

SUMMARY
Endogenous neurotensin (NT) has been implicated in brain processes relevant to schizophrenia as well as the therapeutic effects of antipsychotic drugs (APDs) used to treat this disorder. Converging evidence suggests that NT1 receptors mediate the antipsychotic-like effects of NT, such as prepulse inhibition (PPI) elevation. However, the role of NT2 receptors in these effects is not known. To investigate the contribution of NT2 receptors to the regulation of PPI, we measured baseline PPI and acoustic startle response (ASR), in male and female wild type (WT) and NT2 knockout (KO) mice. For comparison, we also measured locomotor activity.
Baseline PPI was significantly elevated in both male (P < 0.01) and female (P < 0.01) NT2 KO compared to WT mice, while ASR was significantly decreased in KO mice of both genders (P < 0.01). In contrast, female but not male KO mice exhibited significantly less baseline ambulations (P < 0.05).
These data support the regulation of baseline PPI, ASR and locomotor activity by endogenous NT acting at the NT2 receptor. Further studies investigating the role of NT2 receptors in the modulation of APD-like effects are warranted.
Keywords: schizophrenia, neurotensin, animal model, prepulse inhibition, antipsychotic
INTRODUCTION
Neurotensin (NT) is a tridecapeptide that is highly expressed in the peripheral and the central nervous systems (CNS). NT acts as a neurotransmitter in the CNS modulating DA, serotonin, glutamate and acetylcholine neurotransmission [1]. It also regulates many biological processes including hypotension, hypothermia, analgesia, cell proliferation [2], and neural processes of relevance to behavior and cognition [3]. In addition, converging evidence supports a role for NT in a number of neuropsychiatric conditions, such as schizophrenia [4]. Decreases in the levels of endogenous NT has been associated with the symptoms of schizophrenia suggesting that NT may also exert antipsychotic-like effects on brain processes relevant to this brain disorder [5]. Evidence also suggests that some of the therapeutic effects of antipsychotic drugs may be mediated by increases in endogenous NT [6], and exogenous NT and NT agonists have also been shown to produce antipsychotic-like effects in preclinical models [7–13].
Three NT receptor subtypes, NT1, NT2 and NT3 have been identified and there is evidence that additional subtypes exist [14,15]. NT1 and NT2 are high and low affinity G-protein coupled receptors (GPCRs), respectively. The NT3 receptor contains a single transmembrane domain and has not been as extensively studied [16].
NT1 has been the most studied NT receptor subtype and to date it has been implicated in most of the established central effects of NT [17]. Although it is distributed throughout the brain [18,19], NT2 has not been well studied, in part due to the lack of pharmacological probes selective for NT2. Recent investigations using NT2 receptor knockout (KO) mice have begun to shed light on the role of NT2. Yamauchi et al. [20] found that NT2 KO mice exhibit deficits in fear memory and [2] reported that they exhibit abnormalities in thermal nociception compared to WT mice.
PPI describes the normal suppression of the startle reflex, when a startle-eliciting stimulus, such as a sudden loud noise, is immediately preceded by a weak lead stimulus, such as a soft auditory click. PPI is considered an operational measure of sensorimotor gating, a brain-based process of filtering irrelevant environmental information [21,22]. Reduced PPI is associated with schizophrenia and several other neuropsychiatric conditions [21,23] and this deficit has been linked to cognitive abnormalities associated with the disorder.
NT appears to play a role in regulating PPI as NT and NT agonists attenuate PPI deficits produced pharmacologically or by naturally occurring mutations [6,8]. Moreover, mice lacking NT due to genetic alteration (NT KO mice) exhibit lower basal PPI compared to WT suggesting endogenous NT regulates the tone of PPI [24]. Activation of NT1 receptors appears to enhance PPI as evidenced by the ability of NT1 selective agonists to increase PPI in rats and mice [7,8,25]. However, surprisingly we found PPI levels did not differ between NT1 KO and WT mice suggesting that NT1 may not play an important role in the regulation of baseline PPI levels [25]. The role of NT2 receptors in sensorimotor gating has not been well studied. Therefore, we investigated baseline PPI, acoustic startle in WT mice and in NT2 receptor KO mice. For comparison, we also measured baseline locomotor activity.
MATERIALS AND METHODS
Generation of NT2 knockout mice
Generation of NT2 deficient mice was carried out at Roche Pharmaceuticals, Palo Alto, CA. BAC clones were obtained from a mouse 129/Ola genomic library (Incyte) using a probe corresponding to the first coding exon. To generate a null allele for the mouse NT2 gene, two overlapping genomic DNA fragments (8.85 kb KpnI and 7.5 kb SpeI) were subcloned and sequenced. A targeting vector was created from this DNA fragment such that it includes a 2.53 kb StuI-SpeI fragment as the 5′ homology arm, a Lox P flanked neomycin resistance gene (neo) for positive selection, a 4.67 kb EcoRI-NheI fragment as the 3′homology arm, and the HSV-TK (herpes simplex virus-thymidine kinase) gene for negative selection (Figure 1A). This vector was linearized with NotI and introduced into 129/Ola-derived E14-1 embryonic stem cells by electroporation. Cells were cultured in media containing 310 μg/ml G418 (Life Technologies) and 2 μM gancyclovir (Roche). Twelve independent ES clones with homologous recombination were identified by Southern hybridization using a 5′ external probe on genomic DNA digested with PstI and were confirmed using a 3′ external probe and a neo probe. Homologous recombination resulted in the deletion of a 2.1 kb NT2 genomic sequence, including the ATG-containing exon that encodes the N-terminal 208 amino acids. Two of the ES clones were injected into C57BL/6J (Jackson Laboratory) blastocysts to generate chimeric mice. Germline transmission was achieved by mating the male chimeras with female C57Bl/6J mice. NT2 KO mice were derived from NT2 HET × NT2 HET intercrosses. The reduction or absence of NT2 expression in the NT2 HET or NT2 KO mice, respectively, was verified by RT-PCR (Figure 1B and see below) and Northern hybridization (Figure 1C and see below). The expression of NT1 appeared normal in the NT2 KO mice based on RT-PCR (data not shown). No significant gross or microscopic differences were observed between NT2 WT and NT2 KO animals. In addition, both the male and female NT2 KO mice were viable and fertile.
Figure 1.
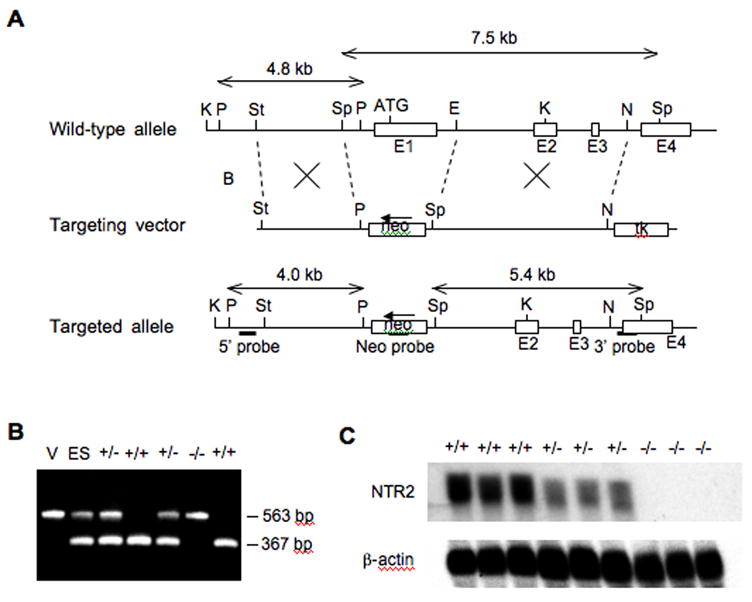
Generation of NT2 knockout mice. (A) Gene targeting strategy. Restriction maps of the wild-type NT2 genomic allele, the targeting vector, and the disrupted allele after homologous recombination. Restriction endonuclease cutting sites are: B, BamHI; E, EcoRI; K, KpnI; N, NheI; P, PstI; Sp, SpeI; St, StuI. E1, E2, E3, E4: the first, second, third and fourth exons. Selective markers: neo, neomycin-resistance gene; tk, thymidine kinase gene. Arrow indicates the transcription direction of neo. The 5′ probe was used to screen for homologous recombinant ES clones while the 3′ probe and the neo probe were used to verify positive clones by Southern blot. (B) Genotyping of the F2 offspring of F1 NT2 +/− × NT2 +/− crosses by PCR. The 367 bp and 563 bp bands are indicative of the wild type and the targeted alleles, respectively. V: vector DNA control. ES: ES DNA. (C) Analysis of NT2 expression in the F2 mice by Northern blot. Genotypes: +/+, homozygous wild-type; +/−, heterozygote; −/−, homozygous knockout.
To produce a purer genetic background for behavioral evaluations, four additional backcrosses to C57Bl/6J were performed. In this respect, five back crosses produces approximately 96 % pure C57Bl/6J background [26]. The N5 NT2 KO and WT control mice were used in subsequent experiments.
RT-PCR and Northern Blot (Brain expression)
Total RNA was extracted from the brains using the TRIzol Reagent (Life Technologies) according to the manufacturer’s instruction. RT-PCR was performed using the SuperScript One-Step RT-PCR System (Life Technologies) and gene-specific primers, including primer 1 (5′-TGA CCG CTG TAT ACC TGG CAC TTT TTG T) and primer 2 (5′-ATG GCC AGC AGC AGG ATG AGC AGG TC) for NT1 and primer 3 (5′-GCG TGC CCA TGG AGC TCT ACA ACT TC) and primer 4 (5′-AGA CCT GGA GCT GGC GCG ACT TAC TA) for NT2. As an internal control, glyceraldehyde 3-phosphate dehydrogenase was amplified. To analyze gene expression by Northern blot, 20 μg of total RNA was separated on a 1% agarose gel containing 6.6% of formaldehyde, transferred onto Nytran membranes (Schleicher & Schuell) and hybridized with 32P-labeled cDNA probes in ExpressHyb hybridization solution (Clontech) at 68°C for 18 hours. The blots were first hybridized with a NT2 probe, corresponding to the first coding exon, and then re-probed for the β-actin transcripts Figure 1C.
In addition, routine genotyping was conducted by PCR on purified tail DNA (DNeasy Kit, QIAGEN) using two NT2-specific primers (5′-GTCCATTCCCCACCTCAGAAG-3′, 5′-GCACCCTCCTGGTATCACACTG-3′) and a neo primer (5′-CCT TCT TGACGAGTTCTTCTGAG-3′). PCR was performed under the following conditions: incubation at 94°C for 2 min, 35 cycles of 94°C for 30 sec, 58°C for 30 sec and 68°C for 1 min, and a final extension at 68°C for 2 min. The expected sizes for the WT and KO alleles were 367 bp and 563 bp, respectively.
Animals
Fifty eight male and 57 female WT, and 36 male and 31 NT2 KO female mice were bred at the UCSD breeding facility, San Diego and housed in groups of 2 – 5 in clear plastic chambers in a climate controlled room on 12:12 hour light/dark cycle (lights on 7:00 AM– 7:00 PM). They were allowed free access to food and water for the extent of the study. All mice were 3–4 months old at testing. Behavioral testing was performed between 8:30 AM and 3:00 PM. All studies described in this publication were carried out in accordance with The Declaration of Helsinki and/or with “Principles of laboratory animal care” (NIH publication No. 86-23, revised 1985) as adopted and promulgated by the National Institutes of Health.
Animal Testing
Behavioral testing was conducted at UCSD. Male and female WT and NT2 KO mice were placed in locomotor chambers for 30 minutes and ambulations measured. They were then immediately placed in startle chambers and tested for baseline PPI and ASR. Baseline PPI data was greater than three standard deviations from the mean for one female KO mouse and baseline locomotor data was greater than three standard deviations from the mean for one female WT mouse and these date were eliminated from baseline analysis.
Startle Testing
Startle testing was performed in four identical startle chambers obtained from San Diego Instruments (San Diego, CA). Each chamber consisted of a clear non-restrictive Plexiglass cylinder resting on a Plexiglass platform inside a ventilated and illuminated enclosure housed in a sound-attenuated room. A continuous background noise of 65 dB, as well as the various acoustic stimuli were produced within each chamber by a high-frequency loudspeaker (Radio Shack Supertweeter, San Diego, CA). The whole-body startle response of each animal produced vibrations of the Plexiglass cylinder, which were transduced into analog signals by a piezoelectric unit, mounted underneath the Plexiglass platform [27]. These analog signals were then digitized and stored by an interface unit connected to a microcomputer. Startle amplitude was defined as the degree of motion detected by the piezoelectric unit.
Animals were tested in startle chambers, where they were subjected to a 10-minute acclimation to the 65 dB background noise, which continued throughout the session. The acclimation was followed by a 14 minute PPI test session. Five trial types were presented during the test session: a 40 msec 120 dB startle pulse (PULSE-ALONE), a 20 msec prepulse (4, 8 or 16 dB above background) preceded the PULSE-ALONE by 100 msec, and a NO-STIMULUS trial. All trial types were presented 10 times in pseudo-random order separated by an average of 15 seconds. In addition, five PULSE-ALONE trials that were not used in the calculation of PPI values were presented at the beginning and at the end of the test session [28].
Locomotor Testing
Locomotor activity testing was performed in twelve identical locomotor boxes obtained from San Diego Instruments (San Diego, CA). Each box was enclosed in a frame that contained eight beams to measure horizontal activity. Two consecutive beam brakes was used to define an ambulation. Animals were placed in boxes and locomotor activity measured for 30 minutes.
Data and Statistical Analysis
A startle response was recorded for all the PULSE-ALONE, and all prepulse trials. From these data, PPI measures and startle magnitude were calculated for each animal. PPI was calculated as a percentage of the pulse-alone startle magnitude using the following formula: [1− (startle magnitude after prepulse-pulse pair/startle magnitude after pulse only] × 100.
A two-factor ANOVA was used to analyze startle with Gender and Genotype as a between subjects factors. We decided a priori to analyze and display male and female results separately. For PPI, Prepulse Intensity was included as a within-subjects factor (three-Factor ANOVA). Significant effects were followed by post hoc t-tests. Locomotor activity was analyzed by a three-factor ANOVA with Gender, Genotype and Time (0 – 10, 10 –20 and 20 – 30 minutes after entering the cage) as between subjects factors. Locomotor activity was also collapsed across the 3 times points providing the total number of ambulations for the entire 30-minute period for each animal. These data were analyzed by two-way ANOVA (Gender and Genotype as between subject factors). Independent t-tests were used to compare ambulations in WT and KO mice in males and females at each of the three time points and for total ambulations. Bonferroni method was used to correct for multiple comparisons.
RESULTS
Baseline PPI
There was a significant main effect of Genotype [F(1,175) = 29.750, P < 0.001], Prepulse Intensity [F(2,350) = 309.524, P < 0.001] and a significant Prepulse Intensity × Genotype Interaction [F(2,350) = 5.994, P < 0.01]. Male KO mice exhibited significantly higher PPI at all three prepulses (pp4 [t(85.997) = −3.546, P < 0.01]; pp8 [t(88.145) = −5.097, P < 0.01; pp16 [t(89.878) = −4.456, P < 0.01]. Female KO mice exhibited significantly higher PPI than WT at pp8 [t(83.916) = −5.044, P < 0.01] and pp16 [t(83.849) = −3.808, P < 0.01] (Figure 2). In contrast, there was no significant difference in female KO PPI levels compared to WT mice at pp4. No other interaction was significant.
Figure 2.
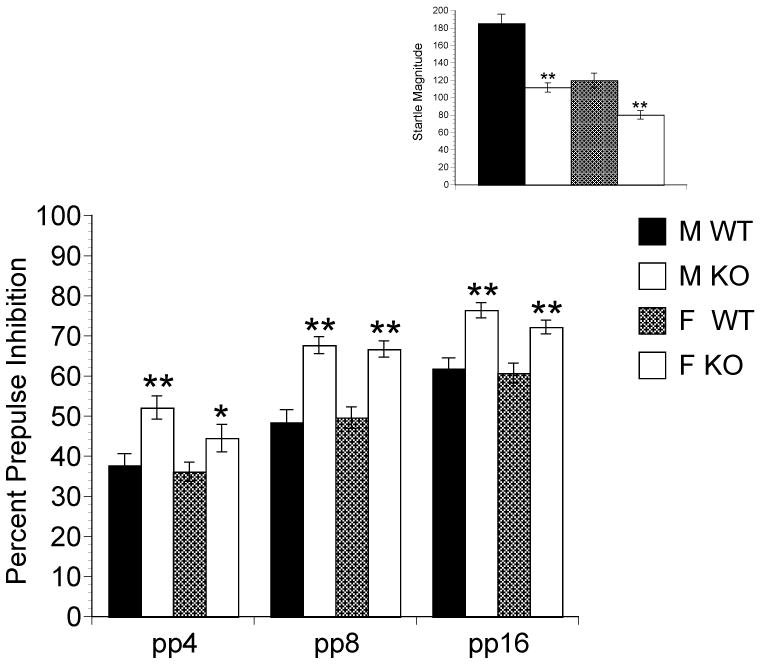
Baseline PPI (Main) at pp4, pp8 and pp16, and startle magnitude (Inset) in WT and NT2 KO mice. NT2 KO mice significantly different from WT mice represented by *P < 0.05, **P < 0.01.
Baseline ASR
There was a main effect of Genotype F[(1,178) = 32.856, P < 0.001] and Gender F[(1,178) =9.550, P < 0.01]. Male [t(67.588)= 6.286, P < 0.001], and female [t(79.969)= 3.674, P < 0.001] KO mice exhibited significantly lower ASR compared to their WT counterparts and this difference was larger in the males (Figure 2 Inset). There were no other significant interactions.
Baseline locomotor activity
The was a main effect of Gender F[(1, 172) = 5.686, 5.686, P < 0.05], Genotype F[(1, 172) = 6.270, P < 0.05] and Time F[(2, 344) = 435.827, P < 0.001]. There were no significant interactions. After correction for multiple comparisons, there were no significant differences in between WT and KO mice (male and female) at any of the three time points. In regards to the collapsed locomotor data, there was a main effect of Gender [F(1,175) = 6.141, P < 0.005] and Genotype [F(1,175) = 6.276, P < 0.01]. Female but not male KO mice exhibited significantly less ambulations than WT mice during the thirty minute session, [t (81.491) = 2.166, P < 0.05], [t (65.994)= 1.640, NS], respectively (Figure 3).
Figure 3.
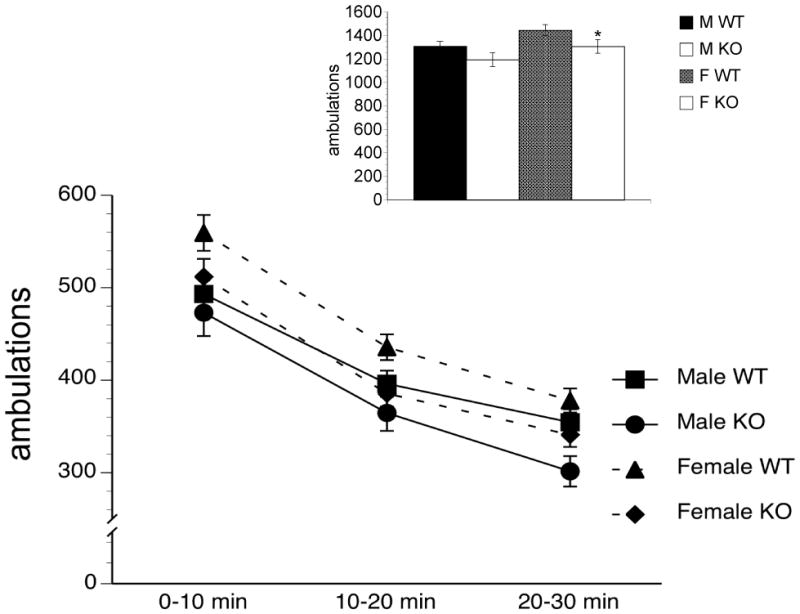
Baseline ambulations in WT and NT2 KO mice, 0 – 10, 10 – 20 and 20 – 30 minutes after entering the cage (Main) and total baseline ambulations during this 30 minute time period (Inset). NT2 KO mice significantly different from WT mice represented by *P < 0.05.
DISCUSSION
We discovered that baseline PPI is elevated in the NT2 KO males and females compared to the WT mice. These data are consistent with endogenous NT regulating PPI by activating the NT2 receptor. To the best of our knowledge this is the first report on the effects of a NT2 deletion on PPI and startle magnitude. We recently reported that PPI did not differ between NT1 KO and WT mice suggesting that endogenous NT affects baseline PPI via another NT receptor, e.g., NT2, consistent with the data presented here. However, pharmacological doses of NT1 [7–13] agonists facilitate PPI. Therefore, it appears that endogenous and exogenous NT may regulate PPI via different mechanisms. In addition, baseline startle was significantly lower in the NT2 KO mice also suggesting a role for the NT2 receptor in the regulation of ASR.
Female but not male NT2 KO mice exhibited significantly lower basal locomotor activity compared to their WT counterparts. However, male KO mice exhibited a strong trend in the same direction. These data for the male KO mice are consistent with those of Yamauchi et al. [20] who reported that male NT2 KO mice did not exhibit altered spontaneous locomotor activity. In this respect, they detected a non-significant 13% decrease in locomotor activity in KO vs. WT mice, which is almost identical to the 9% decrease that we detected. In contrast, Liang et al. [29] recently reported that NT2 KO mice exhibit higher basal locomotion compared to WT mice. The difference in the results between the Liang study and the previously described results is difficult to reconcile but may be due to different environmental conditions in animal facilities.
A recent study reported that a NT2 selective agonist had no effect on baseline PPI but reversed amphetamine-induced hyperactivity and disruption of PPI, an antipsychotic-like effect, similar to that produced by NT1 activation [30]. These results are not easily reconciled with our findings from which we would predict that a NT2 selective agonist might reduce PPI. However, Boules’ et al. results were in rats and Ralph et al. have reported that in some cases, such as the effects of D2 agonists on locomotor activity, mice and rats exhibit different behavioral responses to the same drugs [31]. Another plausible explanation is the differences in the results we observed are an outcome of perturbation of normal development resulting from congenital absence of NT2 receptors. For example, Li et al. [32] reported that NT2 KO mice also have significantly lower levels of striatal glutamate compared to WT mice.
In summary, previous NT2 KO studies have supported a role for NT2 receptors in processing thermal nociception and fear memory. The findings presented in this report suggest that activation of NT2 receptors regulate additional neural mechanisms underlying behaviors relevant to psychiatric disorders. In this respect, we have provided evidence for NT2 receptor regulation of baseline PPI, ASR, and locomotor activity. Future studies are necessary to dissect the role of NT2 receptors in the mechanisms underlying the action of antipsychotic drugs.
Acknowledgments
DF and PDS were partially supported by NIMH grant MH080910.
Footnotes
DISCLOSURES
In the past three years, Dr. Feifel has received funds in return for one or more of the following: conducting contracted research, conducting investigator-initiated research, advisory board participation, speaking from the following pharmaceutical companies: Pharmaceutical Company (Product) Abbott Labs (Divalproex/Depakote), Astra Zeneca (Quietiapine/Seroquel), Bristol Myers Sqibb (Aripiprazole/Abilify), Danipon-Sumitomo (Lurasidone), Eli Lilly (Atomextine/Strattera & Duloxetine/Cymbalta), Janseen (Risperidone/Risperdal & long acting risperidone/Consta & Paliperidone/Invega), Shire (Adderall XR/Mixed amphetamine salts & Intuniv), Wyeth (Venlafaxine/Effexor XT & Desvenlafaxine/Pristiq), Shering-Plough (asenapine/Saphris). ZP is an employee of Sanofi-Aventis, a pharmaceutical company engaged in discovery, development, and distribution of therapeutic solutions. PDS and GM are employees of the University of California, San Diego. RS is an employee of Sepracor. DB has no conflict of interest. He was involved with the work reported in this manuscript while employed as a Research Scientist with the CNS Therapy Area, Department of Neurobehavior and Neuropharmacology, Roche Palo Alto, LLC, Palo Alto, CA, 94304. He is currently employed as Director of Laboratory Research at Acorda Therapeutics, Hawthorne, NY
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.St-Gelais F, Jomphe C, Trudeau LE. The role of neurotensin in central nervous system pathophysiology: what is the evidence? J Psychiatry Neurosci. 2006;31:229–245. [PMC free article] [PubMed] [Google Scholar]
- 2.Maeno H, Yamada K, Santo-Yamada Y, Aoki K, Sun YJ, Sato E, et al. Comparison of mice deficient in the high- or low-affinity neurotensin receptors, Ntsr1 or Ntsr2, reveals a novel function for Ntsr2 in thermal nociception. Brain Res. 2004;998:122–129. doi: 10.1016/j.brainres.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 3.Tirado-Santiago G, Lazaro-Munoz G, Rodriguez-Gonzalez V, Maldonado-Vlaar CS. Microinfusions of neurotensin antagonist SR 48692 within the nucleus accumbens core impair spatial learning in rats. Behav Neurosci. 2006;120:1093–1102. doi: 10.1037/0735-7044.120.5.1093. [DOI] [PubMed] [Google Scholar]
- 4.Manberg PJ, Nemeroff CB, Iversen LL, Rosser MN, Kizer JS, Prange AJ., Jr Human brain distribution of neurotensin in normals, schizophrenics, and Huntington’s choreics. Ann N Y Acad Sci. 1982;400:354–367. doi: 10.1111/j.1749-6632.1982.tb31581.x. [DOI] [PubMed] [Google Scholar]
- 5.Binder EB, Kinkead B, Owens MJ, Nemeroff CB. The role of neurotensin in the pathophysiology of schizophrenia and the mechanism of action of antipsychotic drugs. Biol Psychiatry. 2001;50:856–872. doi: 10.1016/s0006-3223(01)01211-2. [DOI] [PubMed] [Google Scholar]
- 6.Binder EB, Kinkead B, Owens MJ, Kilts CD, Nemeroff CB. Enhanced neurotensin neurotransmission is involved in the clinically relevant behavioral effects of antipsychotic drugs: evidence from animal models of sensorimotor gating. J Neurosci. 2001;21:601–608. doi: 10.1523/JNEUROSCI.21-02-00601.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feifel D, Melendez G, Priebe K, Shilling PD. The effects of chronic administration of established and putative antipsychotics on natural prepulse inhibition deficits in Brattleboro rats. Behav Brain Res. 2007;181:278–286. doi: 10.1016/j.bbr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 8.Feifel D, Melendez G, Shilling PD. Reversal of sensorimotor gating deficits in Brattleboro rats by acute administration of clozapine and a neurotensin agonist, but not haloperidol: a potential predictive model for novel antipsychotic effects. Neuropsychopharmacology. 2004;29:731–738. doi: 10.1038/sj.npp.1300378. [DOI] [PubMed] [Google Scholar]
- 9.Feifel D, Reza TL, Wustrow DJ, Davis MD. Novel antipsychotic-like effects on prepulse inhibition of startle produced by a neurotensin agonist. Journal of Pharmacology and Experimental Therapeutics. 1999;288:710–713. [PubMed] [Google Scholar]
- 10.Shilling PD, Melendez G, Priebe K, Richelson E, Feifel D. Neurotensin agonists block the prepulse inhibition deficits produced by a 5-HT2A and an alpha1 agonist. Psychopharmacology (Berl) 2004;175:353–359. doi: 10.1007/s00213-004-1835-5. [DOI] [PubMed] [Google Scholar]
- 11.Shilling PD, Richelson E, Feifel D. The effects of systemic NT69L, a neurotensin agonist, on baseline and drug-disrupted prepulse inhibition. Behav Brain Res. 2003;143:7–14. doi: 10.1016/s0166-4328(03)00037-8. [DOI] [PubMed] [Google Scholar]
- 12.Boules M, Fredrickson P, Richelson E. Neurotensin agonists as an alternative to antipsychotics. Expert Opin Investig Drugs. 2005;14:359–369. doi: 10.1517/13543784.14.4.359. [DOI] [PubMed] [Google Scholar]
- 13.Boules M, Shaw A, Fredrickson P, Richelson E. Neurotensin agonists: potential in the treatment of schizophrenia. CNS Drugs. 2007;21:13–23. doi: 10.2165/00023210-200721010-00002. [DOI] [PubMed] [Google Scholar]
- 14.Li JH, Sicard F, Salam MA, Baek M, LePrince J, Vaudry H, et al. Molecular cloning and functional characterization of a type-I neurotensin receptor (NTR) and a novel NTR from the bullfrog brain. J Mol Endocrinol. 2005;34:793–807. doi: 10.1677/jme.1.01709. [DOI] [PubMed] [Google Scholar]
- 15.Mazella J, Vincent JP. Functional roles of the NTS2 and NTS3 receptors. Peptides. 2006;27:2469–2475. doi: 10.1016/j.peptides.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 16.Pelaprat D. Interactions between neurotensin receptors and G proteins. Peptides. 2006;27:2476–2487. doi: 10.1016/j.peptides.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 17.Kinkead B, Nemeroff CB. Novel treatments of schizophrenia: targeting the neurotensin system. CNS Neurol Disord Drug Targets. 2006;5:205–218. doi: 10.2174/187152706776359655. [DOI] [PubMed] [Google Scholar]
- 18.Lépée-Lorgeoux I, Betancur C, Rostène W, Pélaprat D. Differential ontogenetic patterns of levocabastine-sensitive neurotensin NT2 receptors and of NT1 receptors in the rat brain revealed by in situ hybridization. Brain Research Developmental Brain Research. 1999;113:115–131. doi: 10.1016/s0165-3806(99)00009-7. [DOI] [PubMed] [Google Scholar]
- 19.Sarret P, Perron A, Stroh T, Beaudet A. Immunohistochemical distribution of NTS2 neurotensin receptors in the rat central nervous system. J Comp Neurol. 2003;461:520–538. doi: 10.1002/cne.10718. [DOI] [PubMed] [Google Scholar]
- 20.Yamauchi R, Wada E, Kamichi S, Yamada D, Maeno H, Delawary M, et al. Neurotensin type 2 receptor is involved in fear memory in mice. J Neurochem. 2007;102:1669–1676. doi: 10.1111/j.1471-4159.2007.04805.x. [DOI] [PubMed] [Google Scholar]
- 21.Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies [see comments] Archives of General Psychiatry. 1990;47:181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- 22.Swerdlow NR, Geyer MA. Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophrenia Bulletin. 1998;24:285–301. doi: 10.1093/oxfordjournals.schbul.a033326. [DOI] [PubMed] [Google Scholar]
- 23.Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 24.Kinkead B, Dobner PR, Egnatashvili V, Murray T, Deitemeyer N, Nemeroff CB. Neurotensin-deficient mice have deficits in prepulse inhibition: restoration by clozapine but not haloperidol, olanzapine, or quetiapine. J Pharmacol Exp Ther. 2005;315:256–264. doi: 10.1124/jpet.105.087437. [DOI] [PubMed] [Google Scholar]
- 25.Feifel D, Pang Z, Shilling PD, Melendez G, Schreiber R, Button D. Sensorimotor gating in neurotensin-1 receptor null mice. Neuropharmacology. 2009;58:173–178. doi: 10.1016/j.neuropharm.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silver LM. Mouse Genetics: Concepts and Applications. Bar Harbor, Maine: Oxford University Press; 1995. [Google Scholar]
- 27.Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology. 1988;94:507–514. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- 28.Bakshi VP, Geyer MA. Multiple limbic regions mediate the disruption of prepulse inhibition produced in rats by the noncompetitive NMDA antagonist dizocilpine. J Neurosci. 1998;18:8394–8401. doi: 10.1523/JNEUROSCI.18-20-08394.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang Y, Boules M, Li Z, Williams K, Miura T, Oliveros A, et al. Hyperactivity of the dopaminergic system in NTS1 and NTS2 null mice. Neuropharmacology. doi: 10.1016/j.neuropharm.2010.02.015. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boules M, Liang Y, Briody S, Miura T, Fauq I, Oliveros A, et al. NT79: A novel neurotensin analog with selective behavioral effects. Brain Res. 2009;1308:35–46. doi: 10.1016/j.brainres.2009.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ralph RJ, Caine SB. Dopamine D1 and D2 agonist effects on prepulse inhibition and locomotion: comparison of Sprague-Dawley rats to Swiss-Webster, 129X1/SvJ, C57BL/6J, and DBA/2J mice. J Pharmacol Exp Ther. 2005;312:733–741. doi: 10.1124/jpet.104.074468. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Liang Y, Boules M, Gordillo A, Richelson E. Effect of amphetamine on extracellular concentrations of amino acids in striatum in neurotensin subtype 1 and 2 receptor null mice: A possible interaction between neurotensin receptors and amino acid systems for study of schizophrenia. Neuropharmacology. doi: 10.1016/j.neuropharm.2010.02.016. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]