

Abstract
Allostery plays a key role in the regulation of the activity and function of many biomolecules. And although many ligands act through allostery, no systematic use is made of it in drug design strategies. Here we describe a procedure for identifying the regions of a protein that can be used to control its activity through allostery. This procedure is based on the construction of a plausible conformational path, which describes protein transition between known active and inactive conformations. The path is calculated by using a framework approach that steers and markedly improves the conjugate peak refinement method. The evolution of conformations along this path was used to identify a putative allosteric site that could regulate activation of Bacillus anthracis adenylyl cyclase toxin (EF) by calmodulin. Conformations of the allosteric site at different steps along the path from the inactive (free) to the active (bound to calmodulin) forms of EF were used to perform virtual screenings and propose candidate EF inhibitors. Several candidates then proved to inhibit calmodulin-induced activation in an in vitro assay. The most potent compound fully inhibited EF at a concentration of 10 μM. The compounds also inhibited the related adenylyl cyclase toxin from Bordetella pertussis (CyaA). The specific homology between the putative allosteric sites in both toxins supports that these pockets are the actual binding sites of the selected inhibitors.
Keywords: anthrax, transition path calculation, molecular dynamics, drug discovery, inhibition of protein–protein association
Structure-based drug design is increasingly used and has impacted the development of many drugs (1–3). Identifying inhibitors by virtual screening involves multiple steps, and its success depends on several factors: the efficiency of ligand and receptor conformational sampling (4–11), the quality of the scoring function (12, 13), the choice of docking pocket, and finally, the availability of a relevant assay to test the virtual screening candidates.
Conventionally, virtual screening uses experimentally validated binding sites such as an enzyme catalytic site or a pocket already containing a ligand. However, targeting the active site of enzymes that act on a common substrate such as ATP (e.g., ATPases, kinases, or the toxin studied here, etc.) with a high enough degree of selectivity to avoid general toxicity remains a challenging endeavor. Targeting different pockets could lead to the identification of more specific inhibitor families and, thus, can help to overcome such issues. Rational approaches allowing efficient identification of such “druggable” sites would therefore be highly desirable in the difficult and costly process of drug discovery.
Pockets can be detected with purely geometrical considerations, for instance, by analyzing local surface curvature (14, 15), or can be filtered by means of other geometric and chemical criteria (16). However, the detected sites may be irrelevant to inactivate the targeted function, and some noncanonical sites (17) may be missed with such criteria.
A promising method to alter biochemical and cellular processes is to target interactions between biomolecules. Because the assembly of partners can be affected by allostery, through a shift in their conformational equilibrium (18–22), a promising strategy would be to find and target allosteric pockets. The range of biomolecules that can be targeted would be broadened because their entire surface can potentially be screened.
Identifying pockets that potentially play an allosteric role is still a major challenge but should pave the way to wide opportunities in rational drug design (23). For such purposes, we develop here an approach in which a plausible description of the conformational path between known active and inactive conformations is built and then used to identify such putative pockets. The path was built with a method inspired by that presented in ref. 24, but including a number of radical improvements. This approach was validated by the discovery of an inhibitor scaffold for adenylyl cyclase (EF) of Bacillus anthracis, the etiologic agent of anthrax.
EF is an essential toxin for B. anthracis. It is able to enter into eukaryotic cells where it is activated upon binding to endogenous calmodulin (CaM) to produce supraphysiological levels of cAMP that alter the cell physiology. By targeting immune cells, EF contributes to the virulence of B. anthracis and is therefore considered as a target for anti-anthrax drugs (25–28).
The binding of CaM to EF induces a major transition from a closed to an open form (29). This reorganizes the catalytic site into its active configuration that can convert ATP into cAMP. The dynamics and energetics of EF-CaM complex were analyzed by extensive molecular dynamics (MD) (30, 31). A path calculation was used for further analysis and, as described above, to devise a rational drug design strategy. A pocket that underwent major and early reorganization along the path describing EF activation transition was identified (Fig. 1). This pocket was then used to screen in silico for inhibitors of CaM-induced EF activation. A number of thiophen ureidoacids thus selected were shown to inhibit EF activity in vitro with affinities in the low micromolecular range. Compounds from this series were also active against CyaA, the adenylyl cyclase toxin of Bordetella pertussis, the causative agent of whooping cough (32, 33).
Fig. 1.

Scheme of the virtual screening approach used to prevent the CaM activation of EF. The pocket to which the ligand was docked is shown in yellow (483 and 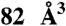 in the inactive and active forms, respectively), EF and CaM are drawn in cartoon format. Switches A, B, and C in EF are highlighted in blue, orange, and magenta, respectively.
in the inactive and active forms, respectively), EF and CaM are drawn in cartoon format. Switches A, B, and C in EF are highlighted in blue, orange, and magenta, respectively.
Results
Conformational Transition Path.
A plausible transition path between the open (active EF-CaM complex) and closed (inactive apo-EF) form was calculated with a method radically improving that presented in ref. 24 to calculate topologically complex transitions (see Materials and Methods). The method markedly extends the power of the conjugate peak refinement (CPR) method (34). Each refinement cycle consisted of one path reduction and one step in subpath generation. After 45 refinement cycles, electrostatic energy was switched from a sigmoidal dielectric constant to the more comprehensive ACE2 model (35–37), to simulate correctly cavities in the protein. CPR refinements then took about 300 hours of CPU on a 2,666 MHz processor. The calculation was halted when the number of conformations in the path was down to 80 and the total curvilinear length was down to 36.9 Å.
Fig. S1 illustrates the path refinement process. The comprehensive electrostatic model yielded energies that were about -25 Mcal/mol lower than the sigmoidal model, which is a typical value for such a change of electrostatic model (35–37). The number of conformations and curvilinear length (Dcurv) first increased as the path meandered to avoid barriers. They then tended to decrease as lower energies and straighter paths shortcuts were found during the search process.
The path was characterized by its maximum energy, coordinate distance (rmsd), and Dcurv from the first conformation along the transition (Fig. S2). The very low energy along the transition showed that the intermediate states are not distorted and have plausible conformations. Furthermore, the curvilinear length of the transition was comparatively short (36.9 Å) for a major conformational change (atomic coordinate rmsd: 9.3 Å) involving secondary structure reshuffling (CB, switch C, and the L-M loop in the helical domain). These figures show a far simpler path than those previously described for a transition involving comparable reorganization (24). Thus, the resulting path appeared to be both comparatively straight and low in energy and therefore a plausible model for the transition.
The transition path was analyzed by principal component analysis (PCA). The path and MD trajectories were projected on the first three PCA eigenvectors (Fig. 2). MD trajectories (ellipses in Fig. 2) remained in the vicinity of their initial conformations (beginning and end of the path) and failed to sample intermediate states such as conformations 8, 28, and 47, which were used to counterselect compounds in the virtual screening strategy (see below). Thus, the path provided information that could not be furnished by MD.
Fig. 2.

Projections onto the first normal modes of the PCA analysis of the conformational transition path. Projections of successive path conformations from the EF open to the closed conformation are colored from light gray to black. The conformers 8, 28, and 47 are labeled in red, as is the starting point (1). Projections of the EF conformations sampled during MD simulations recorded on the EF-CaM complex in the presence of 0, 2, and 4 Ca2+ and on free EF are shown in cyan, green, orange, and purple, respectively. The mean MD conformation projection (Filled Square) is shown, surrounded by an ellipsoid illustrating the scattering of the projections along the trajectory. Considering the number of built intermediate points or the number of energy evaluations as an equivalent of a MD step, the total sampling used to build the path correspond to 300 ns or 8 μs of MD, respectively.
The path described here is represented by series of individual conformations. This representation proved to be convenient, comprehensive, and sufficiently precise to describe the transition in details and to be used in virtual screening.
In Silico Selection of Ligands.
The transition path conformations were systematically analyzed with the PocketFinder module of ICM (16). Ten potential binding sites, including the catalytic site, with volumes ranging from 100 to 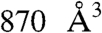 were identified. The second largest pocket in conformation 1 enclosed a cavity of
were identified. The second largest pocket in conformation 1 enclosed a cavity of 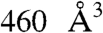 (Table 1). This pocket was formed by residues, A496, P499, I538, E539, P542, S544, S550, W552, Q553, T579, Q581, L625, Y626, Y627, N629, and N709, from three polypeptide segments, defined as switches A, B, and C by Drum et al. (29). The pocket shall henceforth be called the “SABC pocket”. A small pocket between switches B and C, SBC, only separated from SABC by the Q581 side chain can be viewed as an extension. The three switches, A, B, and C, play a critical role in EF activation because they either contact CaM or stabilize the catalytic site (29, 38). The path calculation required careful building of the missing loop 580–590. The pocket definitions were thus refined and modified from those present in 1K8T (Materials and Methods and Fig. S3). The substantial rearrangements during the early steps of the transition greatly distorted and reduced the SABC pocket (Figs. 1 and 3) as a result of: (i) a widening gap between the catalytic region and the helical domain (residues 291–656 and 657–768, respectively), (ii) backbone rearrangements of switches A and C, (iii) reorientation of side chains contacting CaM, and (iv) transient reorientation of the hydrogen network (Fig. S4). Therefore a small molecule binding inside the SABC cavity would very likely interfere with EF structural remodeling leading to activation.
(Table 1). This pocket was formed by residues, A496, P499, I538, E539, P542, S544, S550, W552, Q553, T579, Q581, L625, Y626, Y627, N629, and N709, from three polypeptide segments, defined as switches A, B, and C by Drum et al. (29). The pocket shall henceforth be called the “SABC pocket”. A small pocket between switches B and C, SBC, only separated from SABC by the Q581 side chain can be viewed as an extension. The three switches, A, B, and C, play a critical role in EF activation because they either contact CaM or stabilize the catalytic site (29, 38). The path calculation required careful building of the missing loop 580–590. The pocket definitions were thus refined and modified from those present in 1K8T (Materials and Methods and Fig. S3). The substantial rearrangements during the early steps of the transition greatly distorted and reduced the SABC pocket (Figs. 1 and 3) as a result of: (i) a widening gap between the catalytic region and the helical domain (residues 291–656 and 657–768, respectively), (ii) backbone rearrangements of switches A and C, (iii) reorientation of side chains contacting CaM, and (iv) transient reorientation of the hydrogen network (Fig. S4). Therefore a small molecule binding inside the SABC cavity would very likely interfere with EF structural remodeling leading to activation.
Table 1.
Description of the pockets detected in conformation 1 of the EF transition path
| Pocket number | Volume (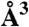 ) ) |
Number of residues | Number of counterparts in CyaA | |||
| Total | Conserved | Similar profile | Similar shape only | |||
| 1 (catalytic) | 871 | 23 | 23 | 17 | 2 | 1 |
| 2 (SABC) | 463 | 16 | 15 | 6 | 3 | 2 |
| 3 | 227 | 12 | 8 | 1 | 1 | 0 |
| 4 | 188 | 10 | 4 | 2 | 0 | 0 |
| 5 (SBC) | 171 | 9 | 8 | 6 | 0 | 0 |
| 6 | 160 | 12 | 12 | 1 | 0 | 1 |
| 7 | 156 | 11 | 11 | 1 | 1 | 1 |
| 8 | 129 | 9 | 3 | 0 | 0 | 0 |
| 9 | 123 | 10 | 0 | 0 | 0 | 0 |
| 10 | 110 | 8 | 8 | 3 | 0 | 0 |
| 11 | 104 | 11 | 9 | 1 | 0 | 0 |
Similar profiles correspond to residues belonging to the same physicochemical family, whereas similar shapes correspond to residues with the same steric hindrance.
Fig. 3.

SABC deformation along the EF transition path. The SABC pocket was detected at the interface of switches A (in green to cyan), B (in blue to indigo), and C (in magenta to red): (A) top view of the surface of SABC in conf 1 (cartoon representation); (B–E) licorice representation of SABC in conf 1 (B), 8 (C), 28 (D), and 47 (E) with the surface of the pocket displayed in transparent gray. Pocket residues are colored according to their numbers in the protein sequence, from green through blue to red. The rmsd from the initial conformation of the residues defining the SABC pocket was 0, 3.1, 6.4, and 7.3 Å, for conf 1, 8, 28, and 47, respectively.
The robustness of the SABC pocket was probed by analyzing cavities on representative conformations (39) of a 15-ns MD of conformation 1. They varied significantly with splitting and merging events (Fig. S5 and Table S1). Although MD conformations appeared improper for virtual screening, each one contained at least one pocket involving more than half of the 16 SABC residues, and conversely, each residue was identified in at least one of these conformations.
All together, the SABC pocket identified with the transition path model appeared to shrink strongly and early in the transition, to be sufficiently large (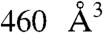 ), and to have a good balance between charged/polar and hydrophobic residues. It therefore appeared a promising target site for virtual screening.
), and to have a good balance between charged/polar and hydrophobic residues. It therefore appeared a promising target site for virtual screening.
The French National Chemical Library [about 28,000 molecules (40)] was screened for ligands able to bind the SABC pocket with the FlexX program (41). Initial conformation 1 was used to select the top 1% best-score candidates. Intermediate conformations 8, 28, and 47 (Figs. 2 and 3) were then used to exclude compounds that could be compatible with the initiation of the activation process (Table S2). The pocket conformations shift was already more than 3 Å rmsd between conformations 1 and 8, suggesting an efficient discrimination. ICM software (42), which arguably uses a more accurate force field (5), was employed to refine the selection (ICM scores lower than -10 kcal/mol). Of the 28 ligands thus selected, 18 were available and were tested in an in vitro assay.
In Vitro Characterization of the Putative EF Inhibitors.
The 18 compounds were first tested at 100 μM in the adenylyl cyclase enzymatic assay described in Materials and Methods. As shown in Fig. 4 and Table 2, a series of 6 highly related compounds, sharing a common thiophen ureidoacid (TUA) moiety, exerted potent inhibitory activity under these conditions. Among them, TUAdiCl still exhibited a strong inhibition at 10 μM (> 90% inhibition), corresponding to 50% inhibition at 2–3 μM. Interestingly, a single Cl atom caused a marked increase of activity for TUAdiCl over TUACl.
Fig. 4.

Inhibition of EF activity by various thiophen ureidoacid (TUA) derivatives. EF (0.5 nM) was preincubated for 20 min with the indicated concentrations of TUA compounds, then 2 μM CaM was added and enzyme activity was measured 10 minutes later, as described in SI Materials. Data in the second panel (EF-CaM) correspond to enzyme activities measured when EF was preincubated with CaM prior to the addition of 100 μM TUA derivatives. Data in the third panel (CyaA) correspond to CyaA (0.2 nM) preincubated 20 min with 100 μM of TUA compounds before addition of 2 μM CaM and measure of the activity 10 min later.
Table 2.
Chemical formula for the six validated compounds and their inhibition rate on EF activity
| Name | R | IC50 (μM) | |
| TUA | 4-H | 16 ± 7 | 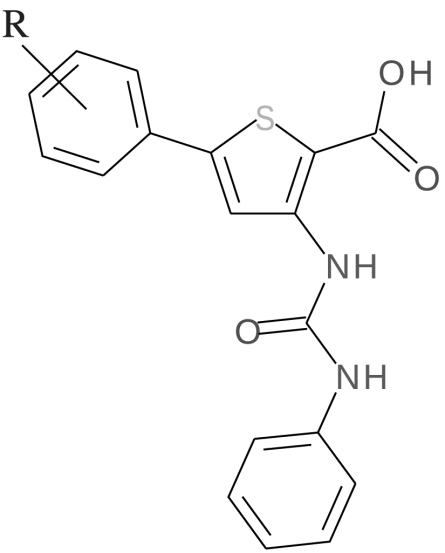 |
| TUABr | 4-Br | 10 ± 3 | |
| TUACl | 4-Cl | 10 ± 2 | |
| TUAF | 4-F | 10 ± 2 | |
| TUAOCH3 | 4-OCH3 | 12 ± 6 | |
| TUAdiCl | 3,4-diCl | 2 ± 1 |
IC50 values were obtained by data fitting with a single binding isotherm.
Given the strong affinity of CaM for EF (32, 43), we anticipated that molecules that could prevent CaM-induced activation of EF might not be able to inhibit the preformed EF-CaM complex. Indeed, the five weakest inhibitors were ineffective on preformed EF-CaM, but the strongest, TUAdiCl could partly inhibit the complex (Fig. 4).
The 6 compounds were also tested on a related enzyme, adenylyl cyclase toxin (CyaA) from B. pertussis. CyaA is also activated by CaM and the structure of its catalytic site is highly similar to that of EF despite a low overall sequence homology (32). The compounds inhibited CyaA-CaM at 100 μM, but not at 10 μM, indicating that they were moderate inhibitors.
Many small hydrophobic molecules can block CaM interaction with target enzymes (44). Thus, the binding of TUAdiCl on CaM rather than on EF was tested. The association of CaM with a CyaA-derived peptide P233-254 (33) was monitored by its fluorescence anisotropy signal (Materials and Methods). The signal increased from 0.03 for the free peptide at 2 μM in 0.2 mM CaCl2 to 0.094 upon addition of 2 μM CaM. Addition of 10 μM TUAdiCl did not affect the signals (0.027 and 0.095 without and with CaM, respectively). Hence, TUAdiCl at a concentration that almost completely inhibited CaM-EF activity did not affect CaM binding to a specific peptide.
TUAdiCl compares favorably with several compounds previously reported to inhibit EF-CaM association in vitro or in vivo with IC50 values in the 10–100 μM range (25, 27, 28) and represents a new chemical family.
Discussion
EF activation mechanism was modeled by construction of a transition path describing the structural motions required to bind CaM, with an approach markedly improving the power of the CPR algorithm (34). An analysis of the full path suggested that the SABC pocket should be a suitable target for virtual screening. Eighteen candidates selected from the French National Chemical Library (40, 45) for their ability to fit in this pocket were tested, and six of them were found to inhibit catalysis in the medium and low micromolar range. These molecules share a common thiophen ureidoacid (TUA) scaffold that hitherto was not known to inhibit EF. These compounds were also found to inhibit the related adenylyl cyclase CyaA from B. pertussis. The inhibition proved to be specific as the compounds did not affect neither CaM nor the pyrophosphatase used in the assay (see SI Text). The presence of detergent in the assay also prevented promiscuous inhibitory effects (46). These results showed that modeling transition paths is a useful tool for drug design to enlarge the search for inhibitors to new chemical families.
Transition Path Determination Is Central to the Approach.
The originality of the approach lies in the determination of a plausible conformational transition path and its use in combination with drug design methods.
The procedure to generate and sort alternative path shortcuts and the topological definition of path connectivity are essential characteristics of the path calculation method presented here. As a consequence, the path is made of a limited series of completely connected and ordered conformations. This contrasts with most previous methods developed for extensive conformational sampling, which fail to produce such limited series of ordered conformations (47–49) and for which sorting conformations along a transition path or defining a reaction coordinate are still major challenges (50).
The order of events was used to identify a suitable pocket for virtual screening. Indeed, our aim was to lock EF in its initial inactive form, and, for this to occur, the targeted pocket had to disappear in the early stages of the conformational transition leading to activation. Inhibitors filling such pocket(s) would be expected to maintain EF in initial conformations unable to bind CaM and, thus, would provide a stronger structural lever to inhibit the activation process. Among all the pockets detected in the inactive EF form, the SABC pocket best matched this condition with large rmsd reached in early stage of the transition. Noticeably, several possible paths can connect the inactive and active forms of EF. Thus, using a conformation located close to one end of the path avoids relying too critically on the path accuracy, even though the path determined possessed excellent energetic and geometrical properties. Altogether, screening with the SABC pocket produced an excellent level of enrichment in the identification of inhibitors: Out of 18 in silico candidates, 6 inhibited EF in the low micromolar range.
Evidences Supporting the Binding of TUA Inhibitors to the Targeted SABC Pocket.
The rationale of the approach led us to the identification of a plausible binding site, the SABC pocket, and then, after virtual screening, to the identification of effective inhibitors. All our results were consistent with the binding of the TUA inhibitors in the SABC pocket, although this could only be unequivocally establish by a crystal structure of the complex. Yet strong experimental evidence in favor of this model was provided by the observation that the TUA compounds were also able to inhibit the related adenylyl cyclase from B. pertussis, CyaA. Catalytic domains of CyaA and EF only share 25% overall sequence identity, and the structural and thermodynamic features of their interaction with CaM differ significantly (32, 43). Analysis of EF pockets (Table 1) revealed that the catalytic site and the SABC pocket and/or its SBC extension were the only ones to share significant homology with their CyaA counterparts. Thus, TUA inhibitors should most likely bind one of these conserved pockets.
The catalytic site is the largest and most conserved pocket with 19 identical or similar residues out of 23. However, the fact that most of the effective compounds could not inhibit the preformed EF-CaM complex clearly ruled out their binding to the active site. It is also unlikely that these inhibitors could bind to the inactive configuration of the catalytic site (i.e., in CaM-free EF) to lock EF in conformations unable to bind CaM, given the similarity between the active and inactive backbone conformations of the catalytic site (Fig. S6).
The second conserved site, the SABC pocket, involves switches A, B, and C of EF that correspond in CyaA to F-G-H-H’ helices, T300-K312 loop, and C-tail, respectively, and interact with CaM to stabilize the catalytic site in a similar way (Fig. S7) (32, 51). Of the 16 residues in the SABC pocket, 6 are identical, 3 are homologous, and 2 are similar to their counterparts in CyaA (Table S3) and could clearly make similar interactions with the ligand (Fig. S8). The similarity of these sequences and potentially conserved binding modes strongly support the common binding of TUA derivatives into the SABC pocket or into its CyaA counterpart.
It is noteworthy that TUAdiCl, unlike the other TUA inhibitors, could significantly inhibit the activity of the preformed EF-CaM complex. This activity may be because of a displacement of the conformational equilibrium of EF-CaM upon binding of TUAdiCl. An MD simulation of EF-CaM was performed in the absence of Ca2+, conditions where it is unstable and weakly active (52). Interestingly, in transient conformations such as 9335, the active site collapsed and a 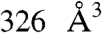 pocket involving 6 SABC residues appeared (Table 3). This correlation, similar to that proposed in ref. 53, suggests that the inhibitor could bind such transient conformations and displace the EF-CaM conformational equilibrium towards inactive forms. Although different from that initially imagined, this mechanism would still be an allosteric effect originating from the SABC pocket.
pocket involving 6 SABC residues appeared (Table 3). This correlation, similar to that proposed in ref. 53, suggests that the inhibitor could bind such transient conformations and displace the EF-CaM conformational equilibrium towards inactive forms. Although different from that initially imagined, this mechanism would still be an allosteric effect originating from the SABC pocket.
Table 3.
Pockets identified at the SA, SB, and SC interface in the EF-CaM complex
| Conformation (ps) | Volume (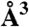 ) ) |
Residues in common with SABC | SASA of catalytic site (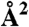 ) ) |
|
| 0Ca | 5,906 | 192 | (3/7 = 43%) I538,Q581,Y627 | 475 |
| 9,335 | 326 | (6/12 = 50%) I538,W552,Q553,Q581,L625,Y627 | 504 | |
| 12,150 | 202 | (3/10 = 30%) E539,Q581,N629 | 422 | |
| 2Ca | 5,016 | 104 | (1/7 = 14%) Q581 | 919 |
| 7,950 | - | - | 916 | |
| 12,262 | 144 | (0/6 = 0%) | 947 | |
| 4Ca | 3,529 | 257 | (0/10 = 0%) | 791 |
| 4,983 | 126 | (2/7 = 29%) Q290,N338 | 886 | |
| 5,471 | 191 | (2/8 = 25%) E248,Q290 | 845 | |
| 11,084 | - | - | 824 | |
| 13,515 | - | - | 910 | |
Pockets were detected at the SA, SB, and SC interface on the EF surface in representative conformations (ref. 39) extracted from MD simulations of the complex loaded with 0, 2, and 4 Ca2+ ions, respectively. The volume of each pocket is given together with the relative number and name of residues in common with SABC. The SASA (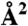 ) of the catalytic site in the same conformation is also given.
) of the catalytic site in the same conformation is also given.
Conclusion
An in silico approach is described that can be employed to target the functional activation of biomolecules. It is based on the calculation of an activation transition path to identify putative allosteric pockets. This opens up avenues for rational drug design because it radically expands pocket identification tools and therefore the opportunities to find ligands. The approach has only been made possible by the development of specific and powerful molecular modeling approaches.
Materials and Methods
Bioinformatics.
Crystallographic structures and equilibrium MD simulations are described in SI Materials (Structure analysis and Molecular dynamics simulations). The protocol used for inhibitor prediction is given in “In silico screening and analysis.”
Refinement of the Conformational Transition Path.
Calculations on the EF chain alone were performed by using CHARMM version 29 (54) and the PARM19 force field. Two levels of approximation were used to model the solvent. First, a simple distance-dependent dielectric (1.416 × r) with force shift at 8 Å globally fitted a sigmoidal dielectric constant (55). Van der Waals interactions were switched between 7 and 8 Å. In the second phase, analytical continuum electrostatics (ACE2) (35–37) was used, with dielectric constants of 1 in the protein, and 80 outside, and a force shift at 12 Å. Van der Waals interactions were switched between 10 and 12 Å.
Coordinates for 1K8T Protein Data Bank entry (apo inactive conformation), and for chain C in 1K93 Protein Data Bank entry (EF-CaM with two Ca2+ ions: active conformation) were used as the initial and final points in the conformational transition. Loop 580-590 (switch B), which is absent in 1K8T, was built by translation of the corresponding loop in 1K93. The internal coordinates of the two conformations were then compared to detect side chain flips and crankshaft motions. Any unnecessary flips of symmetric (F, Y, E, D) and pseudo symmetric (H, N, Q) amino acids were removed after visual inspection of the conformations and putative hydrogen bonds.
A steered molecular dynamics simulation consisting of 100,000 1-fs time steps was driven from the first structure towards the second with a force constant of 0.5 kcal/mol/Å by using a 300 K Langevin thermostat (56). Conformations were recorded every 200 steps, and the 500 frames were used to initiate the transition path calculation.
The path was then refined, by applying iteratively two types of procedure: (i) CPR (34) followed by a reduction of the number of conformations in the path and (ii) sampling of the possible subpaths located between pairs of path conformations. A maximum atom displacement of 0.5 Å was used in CPR. An energy threshold criterion was defined as follows: The energy of any conformation obtained by linear interpolation between two consecutive path conformations must be lower than the threshold.
Reduction was performed by iterative truncation of a given path starting from the first conformation (n1 = 1). At iteration k, the largest index nk+1 for which the energy threshold criterion is fulfilled between conformations nk and nk+1 is searched. Iteration stops when the end of the path is reached.
Procedure (ii) was performed by using CPR in parallel between all pairs of conformations nk and nk′ and in compliance with several criteria: (a) the number of conformers in the path between conformations nk and nk′ is larger than a given threshold Nmin, (b) the coordinate rmsd between the conformations is smaller than a given threshold Dmax, and (c) the ratio between rmsd and Dcurv is smaller than a given threshold Rmax. Dcurv, the curvilinear distance between the conformations nk and nk′, is defined as
 |
[1] |
where Xns are the coordinates of the nsth conformer, and rmsd(Xns,Xns+1) is the atomic coordinate rmsd between the nsth and the ns+1th conformers.
Criteria (a) and (c) intend to focus the search on regions of the path involving many swerves, whereas criterion (b) limits the search to intervals for which the algorithm has a sufficient probability to produce improved solutions. These criteria were tuned in view of previous cycles results to reduce CPU usage.
Experimental.
The Synthesis of identified inhibitors section in SI Text shows the synthesis schemes. All inhibition assays are described in Biochemical assay of compounds and Adenylyl cyclase enzymatic assay. The CaM-binding properties probed by steady-state fluorescence anisotropy section described the corresponding measurements setup.
Supplementary Material
Acknowledgments.
The French National Chemical Library and, in particular, Prof. Marcel Hibert, Dr. Bruno Didier, Dr. Philippe Jauffret, and Mr. Kiet Tran are gratefully acknowledged. Dr. David Giganti indicated useful references. This work was supported by the French Ministry of Defense (Direction Générale de l’Armement - Mission pour la Recherche et l’Innovation Scientifique), the Centre National de la Recherche Scientifique, and Institut Pasteur. Molecular dynamics calculations were performed by means of an allocation from the Centre National de la Recherche Scientifique Institut du Développement et des Ressources en Informatique Scientifique supercomputing center.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0914611107/-/DCSupplemental.
References
- 1.Kitchen D, Decornez H, Furr J, Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Z, Felts A, Friesner R, Levy R. Comparative performance of several flexible docking programs and scoring functions: Enrichment studies for a diverse set of pharmaceutically relevant targets. J Chem Inf Model. 2007;47:1599–1608. doi: 10.1021/ci7000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pauli I, Timmers L, Caceres R, Soares M, de Azevedo W. In silico and in vitro: Identifying new drugs. Curr Drug Targets. 2008;9:1054–1061. doi: 10.2174/138945008786949397. [DOI] [PubMed] [Google Scholar]
- 4.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 5.Abagyan R, Totrov M, Kuznetsov D. ICM: A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- 6.Friesner R, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 7.Verdonk M, Cole J, Hartshorn M, Murray C, Taylor R. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 8.Morris G, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 9.McCammon J. Target flexibility in molecular recognition. Biochim Biophys Acta. 2005;1754:221–224. doi: 10.1016/j.bbapap.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 10.Bastard K, Prévost C, Zacharias M. Accounting for loop flexibility during protein-protein docking. Proteins. 2006;62:956–969. doi: 10.1002/prot.20770. [DOI] [PubMed] [Google Scholar]
- 11.Totrov M, Abagyan A. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr Opin Struct Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 13.Joseph-McCarthy D, Baber J, Feyfant E, Thompson D, Humblet C. Lead optimization via high-throughput molecular docking. Curr Opin Drug Discov Devel. 2007;10:264–274. [PubMed] [Google Scholar]
- 14.Brady G. Fast prediction and visualization of protein binding pockets with pass. J Comput Aided Mol Des. 2000;14:383–401. doi: 10.1023/a:1008124202956. [DOI] [PubMed] [Google Scholar]
- 15.Huang B, Schroeder M. Ligsitecsc: Predicting protein binding sites using the Connolly surface and degree of conservation. BMC Struct Biol. 2006;6:19. doi: 10.1186/1472-6807-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.An J, Totrov M, Abagyan R. Comprehensive identification of druggable protein ligand binding sites. Genome Inform. 2004;15:31–41. [PubMed] [Google Scholar]
- 17.Buschiazzo A, et al. Crystal structure, catalytic mechanism, and mitogenic properties of trypanosoma cruzi proline racemase. Proc Natl Acad Sci USA. 2006;103:1705–1710. doi: 10.1073/pnas.0509010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shan Y, et al. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc Natl Acad Sci USA. 2009;106:139–144. doi: 10.1073/pnas.0811223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freedman T, et al. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc Natl Acad Sci USA. 2006;103:16692–16697. doi: 10.1073/pnas.0608127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunasekaran C, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 21.Tsai C, del Sol A, Nussinov R. Allostery: Absence of a change in shape does not imply that allostery is not at play. J Mol Biol. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savir Y, Tlusty T. Conformational proofreading: The impact of conformational changes on the specificity of molecular recognition. PLoS One. 2007;5:e468. doi: 10.1371/journal.pone.0000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee G, Craik C. Trapping moving targets with small molecules. Science. 2009;324:213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blondel A, Renaud J, Fischer S, Moras D, Karplus M. Retinoic acid receptor: A simulation analysis of retinoic acid binding and the resulting conformational changes. J Mol Biol. 1999;291:101–115. doi: 10.1006/jmbi.1999.2879. [DOI] [PubMed] [Google Scholar]
- 25.Soelaiman S, et al. Structure-based inhibitor discovery against adenylyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J Biol Chem. 2003;278:25990–25997. doi: 10.1074/jbc.M301232200. [DOI] [PubMed] [Google Scholar]
- 26.Shen Y, et al. Selective inhibition of Anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc Natl Acad Sci USA. 2004;101:3242–3247. doi: 10.1073/pnas.0306552101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee Y, Bergson P, He W, Mrksich M, Tang W. Discovery of a small molecule that inhibits the interaction of Anthrax edema factor with its cellular activator, calmodulin. Chem Biol. 2004;11:1139–1146. doi: 10.1016/j.chembiol.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 28.Chen D, et al. Novel inhibitors of Anthrax edema factor. Bioorg Med Chem. 2008;16:7225–7233. doi: 10.1016/j.bmc.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drum C, et al. Structural basis for the activation of Anthrax adenylyl cyclase exotoxin by calmodulin. Nature. 2002;415:396–402. doi: 10.1038/415396a. [DOI] [PubMed] [Google Scholar]
- 30.Laine E, Yoneda J, Blondel A, Malliavin T. The conformational plasticity of calmodulin upon calcium complexation gives a model of its interaction with the oedema factor of Bacillus anthracis. Proteins. 2008;71:1813–1829. doi: 10.1002/prot.21862. [DOI] [PubMed] [Google Scholar]
- 31.Laine E, Blondel A, Malliavin T. Dynamics and energetics: A consensus analysis of the impact of calcium on EF-CaM protein complex. Biophys J. 2009;96:1249–1263. doi: 10.1016/j.bpj.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo Q, et al. Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 2005;24:3190–3201. doi: 10.1038/sj.emboj.7600800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ladant D, et al. Characterization of the calmodulin-binding and of the catalytic domain of Bordetella pertussis adenylate cyclase. J Biol Chem. 1989;264:4015–4020. [PubMed] [Google Scholar]
- 34.Fischer S, Karplus M. Conjugate peak refinement: An algorithm for finding reaction paths and accurate transition states in systems with many degrees of freedom. Chem Phys Lett. 1992;194:252–261. [Google Scholar]
- 35.Schaefer M, Karplus M. A comprehensive analytical treatment of continuum electrostatics. J Phys Chem. 1996;100:1578–1599. [Google Scholar]
- 36.Schaefer M, Bartels C, Karplus M. Solution conformations and thermodynamics of structured peptides: Molecular dynamics simulation with an implicit solvation model. J Mol Biol. 1998;284:835–848. doi: 10.1006/jmbi.1998.2172. [DOI] [PubMed] [Google Scholar]
- 37.Schaefer M, Bartels C, Leclerc F, Karplus M. Volumes for implicit solvent models: Comparison between Voronoi volumes and minimum fluctuation volumes. J Comput Chem. 2001;22:1857–1879. doi: 10.1002/jcc.1137. [DOI] [PubMed] [Google Scholar]
- 38.Shen Y, Zhukovskaya N, Guo Q, Florin J, Tang W. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of Anthrax edema factor. EMBO J. 2005;24:929–941. doi: 10.1038/sj.emboj.7600574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyman E, Zuckerman D. Ensemble-based convergence analysis of biomolecular trajectories. Biophys J. 2006;91:164–172. doi: 10.1529/biophysj.106.082941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.2008. Chimiothèque nationale du cnrs, http://chimiotheque-nationale.enscm.fr.
- 41.Rarey M. 2007. FlexX release 2.1 User Guide, Technical report, BioSolveIt.
- 42.Abagyan R. ICM Manual v.3.1. La Jolla, CA: Molsoft LLC; 2005. [Google Scholar]
- 43.Shen Y, et al. Physiological calcium concentrations regulate calmodulin binding and catalysis of adenylyl cyclase exotoxins. EMBO J. 2002;21:6721–6732. doi: 10.1093/emboj/cdf681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vertessy B, et al. Simultaneous binding of drugs with different chemical structures to Ca2+-calmodulin: Crystallographic and spectroscopic studies. Biochemistry. 1998;37:15300–15310. doi: 10.1021/bi980795a. [DOI] [PubMed] [Google Scholar]
- 45.Hibert M. French/European academic compound library initiative. Drug Discov Today. 2009;14:723–725. doi: 10.1016/j.drudis.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 46.Feng B, Shelat A, Doman T, Guy R, Shoichet B. High-throughput assays for promiscuous inhibitors. Nature Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 47.Bolhuis P, Chandler D, Dellago C, Geissler P. Transition path sampling: Throwing ropes over dark mountain passes. Ann Rev Phys Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 48.Laio A, Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci USA. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu X, Brooks B. Self-guided Langevin dynamics simulation method. Chem Phys Lett. 2003;381:512–518. [Google Scholar]
- 50.Maragliano L, Fischer A, Vanden-Eijnden E, Ciccotti G. String method in collective variables: Minimum free energy paths and isocommittor surfaces. J Chem Phys. 2006;125:024106. doi: 10.1063/1.2212942. [DOI] [PubMed] [Google Scholar]
- 51.Guo Q, et al. Protein-protein docking and analysis reveal that two homologous bacterial adenylyl cyclase toxins interact with calmodulin differently. J Biol Chem. 2008;283:23836–23845. doi: 10.1074/jbc.M802168200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ulmer T, et al. Calcium dependence of the interaction between calmodulin and Anthrax edema factor. J Biol Chem. 2003;278:29261–29266. doi: 10.1074/jbc.M302837200. [DOI] [PubMed] [Google Scholar]
- 53.Jin Liu, Ruth Nussinov. Allosteric effects in the marginally stable von Hippel-Lindau tumor suppressor protein and allostery-based rescue mutant design. Proc Natl Acad Sci USA. 2008;105:901–906. doi: 10.1073/pnas.0707401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brooks B, et al. CHARMM—A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 55.Lavery R, Sklenar H, Zakrzewska K, Pullman B. The flexibility of the nucleic acids: (II). The calculation of internal energy and applications to mononucleotide repeat DNA. J Biomol Struct Dyn. 1986;3:989–1014. doi: 10.1080/07391102.1986.10508478. [DOI] [PubMed] [Google Scholar]
- 56.Loncharich R, Brooks B, Pastor R. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers. 1992;32:523–535. doi: 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.