

Abstract
 A very elegant Fraser-Reid's armed-disarmed approach recently expanded to the building blocks of the superarmed and superdisarmed series shows very high utility in chemoselective oligosaccharide synthesis. Although a number of studies dedicated to the chemoselective activation of 2-amino-2-deoxysugars have emerged, little remains known how the reactivity of the armed/disarmed building blocks of the neutral sugars directly compare to that of their 2-aminosugar counterparts. A preliminary study of this comparative reactivity is presented.
A very elegant Fraser-Reid's armed-disarmed approach recently expanded to the building blocks of the superarmed and superdisarmed series shows very high utility in chemoselective oligosaccharide synthesis. Although a number of studies dedicated to the chemoselective activation of 2-amino-2-deoxysugars have emerged, little remains known how the reactivity of the armed/disarmed building blocks of the neutral sugars directly compare to that of their 2-aminosugar counterparts. A preliminary study of this comparative reactivity is presented.
The involvement of complex carbohydrates in a wide variety of disease-related cellular processes has given this class of natural compounds tremendous diagnostic and therapeutic potential. While scientists have been able to successfully isolate certain classes of natural polysaccharides and glycoconjugates, the availability of pure natural isolates is still inadequate to address the challenges of modern glycoscience. As a consequence, chemical synthesis has become a viable means to obtain both natural complex carbohydrates and unnatural analogues thereof. However, chemical synthesis of oligosaccharides of even moderate complexity still remains a considerable challenge. As such, the development of efficient methods for expeditious oligosaccharide synthesis remains a demanding area of research.1
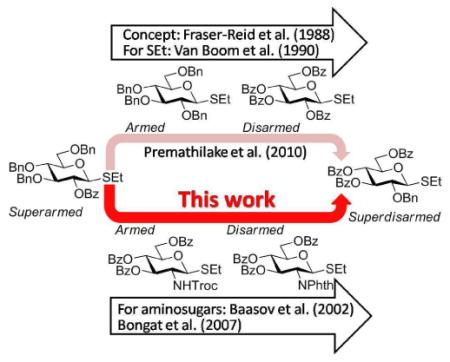
Fraser-Reid's rationalization of the fact that “protecting groups do more than protect”2 opened the entire new direction in oligosaccharide synthesis. The chemoselective armed-disarmed approach makes use of only one class of leaving group for both the glycosyl donor and glycosyl acceptor, which is either activated (armed) or deactivated (disarmed), respectively, by the influence of the protecting groups.3,4 Usually, protecting groups in both reaction components have to be taken into consideration. This allows for direct coupling between the armed glycosyl donor over the disarmed glycosyl acceptor in the presence of a suitable promoter. The disaccharide obtained can then be used for subsequent direct glycosylation in the presence of a more powerful promoter capable of activating the disarmed leaving group. Recently, we expanded the scope of the classic Fraser-Reid's armed-disarmed concept for chemoselective oligosaccharide synthesis by developing a series of building blocks of the superarmed5 and superdisarmed series for sequential activation.6 This discovery was based on the phenomenon that we call the O-2/O-5 cooperative effect,7 and the superarming/disarming was achieved by simple strategic placement of protecting groups. Other approach to superarming using conformationally modified derivatives was introduced by Bols et al.8-10
While the armed - disarmed concept has been developed in application to a broad range of neutral sugar derivatives,1 significantly less information has been acquired with the building blocks of the 2-amino-2-deoxy series.11 While a significant disparity in reaction rates between various aminosugar derivatives have been observed,12-15 no systematic studies have yet become available.16 17 18 Baasov et al19 and more recently our group demonstrated that N-(2,2,2-trichloroethyloxy)carbamoyl (Troc) protection activates (arms) 2-aminosugars in comparison to that of the disarming effect of the N-phthalimido group.20 However, very little remains known how the reactivity of the armed/disarmed building blocks of the aminosugar series compares to that of the corresponding armed/disarmed building blocks of the neutral sugars.
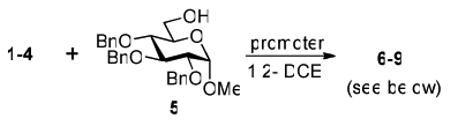
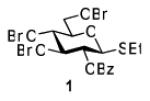
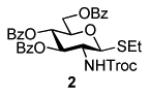
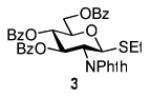
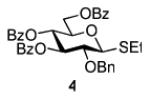
Herein we present our preliminary study focused on the comparison of the differently protected building blocks of the D-gluco and D-glucosamino series. Thioglycosides remain among the most common glycosyl donors and by far the most investigated building blocks in various expeditious strategies including: two-step activation,21 armed-disarmed,22 active-latent,23 orthogonal,24,25 one-pot,26,27 etc.28 Therefore, we chose to base this study on S-ethyl glycosides and for the comparative reactivity studies obtained the superarmed donor 1,6 two N-substituted SEt glycosides (refer to the SI for their synthesis) armed 2 and disarmed 3, as well as the superdisarmed thioglycoside 4 (Figure 1).29
Figure 1.

Glycosyl donors of the armed (1 and 2) and disarmed series (3 and 4).
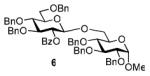
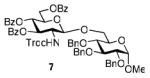
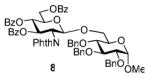
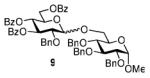
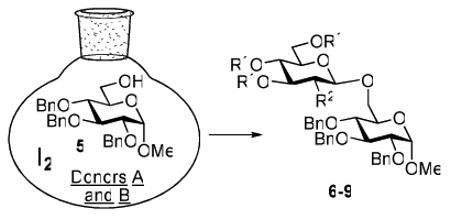
The key requirement for any chemoselective activation to take place is the availability of a suitable promoter that can differentiate between the armed and disarmed building blocks. Therefore, having obtained glycosides 1-4, our next aim was to find a promoter (or promoters) that would be well suited for the activation of each of these glycosyl donors. It is well established that thioglycosides can be activated under a variety of reaction conditions, and the test glycosylations of glycosyl acceptor 530 were initially performed using common promoters. Not unexpectedly, NIS/TfOH was very effective as the promoter, however, these activations were too fast, even at low temperature. As a result, all glycosyl donors reacted at a similar pace providing very good to excellent yields (86-98%) for the formation of disaccharides 6-9 (Table 1, entries 1-4). Subsequently, we chose a significantly milder promoter MeOTf, which was very effective in our previous chemoselective studies with 2-aminosugars.20 Although the differentiation between building blocks of the armed series 1 and 2 in comparison to that of the disarmed series 3 and 4 was notable (2-3 h vs. 6 h, respectively, entries 5-8), and the yields were excellent, we pursued further search of promoters. Recently, we reached success in differentiating superarmed vs. armed building blocks of the D-gluco series.6 The differentiation was particularly efficient in the presence of iodine that was introduced as a mild promoter for thioglycoside activation by Kartha and Field.31 Also herein reactions promoted with iodine (3.3 equiv) showed good differentiation trend ranging from 15 min activation of 1 at rt, 8 h activation of 2 at 50 °C, and activations of 3 and 4 that could not be driven to completion even after 24 h at 50 °C (entries 9-12).
Table 1.
Search for the appropriate promoter for glycosylation reactions.
 | |||||
|---|---|---|---|---|---|
| entry | donor | promoter | time | product | yield (α/β) |
| 1 |
|
NIS/TfOHa | 5 min |
|
98% (β only) |
| 2 |
|
NIS/TfOHa | 5 min |
|
98% (β only) |
| 3 |
|
NIS/TfOHa | 5 min |
|
86% (β only) |
| 4 |
|
NIS/TfOHa | 5 min |
|
89% (1/1) |
| 5 | 1 | MeOTf, rt | 2 h | 6 | 96% (β only) |
| 6 | 2 | MeOTf, rt | 3 h | 7 | 98% (β only) |
| 7 | 3 | MeOTf, rt | 6 h | 8 | 77% (β only) |
| 8 | 4 | MeOTf, rt | 6 h | 9 | 98% (2/1) |
| 9 | 1 | I2, rt | 15 min | 6 | 97% (β only) |
| 10 | 2 | I2, 50 °C | 8 h b | 7 | 72% (β only) |
| 11 | 3 | I2, 50 °C | 24 h b | 8 | 61% (β only) |
| 12 | 4 | I2, 50 °C | 24 h b | 9 | 40% (>19/1) |
no significant difference between reactions at rt (shown) and at −20 °C, but the latter provided better stereoselectivity for the synthesis of 9 (α/β = 2/1, entry 4)
incomplete reaction
We deemed these results sufficient for proof of preliminary differentiation and began studying direct competitive glycosylations of glycosyl donors (1-4) with acceptor 5. These competitive reactions were set up to allow two glycosyl donors compete for the glycosyl acceptor 5 in a single flask. In these experiments, 3.3 equiv. of iodine was used to activate two glycosyl donors (1.3 equiv. each(see Table 2)) over the glycosyl acceptor 5 (1.0 equiv.). As anticipated, the superarmed glycosyl donor 1 outperformed the donor 2: it reacted smoothly and the glycosyl acceptor 5 was entirely consumed within 1 h at rt. As a result, the disaccharide 6 was isolated in 81% yield as the sole product since no trace of disaccharide 7 could be detected and the less reactive glycosyl donor 2 was recovered in 82% yield (entry 1). Subsequently, we investigated relative reactivities of glycosyl donors 2 and 3, and, as expected the armed glycosyl donor 2 outperformed the disarmed donor 3: it reacted smoothly and the glycosyl acceptor 5 was entirely consumed within 24 h at 50 °C. As a result, the disaccharide 7 was isolated in 95% yield as the sole product and no trace of disaccharide 8 was detected (entry 2). The less reactive glycosyl donor 3 was recovered in 67% yield.
Table 2.
Competitive glycosylations using glycosyl donors 1-4.
 | |||||
|---|---|---|---|---|---|
| entry | donors | time | product | s yield | donor recovery |
| 1a | 1 + 2 | 1 h | 6 | 81% | 82% (2) |
| 2 | 2 + 3 | 24 h | 7 | 95% | 67% (3) |
| 3 | 3 + 4 | 24 h | 8+9 | 53%+36% | 49% (4) |
| 4b | 2 + 4 | 9.5 h | 7 | 98% | 87% (4) |
reaction at rt, all others at 50 °C
incomplete reaction
When relative reactivities of glycosyl donors 3 and 4 were compared, both disaccharides 8 and 9 were formed in 53 and 36% yield, respectively, as a result of insufficient reactivity difference of these two classes of disarmed glycosyl donors (entry 3). It should be noted that the reaction was very sluggish and did not go to completion even after 48 h. With the focus of the chemoselective activation of the aminosugars over their neutral counterparts, we also investigated relative reactivities of glycosyl donors 2 and 4, and, to our delight, the aminosugar donor 2 outperformed the disarmed glucosyl donor 4. This reaction proceeded very smoothly, and the glycosyl acceptor 5 was entirely consumed within 9.5 h at 50 °C. As a result, the disaccharide 7 was isolated in 98% yield as the sole product. No formation of disaccharide 9 was detected (entry 4) and the less reactive glycosyl donor 4 was recovered in 87% yield.
The knowledge gained from the competitive glycosylations created a solid foundation for attempting a multi-step synthesis of oligosaccharides with alternating neutral sugar – aminosugar unit, a sequence commonly seen in many natural glycoconjugates and polysaccharides. As verification of the promising results achieved in competition experiments, direct chemoselective activation of the superarmed glycosyl donor 1 over the 2-amino-2-deoxy acceptor 10 (see the SI for the synthesis) was investigated. Initially, when glycosidation of 1 with acceptor 10 was carried out at rt, the requisite disaccharide 11 was isolated in a poor yield (see the SI), and the formation of a number of by-products was noted. However, when the reaction was performed at −20 °C, the disaccharide 11 was isolated in 77% (Scheme 1). As expected, very similar outcome was achieved in reactions with 2-deoxy-2-phthalimido acceptor (see the SI for details). Being encouraged by this promising result, we decided to apply this approach to the synthesis of a trisaccharide. For this purpose, we obtained glycosyl acceptor 12 derived from the superdisarmed glycosyl donor 4 (see the SI for synthesis). Chemoselective activation of the disaccharide donor 11 over superdisarmed acceptor 12 was successful in the presence of iodine at 50°C and the target trisaccharide 13 was isolated in 60% yield.
Scheme 1.

Chemoselective synthesis of trisaccharide 13.
In conclusion, we performed a comparative study of the armed and disarmed building blocks of the D-gluco and glucosamino series. Competitive glycosylations clearly showed the reactivity pattern of the building blocks investigated. The synthesis of trisaccharide 13 performed by a two-step chemoselective activation sequence clearly illustrated the versatility of the developed approach in the context of the synthesis of oligosaccharides with alternating neutral and aminosugar units. It is apparent that the S-ethyl leaving group of the trisaccharide 13 can be directly activated for the subsequent synthesis of larger oligosaccharides. This activation, however, may need to employ a significantly more powerful promoter, such as NIS/TfOH, and therefore is not covered by the scope of this comparative chemoselective activation studies.
Supplementary Material
Acknowledgment
The authors thank NIGMS (GM077170) and AHA (0855743G) for financial support of this work. Dr. Winter and Mr. Kramer (UM - St. Louis) are thanked for HRMS determinations.
Footnotes
Supporting Information Available: Extended experimental data, experimental procedures, 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Smoot JT, Demchenko AV. Adv. Carbohydr. Chem. Biochem. 2009;62:161–250. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]
- 2.Fraser-Reid B, Jayaprakash KN, López JC, Gómez AM, Uriel C. In: ACS Symp. Ser. (Frontiers in Modern Carbohydrate Chemistry) Demchenko AV, editor. Vol. 960. Oxford Univ. Press; 2007. pp. 91–117. [Google Scholar]
- 3.Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J. Am. Chem. Soc. 1988;110:5583–5584. [Google Scholar]
- 4.Fraser-Reid B, Wu Z, Udodong UE, Ottosson H. J. Org. Chem. 1990;55:6068–6070. [Google Scholar]
- 5.Mydock LK, Demchenko AV. Org. Lett. 2008;10:2103–2106. doi: 10.1021/ol800345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Premathilake HD, Mydock LK, Demchenko AV. J. Org. Chem. 2010;75:1095–1100. doi: 10.1021/jo9021474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamat MN, Demchenko AV. Org. Lett. 2005;7:3215–3218. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 8.Jensen HH, Pedersen CM, Bols M. Chem. Eur. J. 2007;13:7576–7582. doi: 10.1002/chem.200700947. [DOI] [PubMed] [Google Scholar]
- 9.Pedersen CM, Nordstrom LU, Bols M. J. Am. Chem. Soc. 2007;129:9222–9235. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen CM, Marinescu LG, Bols M. Chem. Commun. 2008:2465–2467. doi: 10.1039/b801305e. [DOI] [PubMed] [Google Scholar]
- 11.Bongat AFG, Demchenko AV. Carbohydr. Res. 2007;342:374–406. doi: 10.1016/j.carres.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 12.Ellervik U, Magnusson G. Carbohydr. Res. 1996;280:251–260. doi: 10.1016/0008-6215(95)00318-5. [DOI] [PubMed] [Google Scholar]
- 13.Dullenkopf W, Castro-Palomino JC, Manzoni L, Schmidt RR. Carbohydr. Res. 1996;296:135–147. doi: 10.1016/s0008-6215(96)00237-6. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J. Am. Chem. Soc. 1999;121:734–753. [Google Scholar]
- 15.Douglas NL, Ley SV, Lucking U, Warriner SL. J. Chem. Soc., Perkin Trans. 1. 1998:51–65. [Google Scholar]
- 16.Hansson J, Garegg PJ, Oscarson S. J. Org. Chem. 2001;66:6234–6243. doi: 10.1021/jo001302m. [DOI] [PubMed] [Google Scholar]
- 17.Ritter TK, Mong K-KT, Liu H, Nakatani T, Wong C-H. Angew. Chem. Int. Ed. 2003;42:4657–4660. doi: 10.1002/anie.200351534. [DOI] [PubMed] [Google Scholar]
- 18.Yamada T, Kinjyo S, Yoshida J, Yamago S. Chem. Lett. 2005;34:1556–1557. [Google Scholar]
- 19.Fridman M, Solomon D, Yogev S, Baasov T. Org. Lett. 2002;4:281–283. doi: 10.1021/ol017054d. [DOI] [PubMed] [Google Scholar]
- 20.Bongat AFG, Kamat MN, Demchenko AV. J. Org. Chem. 2007;72:1480–1483. doi: 10.1021/jo062171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicolaou KC, Dolle RE, Papahatjis DP, Randall JL. J. Am. Chem. Soc. 1984;106:4189–4192. [Google Scholar]
- 22.Veeneman GH, van Boom JH. Tetrahedron Lett. 1990;31:275–278. [Google Scholar]
- 23.Roy R, Andersson FO, Letellier M. Tetrahedron Lett. 1992;33:6053–6056. [Google Scholar]
- 24.Kanie O, Ito Y, Ogawa T. J. Am. Chem. Soc. 1994;116:12073–12074. [Google Scholar]
- 25.Pornsuriyasak P, Demchenko AV. Chem. Eur. J. 2006;12:6630–6646. doi: 10.1002/chem.200600262. [DOI] [PubMed] [Google Scholar]
- 26.Ye XS, Wong CH. J. Org. Chem. 2000;65:2410–2431. doi: 10.1021/jo991558w. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Huang L, Wang H, Ye XS. Angew Chem., Int. Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 28.Boons GJ, Geurtsen R, Holmes D. Tetrahedron Lett. 1995;36:6325–6328. [Google Scholar]
- 29.Mydock LK. University of Missouri; St. Louis: 2010. Doctoral Dissertation. [Google Scholar]
- 30.Kuester JM, Dyong I. Justus Liebigs Ann. Chem. 1975:2179–2189. [Google Scholar]
- 31.Kartha KPM, Aloui M, Field RA. Tetrahedron Lett. 1996;37:5175–5178. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.