, Haibe-Kains B, Saussoy P, Van Den NE, Michaux L, et al. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood. 2009;113(21):5237–45. doi: 10.1182/blood-2008-11-189407. [DOI] [PubMed] [Google Scholar]
- 25.Tondreau T, Meuleman N, Delforge A, Dejeneffe M, Leroy R, Massy M, et al. Mesenchymal stem cells derived from CD133-positive cells in mobilized peripheral blood and cord blood: proliferation, Oct4 expression, and plasticity. Stem Cells. 2005;23(8):1105–12. doi: 10.1634/stemcells.2004-0330. [DOI] [PubMed] [Google Scholar]
- 26.Najar M, Rouas R, Raicevic G, Boufker HI, Lewalle P, Meuleman N, et al. Mesenchymal stromal cells promote or suppress the proliferation of T lymphocytes from cord blood and peripheral blood: the importance of low cell ratio and role of interleukin-6. Cytotherapy. 2009;11(5):570–83. doi: 10.1080/14653240903079377. [DOI] [PubMed] [Google Scholar]
- 27.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139(2):271–9. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 28.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 29.Ferlini C, Di Cesare S, Rainaldi G, Malorni W, Samoggia P, Biselli R, et al. Flow cytometric analysis of the early phases of apoptosis by cellular and nuclear techniques. Cytometry. 1996;24(2):106–15. doi: 10.1002/(SICI)1097-0320(19960601)24:2<106::AID-CYTO2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 30.Stamatopoulos B, Haibe-Kains B, Equeter C, Meuleman N, Soree A, De Bruyn C, et al. Gene expression profiling reveals differences in microenvironment interaction between patients with chronic lymphocytic leukemia expressing high versus low ZAP70 mRNA. Haematologica. 2009;94(6):790–9. doi: 10.3324/haematol.2008.002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bouzar AB, Boxus M, Defoiche J, Berchem G, Macallan D, Pettengell R, et al. Valproate synergizes with purine nucleoside analogues to induce apoptosis of B-chronic lymphocytic leukaemia cells. Br J Haematol. 2009;144(1):41–52. doi: 10.1111/j.1365-2141.2008.07426.x. [DOI] [PubMed] [Google Scholar]
- 32.Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114(20):4441–50. doi: 10.1182/blood-2009-07-233718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60(18):5165–70. [PubMed] [Google Scholar]
- 34.Huang L, Sowa Y, Sakai T, Pardee AB. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene. 2000;19(50):5712–9. doi: 10.1038/sj.onc.1203963. [DOI] [PubMed] [Google Scholar]
- 35.Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM, et al. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18(50):7016–25. doi: 10.1038/sj.onc.1203176. [DOI] [PubMed] [Google Scholar]
- 36.Kim DK, Cho ES, Um HD. Caspase-dependent and -independent events in apoptosis induced by hydrogen peroxide. Exp Cell Res. 2000;257(1):82–8. doi: 10.1006/excr.2000.4868. [DOI] [PubMed] [Google Scholar]
- 37.Verhoven B, Krahling S, Schlegel RA, Williamson P. Regulation of phosphatidylserine exposure and phagocytosis of apoptotic T lymphocytes. Cell Death Differ. 1999;6(3):262–70. doi: 10.1038/sj.cdd.4400491. [DOI] [PubMed] [Google Scholar]
- 38.Kim R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer. 2005;103(8):1551–60. doi: 10.1002/cncr.20947. [DOI] [PubMed] [Google Scholar]
- 39.Genini D, Adachi S, Chao Q, Rose DW, Carrera CJ, Cottam HB, et al. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood. 2000;96(10):3537–43. [PubMed] [Google Scholar]
- 40.Henderson C, Mizzau M, Paroni G, Maestro R, Schneider C, Brancolini C. Role of caspases, Bid, and p53 in the apoptotic response triggered by histone deacetylase inhibitors trichostatin-A (TSA) and suberoylanilide hydroxamic acid (SAHA) J Biol Chem. 2003;278(14):12579–89. doi: 10.1074/jbc.M213093200. [DOI] [PubMed] [Google Scholar]
- 41.Stamatopoulos B, Meuleman N, De Bruyn C, Mineur P, Martiat P, Bron D, et al. Antileukemic activity of valproic acid in chronic lymphocytic leukemia B cells defined by microarray analysis. Leukemia. 2009;23(12):2281–9. doi: 10.1038/leu.2009.176. [DOI] [PubMed] [Google Scholar]
- 42.Bokelmann I, Mahlknecht U. Valproic acid sensitizes chronic lymphocytic leukemia cells to apoptosis and restores the balance between pro- and antiapoptotic proteins. Mol Med. 2008;14(1–2):20–7. doi: 10.2119/2007-00084.Bokelmann. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siitonen T, Mantymaa P, Saily M, Savolainen E, Koistinen P. Etoposide-induced apoptosis is not associated with the fas pathway in acute myeloblastic leukemia cells. Leuk Res. 2000;24(4):281–8. doi: 10.1016/s0145-2126(99)00176-9. [DOI] [PubMed] [Google Scholar]
- 44.Shao RG, Cao CX, Nieves-Neira W, Dimanche-Boitrel MT, Solary E, Pommier Y. Activation of the Fas pathway independently of Fas ligand during apoptosis induced by camptothecin in p53 mutant human colon carcinoma cells. Oncogene. 2001;20(15):1852–9. doi: 10.1038/sj.onc.1204264. [DOI] [PubMed] [Google Scholar]
- 45.Eischen CM, Kottke TJ, Martins LM, Basi GS, Tung JS, Earnshaw WC, et al. Comparison of apoptosis in wild-type and Fas-resistant cells: chemotherapy-induced apoptosis is not dependent on Fas/Fas ligand interactions. Blood. 1997;90(3):935–43. [PubMed] [Google Scholar]
- 46.Fulda S, Sieverts H, Friesen C, Herr I, Debatin KM. The CD95 (APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res. 1997;57(17):3823–9. [PubMed] [Google Scholar]
- 47.Nagane M, Pan G, Weddle JJ, Dixit VM, Cavenee WK, Huang HJ. Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo. Cancer Res. 2000;60(4):847–53. [PubMed] [Google Scholar]
- 48.Crazzolara R, Johrer K, Johnstone RW, Greil R, Kofler R, Meister B, et al. Histone deacetylase inhibitors potently repress CXCR4 chemokine receptor expression and function in acute lymphoblastic leukaemia. Br J Haematol. 2002;119(4):965–9. doi: 10.1046/j.1365-2141.2002.03955.x. [DOI] [PubMed] [Google Scholar]
- 49.Munk PI, Reed J. Microenvironmental interactions and survival of CLL B-cells. Leuk Lymphoma. 2004;45(12):2365–72. doi: 10.1080/10428190412331272703. [DOI] [PubMed] [Google Scholar]
- 50.Burger M, Hartmann T, Krome M, Rawluk J, Tamamura H, Fujii N, et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood. 2005;106(5):1824–30. doi: 10.1182/blood-2004-12-4918. [DOI] [PubMed] [Google Scholar]