

Abstract
Deletions leading to complete or partial removal of ParB were introduced into the Pseudomonas aeruginosa chromosome. Fluorescence microscopy of fixed cells showed that ParB mutants lacking the C-terminal domain or HTH motif formed multiple, less intense foci scattered irregularly, in contrast to the one to four ParB foci per cell symmetrically distributed in wild-type P. aeruginosa. All parB mutations affected both bacterial growth and swarming and swimming motilities, and increased the production of anucleate cells. Similar effects were observed after inactivation of parA of P. aeruginosa. As complete loss of ParA destabilized its partner ParB it was unclear deficiency of which protein is responsible for the mutant phenotypes. Analysis of four parB mutants showed that complete loss of ParB destabilized ParA whereas three mutants that retained the N-terminal 90 aa of ParB did not. As all four parB mutants demonstrate the same defects it can be concluded that either ParB, or ParA and ParB in combination, plays an important role in nucleoid distribution, growth and motility in P. aeruginosa.
INTRODUCTION
Representatives of two families of bacterial Par proteins, ParA and ParB, are encoded by the majority of bacterial chromosomes in the close vicinity of oriC (Gal-Mor et al., 1998; Ogasawara & Yoshikawa, 1992); the exceptions are Enterobacteriaceae and Pasteurellaceae, which appear to lack these loci. The first Par proteins were studied in low-copy-number plasmids, where they were shown to drive active partitioning of plasmid molecules to the progeny cells (Bignell & Thomas, 2001).
Increasing data from different par systems have established that Par proteins interact with each other to play their role in plasmid or chromosome partitioning (Autret et al., 2001; Bartosik et al., 2004; Ebersbach & Gerdes, 2004; Erdmann et al., 1999; Figge et al., 2003; Kim & Shim, 1999; Lukaszewicz et al., 2002; Ravin et al., 2003). Component A of the partitioning system is an ATPase either of Walker-type (ParA) or actin-like (ParM) and forms dynamic structures within bacterial cells (Adachi et al., 2005; Davey & Funnell, 1994; Davis et al., 1992; Ebersbach & Gerdes, 2004; Easter & Gober, 2002; Jensen & Gerdes, 1997; Marston & Errington, 1999; Quisel & Grossman, 2000). Component B is a DNA-binding protein that recognizes the centromere-like sequence(s), usually called parS, and forms regularly distributed foci (in complex with DNA) in bacterial cells irrespective of whether it is encoded on the chromosome or a plasmid (Biek & Strings, 1995; Bignell et al., 1999; Bouet et al., 2000; Erdmann et al., 1999; Funnell, 1991; Glaser et al., 1997; Gordon et al., 1997; Lewis & Errington, 1997; Niki & Hiraga, 1997). The nucleoprotein complexes formed by the components of the par systems are required for accurate partitioning of plasmids prior to cell division.
Studies on chromosomally encoded Par proteins in Bacillus subtilis, Caulobacter crescentus, Streptomyces coelicolor, Pseudomonas putida, P. aeruginosa and, recently, Helicobacter pylori, Burkholderia cenocepacia, Vibrio cholerae and Mycobacterium smegmatis have indicated their involvement (to different extents in different species) in chromosome partitioning but also in other cell processes (Bartosik et al., 2004; Dubarry et al., 2006; Godfrin-Estevenon et al., 2002; Ireton et al., 1994; Jakimowicz et al., 2007a, b; Kim et al., 2000; Lee et al., 2006; Lewis et al., 2002; Lin & Grossman, 1998; Mohl & Gober, 1997; Saint-Dic et al., 2006).
In most chromosomal par systems studied to date mutations in the par genes can be easily generated, indicating that these genes are not essential for growth. The exception so far is C. crescentus, in which Par proteins are involved in the regulation of cytokinesis and gene knockouts are lethal (Figge et al., 2003; Mohl et al., 2001). In B. subtilis the ParA homologue Soj was first described as the regulator of the entry into sporulation. The ParB homologue Spo0J is required for both initiation of sporulation and normal chromosome partitioning during vegetative growth. Mutations in spo0J lead to an increased number of anucleate cells, defects in separation of replicated origins and impaired regulation of replication initiation during the vegetative cell cycle (Ireton et al., 1994; Lee et al., 2003; Lee & Grossman, 2006). Mutations in soj (parA) have no significant effect on chromosome segregation in B. subtilis although the chromosome becomes decondensed. A soj spo0J double mutant is impaired in chromosome partitioning during sporulation (Sharpe & Errington, 1996). For P. putida parA, parB or double parAB mutants grown in minimal medium 5–10 % of cells were anucleate during the transition from exponential growth to stationary phase and it was suggested that the Par proteins in this species are particularly important for chromosome partitioning when the bacteria are undergoing cell division in the absence of ongoing DNA replication (Lewis et al., 2002; Godfrin-Estevenon et al., 2002).
In the sequenced P. aeruginosa PAO1 genome the parA and parB genes were identified in a region about 8 kb from oriC. As in many other eubacterial chromosomes the P. aeruginosa par genes are situated close to genes implicated in DNA replication, cell division and basic metabolism. Comparative sequence analysis of ParB representatives uncovered conserved motifs within the protein, regions 1 to 4 (Fig. 1a) (Bartosik et al., 2004), in addition to previously identified sequences of unknown function, ParB Box I and Box II (Yamaichi & Niki, 2000). The HTH motif (the putative domain of interaction with DNA) is located in the central part of ParB protein and the dimerization determinants are located in the C terminus (Bartosik et al., 2004). Ten putative parS sites were identified in the P. aeruginosa chromosome and exhibited a hierarchy of binding strengths for ParBP.a. in vitro (Bartosik et al., 2004). In vivo experiments demonstrated that ParBP.a. is able to bind to palindromic parS2/3 and silence the expression of genes in close proximity to the binding site. ParA, ParB and parS2/3 of P. aeruginosa can act as a plasmid active partitioning system (Bartosik et al., 2004). The overproduction of ParA (Lasocki et al., 2007) or ParB (Bartosik et al., 2004) disturbs cell division and nucleoid distribution. The ‘toxicity’ determinants (causing growth retardation when overproduced) have been mapped to the N-terminal 85 aa of ParA (Lasocki et al., 2007) and N-terminal 90 aa of ParB (Bartosik et al., 2004). The inactivation of chromosomal parA is not lethal but leads to a more than 100-fold increase in the number of anucleate cells even in actively dividing cultures. The growth rate is reduced and cells are impaired in two types of motility: drastically in swarming and to a lesser extent in swimming. The observed defects cannot be complemented by parA in trans, suggesting that the amount of Par proteins as well as the ParA : ParB ratio is very important. The ParB produced by parA null mutants is prone to degradation so it was difficult to separate which effects of parA mutation are due to the lack of ParA and which to a decreased level of ParB in the cells (Lasocki et al., 2007). In this study, to distinguish between these effects and to better determine the role of ParB in the P. aeruginosa cell cycle, strains completely or partially deleted for parB were constructed. We found that mutations in parB are not lethal for P. aeruginosa and have pleiotropic effects on chromosome segregation, colony formation and motility similar to the phenotype of parA mutants (Lasocki et al., 2007). Complete deletion of parB resulted in instability of ParA, but as three of the four parB mutants do not affect ParA level we can conclude that impairment or loss of ParB is a key reason for the observed defects in both parB and parA mutants.
Fig. 1.

Effect of parB mutations on growth, colony formation, and swimming and swarming motility of P. aeruginosa. (a) Domain structure of ParB of P. aeruginosa. The conserved regions are marked in grey. The linker region is marked in black. The N-terminal domain of interaction with putative host factors and C-terminal domain involved in dimerization are hatched. (b) The parB gene from P. aeruginosa and its mutant derivatives constructed for allele replacement. The restriction sites shown are those used in manipulations. The tetracycline-resistance cassette (TcR; not to scale) comes from plasmid pKRP12 (Reece & Phillips, 1995). (c) Growth of WT PAO1161 strain, parB mutants and parB+ revertant strain (revB+) on rich medium at 37 °C. Overnight cultures were diluted 100-fold. The OD600 was measured at hourly intervals. (d) Colony morphology of WT and parB mutant strains observed after 24 h incubation on L agar plates at 37 °C using an Olympus IX70 microscope. Photomicrographs were projected, and visualized with DP-Soft (analySIS) software produced by Soft Imaging Systems for Olympus. The final montage was created with Adobe Photoshop, version 6.0. (e) Motility of WT, parB mutants and parB+ revertant strain tested as described in Methods. The test plates were inoculated with a sterile toothpick and incubated for 24–48 h at 30 °C. The parA− strain was included to demonstrate the similarity to the ParB− phenotype.
METHODS
Bacterial strains and growth.
Escherichia coli strains used were K-12 strain DH5α [F− (φ80dlacZΔM15) recA1 endA1 gyrA96 thi-1 hsdR17 (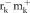 ) supE44 relA1 deoR Δ(lacZYA–argF)U196], B strain BL21 [F−
ompT hsdSB (
) supE44 relA1 deoR Δ(lacZYA–argF)U196], B strain BL21 [F−
ompT hsdSB (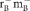 ) gal dcm (λ DE3)] (Novagen) and S17-1 (pro hsdR hsdM recA TpR SmR ΩRP4-Tc : : Mu-Km : : Tn7) (Simon et al., 1986). Pseudomonas aeruginosa PAO1161 (leu r−m+) was kindly provided by B. M. Holloway (Monash University, Clayton, Victoria, Australia). PAO1161 RifR was isolated during growth with 125 μg rifampicin ml−1. Bacteria were grown in L broth (Kahn et al., 1979) at 37 °C or 30 °C or on L agar (L broth with 1.5 %, w/v, agar) supplemented with antibiotics as appropriate: benzylpenicillin sodium salt at 150 μg ml−1 in liquid medium and 300 μg ml−1 on agar plates for penicillin resistance in E. coli, streptomycin sulphate at 30 μg ml−1 for streptomycin resistance in E. coli and 200 μg ml−1 in P. aeruginosa, tetracycline at 10 μg ml−1 for tetracycline resistance in E. coli and 100 μg ml−1 for P. aeruginosa, carbenicillin at 300 μg ml−1 for carbenicillin resistance in P. aeruginosa, rifampicin at 300 μg ml−1 for rifampicin resistance in P. aeruginosa. Some experiments were performed in M9 minimal medium (Sambrook et al., 1989). The L agar used for blue/white screening contained 0.1 mM IPTG and X-Gal at 40 μg ml−1.
) gal dcm (λ DE3)] (Novagen) and S17-1 (pro hsdR hsdM recA TpR SmR ΩRP4-Tc : : Mu-Km : : Tn7) (Simon et al., 1986). Pseudomonas aeruginosa PAO1161 (leu r−m+) was kindly provided by B. M. Holloway (Monash University, Clayton, Victoria, Australia). PAO1161 RifR was isolated during growth with 125 μg rifampicin ml−1. Bacteria were grown in L broth (Kahn et al., 1979) at 37 °C or 30 °C or on L agar (L broth with 1.5 %, w/v, agar) supplemented with antibiotics as appropriate: benzylpenicillin sodium salt at 150 μg ml−1 in liquid medium and 300 μg ml−1 on agar plates for penicillin resistance in E. coli, streptomycin sulphate at 30 μg ml−1 for streptomycin resistance in E. coli and 200 μg ml−1 in P. aeruginosa, tetracycline at 10 μg ml−1 for tetracycline resistance in E. coli and 100 μg ml−1 for P. aeruginosa, carbenicillin at 300 μg ml−1 for carbenicillin resistance in P. aeruginosa, rifampicin at 300 μg ml−1 for rifampicin resistance in P. aeruginosa. Some experiments were performed in M9 minimal medium (Sambrook et al., 1989). The L agar used for blue/white screening contained 0.1 mM IPTG and X-Gal at 40 μg ml−1.
Plasmid DNA isolation, analysis, cloning and manipulation of DNA.
Plasmid DNA was isolated and analysed by standard procedures (Sambrook et al., 1989). The plasmids used in this study are listed in Table 1. Details of the cloning procedures are available as supplementary data with the online version of this paper (Supplementary Methods).
Table 1.
Plasmids used in this work
| Plasmid | Relevant features | Reference/source |
|---|---|---|
| pABB211 | pET28mod derivative with T7p-parB1–229 fusion | Bartosik et al. (2004) |
| pABB330 | pGBT30 derivative with tacp-parBΔ121–183 fusion | Bartosik et al. (2004) |
| pABB811 | pGB2 derivative with parS2/3I of P. aeruginosa | Bartosik et al. (2004) |
| pAKE600 | oriMB1oriTRK2, ApRsacB | El-Sayed et al. (2001) |
| pBGS18 | oriMB1 KmR | Spratt et al. (1986) |
| pET28mod | oriMB1 KmR T7p lacO His6 tag, modified to remove T7 tag | Lukaszewicz et al. (2002) |
| pGBT30 | oriMB1 ApRlacIQtacp, expression vector | Jagura-Burdzy et al. (1991) |
| pGBT400 | oriRSF1010 BHR SmRlacIQtacp, expression vector | Jagura-Burdzy et al. (1999) |
| pGEM-T Easy-egfp | pGEM-T Easy derivative with egfp cassette | Wei Zeng, PhD thesis, University of Birmingham |
| pKLB181 | pUC18 with parS2/3 on the HindIII–SalI fragment | Bartosik et al. (2004) |
| pKLB2 | pGBT30 with tacp-parB transcriptional fusion | Bartosik et al. (2004) |
| pKLB2.4 | pGAD424 with gal4AD-parB translational fusion | Bartosik et al. (2004) |
| pKLB28 | pET28mod derivative with T7p-parB fusion | Bartosik et al. (2004) |
| pKLB3 | pGBT30 with tacp-rbs-parAB transcriptional fusion | Bartosik et al. (2004) |
| pKLB40.1 | pGBT400 with tacp-parA transcriptional fusion | Lasocki et al. (2007) |
| pKLB60.1 | pAKE600 without BamHI site | Lasocki et al. (2007) |
| pKRP12 | oriMB1 ApR TcR cassette | Reece & Phillips (1995) |
| pUC18 | oriMB1 ApR | Yanisch-Perron et al. (1985) |
| Plasmids constructed during this study | ||
| pABB10 | pUC18 with the EcoRI–SalI fragment carrying parB | |
| pABB101 | pGBT400 with the BamHI–SalI fragment of pABB301 carrying lacIQ tacp-egfp-parB | |
| pABB131 | pGBT400 with the BamHI–SalI fragment of pABB331 carrying lacIQ tacp-egfp-parBΔ121–183 | |
| pABB14 | pUC18 with the EcoRI–SalI fragment carrying the parB1–90 : : TcR disruption mutation | |
| pABB18 | pBGS18 with the EcoRI–SalI fragment carrying parAparB1–18 : : TcR from pABB318 | |
| pABB230 | pET28mod with the EcoRI–SalI fragment carrying parBΔ121–183 | |
| pABB24 | pET28mod with the EcoRI–SalI fragment carrying parB1–90 : : TcR from pABB14 | |
| pABB301 | pKLB2 with EcoRI fragment of pGEM-T Easy-egfp to form egfp-parB translational fusion | |
| pABB318 | pGBT30 with parAparB1–18 : : TcR | |
| pABB331 | pABB330 with EcoRI fragment of pGEM-T Easy-egfp to form egfp-parBΔ121–183 translational fusion under tacp control | |
| pABB611 | pAKE600 with the HindIII–SalI fragment carrying gidBparAparB | |
| pABB614 | pAKE600 with the EcoRI–SalI fragment carrying parB1–90 : : TcR from pABB24 | |
| pABB618 | pAKE600 with the EcoRI–SalI fragment carrying parAparB1–18 : : TcR from pABB18 | |
| pABB660 | pKLB60.1 with the EcoRI–HindIII fragment carrying parB1–229 | |
| pABB661 | pABB660 with the BamHI–SalI fragment carrying parB235–290 | |
| pABB662 | pABB661 with the TcR cassette inserted into the BamHI site | |
| pABB663 | pAKE600 with the EcoRI–SalI fragment carrying parBΔ121–183 |
Standard PCR (Mullis et al., 1986) was performed as described previously (Lasocki et al., 2007) with the following pairs of primers for parB or gidBparAparB amplification: ParB1 (5′-CCGAATTCATGGCAGCCAAGAAACGTGG-3′) and ParB2 (5′-CCGTCGACCCGACTACCCGCTACAACCC-3′); ParB1 and ParB3 (5′-GGGGATCCGTCCACTGCCGAACCAGGGC-3′); ParB4 (5′-GGGGATCCCCTGCGGGACCGGTCAAGAG-3′) and ParB2; GidB1 (5′-CCAAGCTTATGTCTGCGGTAACCCA-3′) and ParB2 (restriction enzyme recognition sites underlined; start codons in bold). Chromosomal DNA of P. aeruginosa PAO1161 or PAO1161parB mutant strains was used as the template in appropriate PCRs.
DNA sequencing was performed at an internal sequencing facility (IBB PAS, Warsaw, Poland) by using the dye terminator method in conjunction with an ABI 373 automated DNA sequencer.
Bacterial transformation.
Competent cells of E. coli were prepared by the standard CaCl2 method (Sambrook et al., 1989). Competent cells of P. aeruginosa were prepared by the method of Irani & Rowe (1997).
Introduction of par mutant alleles into P. aeruginosa PAO1161.
E. coli S17-1 was transformed with pAKE600 suicide vector derivatives (El-Sayed et al., 2001). Transformants were conjugated with the recipient strain P. aeruginosa PAO1161RifR in which the suicide vector with a pMBI ori was unable to replicate (see Supplementary Methods). To obtain the PAO1161parB+ revertant strain an E. coli S17-1(pABB611) transformant was conjugated with the recipient strain PAO1161parB1–18 : : TcR. Reversion of parB1–18 : : TcR mutation to wild-type parB was confirmed by PCR, sequencing and Western blot analysis.
Protein analysis.
The growth of bacteria was monitored by measuring the OD600; the cultures were diluted and plated on L agar to determine c.f.u. ml−1. Bacteria were harvested, resuspended in sonication buffer (50 mM phosphate buffer pH 8.0 and 300 mM NaCl) and disrupted by sonication. Cleared extracts after centrifugation were analysed by SDS-PAGE followed by Western blotting performed as described previously (Bartosik et al., 2004).
Motility assays.
The swimming and swarming assays were performed according to Rashid & Kornberg (2000) with modifications described previously (Lasocki et al., 2007).
DAPI staining and immunofluorescence microscopy.
The DAPI staining procedure and immunofluorescence microscopy (using FITC-conjugated secondary antibodies) was carried out as previously described (Bartosik et al., 2004; Bignell et al., 1999). Cells were studied using an Olympus IX70 inverted reflected-light fluorescence microscope fitted with a Sensys CCD camera (Photometrics). Images were captured and manipulated on a Macintosh G3 with the Smartcapture I program (Digital Scientific).
Fluorescence microscopy of living cells.
PAO1161(pABB101) or (pABB131) transformants were grown in selective L broth medium at 24 °C to facilitate observations of the thermolabile EGFP. When cultures reached an OD600 of about 0.2, IPTG (0.2 mM) was added to induce the expression of the fusion proteins. After 2–3 h induction (OD600 0.5–0.9), samples were collected and immediately used to prepare microscope slides. Two microlitres of 1.5 % low-melting agarose (Bio-Rad) solution in L broth was put onto a 76×25×1 mm objective slide. The coated slide was left to solidify and dry in the open air at room temperature for about 10 min. A 5 μl drop of the cell suspension was placed on the surface of the agarose and immediately covered with a coverslip and studied as described above.
RESULTS
Mutations in parB result in the combination of growth, chromosome segregation and motility defects
ParB proteins appear to consist of three segments: an N-terminal segment 1 (containing conserved region 1, Box I and Box II) of as yet ill-defined function, an internal segment 2 with the DNA-recognition domain (containing the HTH motif, and conserved regions 2 and 3) and C-terminal segment 3 with the dimerization domain (highly conserved region 4) (Fig. 1a; Bartosik et al., 2004). Segments 2 and 3 are joined by a variable linker. To investigate P. aeruginosa ParB function, the parB gene was cloned in E. coli and mutant alleles were constructed (Fig. 1b) that produced only the first 18 aa of ParB (parB1–18 : : TcR), or ParB that had lost segments 2 and 3 (parB1–90 : : TcR) or only segment 3 (parB1–229 : : TcR) or part of segment 2 (parBΔ121–183). The latter two mutant proteins were overexpressed in E. coli after cloning into pET28mod and purified with a His6-tag at the N terminus. DNA-binding activity of both proteins and their oligomeric state were determined in vitro by electrophoretic mobility shift assays and glutaraldehyde cross-linking using wild-type (WT) ParB as the control. Loss of the C terminus in ParB1–229 resulted in a monomeric (as shown previously: Bartosik et al., 2004), sequence-specific DNA-binding protein with >5-fold decreased affinity for parS (Supplementary Fig. S1A) whereas deletion of segment 2 (parBΔ121–183) resulted in loss of the ability to bind to DNA (Supplementary Fig. S1A) but retention of the dimeric state typical for WT ParB as expected (Supplementary Fig. S1B).
The mutated alleles of parB were then introduced into the P. aeruginosa PAO1161 chromosome by a two-step allelic exchange as previously described (El-Sayed et al., 2001). The presence of the mutations was confirmed by PCR analysis and the linked selectable tetA marker where applicable. Western blotting with anti-ParB antibodies detected the truncated proteins ParB1–229 and ParBΔ121–183 in the extracts of the respective mutant strains at twofold lower level than observed for WT ParB, whereas the shorter ParB products (in parB1–18 and parB1–90 strains) could not be detected at all (data not shown). At no stage of mutant construction was there great difficulty in achieving success, suggesting that the mutations do not have a major effect on viability.
Growth rate (as mean growth rate) of all the parB mutants was 5–10 % lower than PAO1161 in both L broth (Fig. 1c) and minimal medium (data not shown). Cell size of the parB mutants increased proportionately (2.33 μm for mutant cells versus 2.20 μm for PAO1161 in L broth and 1.86 μm versus 1.60 μm in minimal medium); the corresponding values in stationary phase were 1.88 μm for mutant cells versus 1.81 μm for PAO1161 in L broth and 1.65 μm versus 1.49 μm in minimal medium, respectively (on samples of at least 2000 cells with standard deviation 0.05 μm).
The colony morphology of all parB mutants was irregular, with lobed or jagged edges as compared to the smooth-edged circular colonies of the WT (Fig. 1d). All four parB mutants, despite having either the HTH motif or/and the dimerization domain deleted, exhibited defects in swarming and swimming (Fig. 1e) but not in twitching (data not shown). Similar phenotypes were observed previously for a parA mutant (Lasocki et al., 2007).
Epifluorescence microscopy after staining DNA with DAPI (Supplementary Fig. S2a) showed that parB mutants demonstrated a >100-fold increase in the number of anucleate cells during growth on rich medium: from 0.02 % for WT to about 2 % in mutant cultures among actively growing cells and to 4 % in stationary-phase cultures (on samples of at least 1000 cells). The mutant strains grown on minimal medium contained up to 4 % or 7 % of chromosome-less cells in the exponential and stationary phase of growth, respectively (on samples of at least 1000 cells). In addition about 5 % of the cells appeared to be undergoing division without separation of chromosomes (Supplementary Fig. S2A).
ParB is necessary for ParA stability in P. aeruginosa
Since ParB and ParA interact with each other (Bartosik et al., 2004) and in the light of the observation that ParB is highly unstable in parA null mutants (Lasocki et al., 2007) it was important to determine whether any of the parB mutations affected the level of ParA. Samples of the parB mutants and WT as control were collected at a similar optical density and c.f.u. ml−1 were determined so that extracts from approximately 5×109 cells could be separated by SDS-PAGE and analysed for the presence of ParA by Western blotting (Fig. 2a). For parB1–229, parBΔ121–183 and parB1–90 the level of ParA was not significantly different from the level in WT cells (fluctuations may be due to the use of unsynchronized cultures) whereas it was undetectable in the extract prepared from the parB null mutant (parB1–18). Even a fivefold increase in the number of cells (2.5×1010) from the late exponential phase analysed on the gel gave no signal for ParA (Fig. 2a; last track). As 50 ng ParA is visible under these conditions this means that the level of ParA in PAO1161parB1–18 drops on average to below 40 molecules per cell. The parB mutant phenotypes described in the sections above were similar for all four analysed mutations so it is unlikely that the decrease in stability of ParA was the basis for any of the observed changes; ParA instability occurred in only one mutant, parB1–18.
Fig. 2.

Analysis of ParA and ParB levels in WT and mutants of P. aeruginosa. (a) Samples from early- and late-exponential-phase cultures at the indicated OD600 were collected and sonicated. The cleared supernatants from about 5×109 cells were analysed by 15 % SDS-PAGE, followed by transfer onto nitrocellulose membrane and reaction with semi-purified anti-ParA antibody (Lasocki et al., 2007). (b) and (c) Western blot analysis of ParB and ParA, respectively, in parB1–18 mutant, parB+ revertant (revB+) and WT PAO1161 strains. Cells were collected at three stages of culture growth (indicated by OD600). Cleared extracts from about 109 cells were analysed by 15 % SDS-PAGE followed by transfer onto nitrocellulose and immunodetection with Anti-ParB (b) and Anti-ParA (c) antibodies.
Localization of ParB in P. aeruginosa cells
To determine the effect of the mutations on ParB localization within bacterial cells, two types of fluorescence microscopy were applied. Immunofluorescence microscopy with antibodies to ParB was applied to bacteria from both exponential- and stationary-phase cultures grown in rich medium at 37 °C. In the WT, the signal for DAPI-stained chromosomal DNA was found almost throughout the entire volume of the bacterial cell (Fig. 3a). ParB formed specific foci apparently connected with the nucleoids. The majority of the cells from actively dividing cultures contained two to four ParB foci distributed symmetrically along the cell length (positions 1/4 and 3/4 or 1/8, 3/8, 5/8 and 7/8, respectively: Fig. 3a and f). In a small percentage of cells (5–7 %) there were also multiple small foci of ParB next to the bigger, more compact ParB foci, suggesting that the bigger foci may be formed by condensation of smaller foci.
Fig. 3.

Cellular localization of ParB and its mutant forms in P. aeruginosa. (a–e) Localization of ParB in WT P. aeruginosa, mutant strains parB1–18, parB1–229 and parBΔ121–183, and parB+ revertant strain (revB+). Images show the location of ParB in the cells from the exponential phase of cultures grown in rich medium at 37 °C. Higher magnification of representative cells is also shown. Immunofluorescence and phase-contrast micrographs are overlaid. The dark background is a phase-contrast image, the dark blue is the DAPI-stained chromosome and the green/light blue is the FITC-stained ParB. (f) Bar charts showing the percentage of cells with different numbers of ParB foci for various strains. At least 500 cells were counted for each strain. (g, h) Overlaid phase-contrast and EGFP fluorescence images showing the cellular localization of the EGFP-ParB and EGFP-ParBΔ121–183 fusion proteins in living cells of P. aeruginosa. Cells from the PAO1161(pABB101) and PAO1161(pABB131) cultures, grown in L broth at 24 °C, were collected after 2–3 h induction with 0.2 mM IPTG and immediately prepared for microscopic examination. The dark background is the phase-contrast image and the green colour represents EGFP fused to ParB and ParBΔ121–183, respectively. (i–l) Immunofluorescence–phase-contrast overlay micrographs showing the localization of WT ParB in P. aeruginosa PAO1161(pKLB40.1tacp-parA) overproducing ParA protein. The graph presents the growth curves of PAO1161(pGBT400) and PAO1161(pKLB40.1) cultures grown in rich medium at 37 °C, from which the samples were collected for immunofluorescence microscopy. Different concentrations of IPTG were applied to overproduce ParA. PAO1161(pGBT400) cells grown in the presence of 0.5 mM IPTG were used as a control. The dark background is the phase-contrast image, the dark blue is the DAPI-stained chromosome and the green/light blue is the FITC-stained ParB. Scale bars 2 μm (all panels).
In the stationary-phase cultures the majority of cells had one or two foci of ParB (Supplementary Fig. S2B) and the FITC signals were much weaker in comparison to cells in exponential phase. In late stationary phase an extremely weak or no signal for ParB was detected in most of the cells. As the cells from stationary-phase culture may be more resistant to the fixing conditions a much harsher lysis method was used but the intensity of the ParB signal did not increase (data not shown). The level of ParB in stationary-phase cells of PAO1161 is about fivefold lower in comparison to the exponential phase (M. Andres & G. Jagura-Burdzy, unpublished results), which may explain the weaker signals for ParB in the cells from stationary-phase cultures.
For the parB1–18 mutant strain the signal for ParB was not detected at all, as expected (Fig. 3b and f). In the parB1–90 mutant strain a very weak signal for ParB was observed only in less than 10 % of the cells and it did not exhibit a characteristic pattern of distribution (Fig. 3f). The signal for ParB in two parB mutants, parB1–229 and parBΔ121–183, was weaker than in WT bacteria (Fig. 3c and d). The fluorescent signal of ParB1–229 was more dispersed and in the majority cells it was localized closer to the cell envelope (Fig. 3c and f); ParBΔ121–183 formed multiple foci scattered irregularly in the cells (Fig. 3d and f).
To follow changes in ParB distribution in living P. aeruginosa cells the EGFP protein (a more stable, red-shifted variant of WT green fluorescent protein from Clontech which has been optimized for brighter fluorescence) was fused to the N terminus of WT ParB and also to ParBΔ121–183. To confirm that the hybrid proteins were stable and functionally active they were tested for the previously observed ability of ParBP.a.to silence genes that are linked to parS (Bartosik et al., 2004). The observable effect of repA gene silencing in pGB2-parS2/3I (pABB811) caused by ParB is loss of the reporter plasmid. E. coli DH5α(pABB811) cells were transformed with pGBT30, pKLB2 and pABB301, so that the stability of pABB811 could be checked in the presence of the vector, WT ParB and EGFP-ParB, respectively. Strong destabilization of pABB811 was observed in the presence of excess ParB or EGFP-ParB (Supplementary Fig. S3A). This indicated that the fusion protein EGFP-ParB was functional in DNA binding and silencing. Overproduction of EGFP-ParBΔ121–183 (pABB331), lacking the DNA-binding domain, did not cause destabilization of the reporter plasmid, as was observed previously for ParBΔ121–183 (Bartosik et al., 2004). To study the subcellular localization of ParB in P. aeruginosa cells, the fusions were transferred to the expression vector pGBT400, which is able to propagate in P. aeruginosa. Stability of the hybrid proteins in P. aeruginosa was confirmed by Western blotting (Supplementary Fig. S3B).
Fluorescence–phase-contrast combination microscopy of cells from exponential-phase cultures of PAO1161(pABB101 lacIQ tacp-egfp-parB) showed that EGFP-ParB formed one to four foci, symmetrically distributed in living cells of P. aeruginosa (Fig. 3g). These results corresponded to those of immunofluorescence microscopy on fixed cells. The EGFP-ParBΔ121–183 fusion protein in PAO1161(pABB131 lacIQ tacp-egfp-parBΔ121–183) was unable to form foci, giving a dispersed signal throughout the cell (Fig. 3h), as observed for EGFP on its own (data not shown).
The cells immobilized on the slides stayed alive for a long time so it was possible to observe changes in EGFP-ParB localization using time-lapse microscopy. Fluorescence–phase-contrast micrographs from the same field were collected at 2 min intervals. The experiment presented in Supplementary Fig. S2(C) showed that ParB foci undergo dynamic and rapid changes in approximately one-third of the cells. New foci of ParB were formed by duplication/splitting of pre-existing foci followed by rapid moving apart to give rise to regularly distributed foci in longer cells (four foci in mother cells before the completion of cell division).
ParA overproduction affects formation of ParB foci and their distribution in P. aeruginosa cells
The overproduction of ParAP.a. is toxic for P. aeruginosa (Lasocki et al., 2007). To see how an excess of ParA influences formation of ParB foci, PAO1161(pKLB40.1 =pGBT400tacp-parA) was grown in the absence and presence of IPTG, and cells were then collected and analysed by immunofluorescence microscopy. PAO1161(pGBT400) grown in the presence of 0.5 mM IPTG was used as a control and showed that neither the presence of the vector nor IPTG changed the distribution of ParB foci (Fig. 3i). In fast-dividing cells, chromosomes initiate subsequent rounds of replication before finishing the previous cycle and it is assumed that there are at least two almost fully replicated genomes per cell. In the cells of PAO1161(pKLB40.1) grown without IPTG the ParB distribution was indistinguishable from the control (Fig. 3j). Two or four ParB foci were distributed evenly within shorter and longer cells, respectively.
Addition of 0.2 mM or 0.5 mM IPTG to PAO1161(pKLB40.1) significantly reduced the growth rate (graph in Fig. 3) while cells appeared to be at least double the length of the control cells collected at the same growth phase (Fig. 3k and l) with frequently occurring filamentous cells, some of them very long. Occasionally the nucleoids did not fill the whole cell, especially in the long filaments, leaving ‘empty’ spaces at the poles. In the elongated cells ParB did form foci but they were of much less regular pattern. The shortest observed cells exhibited ‘normal’ ParB foci but accompanied by a large number of smaller ParB foci, spread throughout the cell as the condensation of ParB foci was disturbed. In the fraction of the very long cells the larger ParB foci were evenly distributed along the filaments, but in the majority of the cells (more than 80 %) numerous ParB foci differing in size and number were observed that did not conform to the WT pattern of distribution. Some of the small ParB foci appeared to be preferentially positioned at the cell poles whereas in other cells they were localized in the middle of the cell (Fig. 3l).
Replacement of the mutated parB1–18 : : TcR allele by WT parB sequence restores the WT phenotypes
Introduction of mutations into parB might have generated secondary mutations in the genome, but since all four independent parB mutant strains had similar phenotypes (with the exception of the ParA level in the cells of the parB1–18 mutant) this seemed unlikely. However, if we could complement the defects by providing parB from a different location then this would clearly demonstrate that the defect was the direct result of the introduced mutation.
To check if the observed phenotypes caused by mutations in parB could be complemented by ParB protein delivered in trans the WT parB with its ribosome-binding site was cloned under control of tacp in a medium-copy-number vector able to propagate in P. aeruginosa. None of the defective parB alleles could be complemented by the presence of parB delivered in trans (data not shown). Complementation tests for parB mutants were also carried out with both parA and parB genes delivered in trans but without success (data not shown). Western blot analysis showed that the amount of Par proteins delivered in trans (even without inducer) in a selection of transformants was about twofold higher than the level of Par proteins in the PAO1161 (data not shown).
Our preliminary data indicated that parAparB genes can be expressed at low level from a promoter internal to gidB. The region encompassing gidBparAparB was cloned into the suicide vector (pABB611) and forced to integrate into PAO1161parB1–18 using homologous recombination. The levels of both ParB and ParA were found to be comparable with WT (data not shown); however, the integrants did not demonstrate the WT phenotypes. They grew slightly better than the parB1–18 strain but still produced anucleate cells and were impaired in swarming and swimming motilities.
Finally it was decided to exchange the mutated allele parB1–18 for the WT parB using the allelic exchange method on the integrants of PAO1161parB1–18 : :pABB611 (El-Sayed et al., 2001) and in this way to confirm the role of the parB mutation in the observed phenotypes. The production of full-length ParB at a level comparable to WT PAO1161 in the parB+ revertant strain was demonstrated by Western blotting (Fig. 2b). The growth rates of the tested parB+ revertants were the same as that of PAO1161 (Fig. 1c). The swarming and swimming motilities were also restored to normal in parB+ strain (Fig. 1e). The ParA content in cellular extracts of the revertant, verified by immunodetection, returned to the WT level (Fig. 2c). Microscopic observations did not detect anucleate cells in the parB+ revertant strain when approximately 5000 cells were examined (less than 0.02 %) (data not shown). Moreover the characteristic pattern of ParB localization was demonstrated by immunofluorescence microscopy (Fig. 3e and f). These results confirmed that the pleiotropic defects observed in the parB mutant were the direct result of the parB allele and were not due to secondary mutations generated elsewhere in the chromosome.
DISCUSSION
The primary focus of this study was to test the hypothesis that some or all of the phenotypes of a complete parA deletion mutant of P. aeruginosa could be due to destabilization of ParB. Because of the previous finding that loss of ParA results in disappearance of ParB (Lasocki et al., 2007), we deliberately made both complete and partial knockouts of parB. The in vitro properties of the deletion mutant proteins, prior to studies in vivo, confirmed the growing body of data on this family of proteins. Thus removal of the C-terminal domain affected dimerization and reduced the affinity but not the specificity for parS sequences, while deletion of amino acids 121–183, which encompass the predicted HTH motif from amino acids 152 to 171, did not affect dimerization but did abolish DNA binding. When ParA and ParB levels were checked in the four constructed parB mutants it was discovered that ParA stability depends on ParB, suggesting that interaction between these two proteins forms a complex that stabilizes both proteins. Comparison of the parB mutants indicated that the N-terminal 90 aa is necessary for this stabilization. Thus the mutants of ParB that retain this region and therefore still produce normal ParA levels, but inactivate ParB function, enabled us to test our hypothesis.
ParA–ParB interactions seem to be a general rule for Par systems but the location of the ParB domain responsible for these interactions appears to vary in different systems (Autret et al., 2001; Figge et al., 2003; Kim & Shim, 1999; Lukaszewicz et al., 2002; Ravin et al., 2003; Surtees & Funnell, 1999). The finding that P. aeruginosa ParB1–90 was sufficient to preserve the normal level of ParA suggests that the N-terminal region of ParB interacts with ParA. This would be consistent with the observation that the ATPase activity of Soj (ParA) from Thermus thermophilus is stimulated by the presence of the first 20 aa of Spo0J (ParB) (Leonard et al., 2005). This contrasts with our previous studies, where the C-terminal 56 aa of ParB was shown to be responsible for interactions with ParA (Bartosik et al., 2004). It is possible that there are two separate domains within ParB that are important in ParB–ParA interactions – one in the N-tip and the other in the C-tip – although the removal of ParB sequence from the C terminus up to the N-terminal 90 aa did not reveal the second interaction domain with ParA in yeast two-hybrid studies (Bartosik et al., 2004). Future detailed analysis will be necessary to resolve this, but for the purposes of this study the discrepancy is not important.
The ability to introduce parB mutations into the chromosome of P. aeruginosa indicates that this species, like the majority of other bacteria studied to date, e.g. P. putida (Godfrin-Estevenon et al., 2002; Lewis et al., 2002), B. subtilis (Ireton et al., 1994; Lee et al., 2003) and Streptomyces coelicolor (Kim et al., 2000), remains viable without a functional parB gene, the exception being C. crescentus, where a lethal effect is observed (Mohl & Gober, 1997).
The parB mutants of P. aeruginosa grew more slowly than the WT (5–10 % longer division time) and the cells tended to be longer than WT. As only three out of four had insertion of the TcR cassette we can exclude the trivial explanation that the presence of the cassette induces the observed changes. All four parB mutants were impaired in nucleoid segregation, with more than 100-fold increase in the frequency of anucleate cells appearing (from <0.02 % for WT to 2–4 % in rich medium and 7 % in minimal medium), with many more cells demonstrating a guillotining effect during cell division without nucleoid separation. The perturbation in nucleoid separation was clearly visible in the exponential phase of growth, in contrast to the defect in P. putida, which was only seen during the transition into stationary phase (Godfrin-Estevenon et al., 2002; Lewis et al., 2002).
The parB mutants were also drastically impaired in swarming and slightly in swimming activity, and formed irregular colonies with lamellar, jagged edges in contrast to the circular, regular colonies of the WT P. aeruginosa strain. The defects in motility may be the result of changes in cell-to-cell communication or flagellar structure/function, as flagellar function determines swarming and swimming abilities (Kohler et al., 2000). Microscopic observation of PAO1161parB1–18 cells showed the presence of flagella attached to one pole of the cell (data not shown) although at this stage the impairment of their rotary function cannot be excluded. Flagellar biogenesis is precisely controlled, and genes whose products participate in this process are clustered in different regions of the chromosome (PAO1 NC_002516; Dasgupta et al., 2003). One of the predicted parS sites is adjacent to pilGHIJK–chpABCD whereas others are located 10–15 kb from flgM flgN (parS8), motA motB (parS9), algZ algR (parS10), operons involved in flagellar biogenesis, quorum-sensing regulation, signal transduction and chemotaxis necessary for twitching and swarming motilities, cell adherence and colonization (Darzins, 1994; Kohler et al., 2000). Since ParBP.a. is a DNA-binding protein and its interactions with particular parS sites can cause silencing of neighbouring genes if overproduced (Bartosik et al., 2004), it is feasible that the observed defects in P. aeruginosa parB mutants are due to the overexpression of some loci when ParB is depleted. Spo0J, the ParB homologue of B. subtilis, was shown to spread laterally along DNA from parS nucleation sites covering more than 8 kb from particular parS sites (Murray et al., 2006) although the latest studies did not confirm the significance of spreading in gene expression (Breier & Grossman, 2007). It is also likely that ParB may directly interact with the proteins involved in flagellar biogenesis or cell-to-cell signalling (model in Fig. 4); this hypothesis is currently being investigated.
Fig. 4.
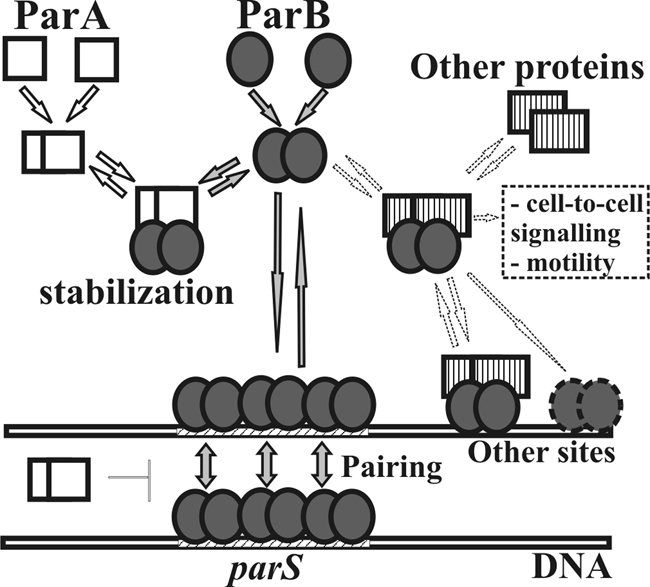
Model of action of Par proteins in P. aeruginosa. ParA and ParB proteins are able to interact with each other. ParB binds to parS sequences within the chromosome and spreads along DNA starting from parS. ParA may control the pairing of ParB–DNA complexes. ParB may also recognize other binding sites through interactions with other partners. Different complexes are involved in different cell processes such as chromosome segregation, cell division and motility.
Two visualization methods (immunofluorescence and EGFP) demonstrated that ParB in WT cells of P. aeruginosa forms regularly distributed foci (two in shorter cells and four in longer cells) located at the edges of the nucleoid. EGFP-ParBP.a fusion proteins in living cells showed that the new foci of ParB are formed by duplication of pre-existing foci and these quickly move apart (Supplementary Fig. S2C).
The ParB mutants defective in either the DNA-binding domain (but able to dimerize) or the C terminus, responsible for dimerization (still able to bind DNA but with lower specificity), lost the ability to form condensed specifically localized foci. ParBΔ121–183 formed several foci, random in number, whereas ParB1–229 was visible as tiny, multiple foci close to the cell poles, suggesting retention of some ability to interact and localize. Significantly, both of these ParB mutant forms possess an intact N terminus, suggested previously to be involved in interaction with cellular partners (Bartosik et al., 2004). The recognition of cellular receptors may be required to promote the condensation and symmetrical distribution of ParB foci (Ebersbach et al., 2008; Bowman et al., 2008). The difference in ParBΔ121–183 signal depending on the visualization method used (immunofluorescence or EGFP) may be due to the presence of WT ParB. If there are receptors for ParB located in the cell membrane close to the poles they might be recognized by ParBΔ121–183 in the PAO1161parBΔ121–183 mutant but have been preferentially occupied by the WT ParB in P. aeruginosa cells carrying both proteins (EGFP-ParBΔ121–183 and WT ParB), leading to the dispersion of the signal produced by EGFP-ParBΔ121–183.
Excess of ParAP.a. leads to formation of elongated cells with abnormal ParB foci of different size and number. If the normal ratio of ParB to ParA allows ParB to spread from its binding sites and condense to form foci that can be separated by ParA, then excess ParA may interfere with this process because of a preponderance of ParA–ParB complexes and may also block other ParB interactions that are necessary for normal cellular functions.
The characterization of parB mutants of P. aeruginosa indicates the vital role of ParB in determining the functions impaired in both parA and parB mutants but it does not exclude the possibility that both ParA and ParB are involved in proper nucleoid segregation, growth rate and motility. It clearly demonstrates that ParA on its own is not sufficient to control those processes. Complementation studies of par mutants (Lasocki et al., 2007 and this work) are very difficult as the small changes in the level of expression of parA and parB or slightly unbalanced production of both partners leads to the same phenotypes as observed for parA or parB mutants. We are aiming to isolate parA mutants producing ParB at WT level but unable to form an active ParA–ParB complex to discriminate between these possibilities.
On the basis of our current results we propose a working model for the action of Par proteins in P. aeruginosa cells (Fig. 4). In the cell ParA and ParB are expressed at a balanced level. Both proteins are able to form dimers and as such interact with each other (Bartosik et al., 2004; K. Lasocki & G. Jagura-Burdzy unpublished). These interactions play an important stabilizing role for both proteins, and in the case of ParB the N-terminal 90 aa is vital for protection of ParA against degradation. ParB binds to parS sequences within the P. aeruginosa chromosome and is able to spread along DNA starting from parS. Our in vivo silencing studies in E. coli suggest that ParA is able to modulate ParB interactions with parS and formation of ParB–DNA complexes (A. Bartosik & G. Jagura-Burdzy, unpublished data), and these may be directly involved in nucleoid segregation. The model also predicts that ParB may interact with other cellular partners, e.g. the proteins involved in flagellar biogenesis or cell-to-cell signalling, and in complex with them or on its own may interact with sequences other than parS within the genome. Identifying such partners if they exist and defining the protein–protein interactions involved in this network should provide a much better understanding of the role of ParB in the cell biology of its host and will be the subject of future work.
Acknowledgments
We thank Wei Zeng for instruction in fluorescence microscopy and technical assistance. This work was funded by Wellcome Trust Collaborative Research Initiative grants 056022/Z/98/Z, 067068/Z/02/Z and in part by MNiSW grant 2913/B/PO1/2008/34, an EMBO short-term fellowship (ASF 9976) awarded to A. A. B. and BBSRC grant 6/G10277.
Abbreviations
DAPI, 4′,6-diamidino-2-phenylindole
Footnotes
Supplementary methods and three supplementary figures are available with the online version of this paper.
References
- Adachi, S., Hori, K. & Hiraga, S. (2005). Subcellular positioning of F plasmid mediated by dynamic localization of SopA and SopB. J Mol Biol 356, 850–863. [DOI] [PubMed] [Google Scholar]
- Autret, S., Nair, R. & Errington, J. (2001). Genetic analysis of the chromosome segregation protein Spo0J of Bacillus subtilis: evidence for separate domains involved in DNA binding and interactions with Soj protein. Mol Microbiol 41, 743–755. [DOI] [PubMed] [Google Scholar]
- Bartosik, A. A., Lasocki, K., Mierzejewska, J., Thomas, C. M. & Jagura-Burdzy, G. (2004). ParB of Pseudomonas aeruginosa: interactions with its partner ParA and its target parS and specific effects on bacterial growth. J Bacteriol 186, 6983–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biek, D. P. & Strings, J. (1995). Partition functions of mini-F affect plasmid DNA topology in Escherichia coli. J Mol Biol 246, 388–400. [DOI] [PubMed] [Google Scholar]
- Bignell, C. & Thomas, C. M. (2001). The bacterial ParA-ParB partitioning proteins. J Biotechnol 91, 1–34. [DOI] [PubMed] [Google Scholar]
- Bignell, C. R., Haines, A. S., Khare, D. & Thomas, C. M. (1999). Effect of growth rate and incC mutation on symmetric plasmid distribution by the IncP-1 partitioning apparatus. Mol Microbiol 34, 205–216. [DOI] [PubMed] [Google Scholar]
- Bouet, J. Y., Suretees, J. A. & Funnell, B. E. (2000). Stoichiometry of P1 plasmid partition complexes. J Biol Chem 275, 8213–8219. [DOI] [PubMed] [Google Scholar]
- Bowman, G. R., Comolli, L. R., Zhu, J., Eckart, M., Koenig, M., Downing, K. H., Moerner, W. E., Earnest, T. & Shapiro, L. (2008). A polymeric protein anchors the chromosomal origin/ParB complex at a bacterial cell pole. Cell 134, 945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breier, A. M. & Grossman, A. (2007). Whole-genome analysis of the chromosome partitioning and sporulation protein Spo0J (ParB) reveals spreading and origin-distal sites on the Bacillus subtilis chromosome. Mol Microbiol 64, 703–718. [DOI] [PubMed] [Google Scholar]
- Darzins, A. (1994). Characterization of a Pseudomonas aeruginosa gene cluster involved in pilus biosynthesis and twitching motility: sequence similarity to the chemotaxis proteins of enterics and gliding bacterium Myxococcus xanthus. Mol Microbiol 11, 137–153. [DOI] [PubMed] [Google Scholar]
- Dasgupta, N., Wolfgang, M. C., Goodman, A. L., Arora, S. K., Jyot, S., Lory, J. & Ramphal, R. (2003). A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol Microbiol 50, 809–824. [DOI] [PubMed] [Google Scholar]
- Davey, M. J. & Funnell, B. E. (1994). The P1 plasmid partition protein ParA. A role for ATP in site-specific DNA binding. J Biol Chem 269, 29908–29913. [PubMed] [Google Scholar]
- Davis, M. A., Martin, K. A. & Austin, S. J. (1992). Biochemical activities of the ParA partition protein of the P1 plasmid. Mol Microbiol 6, 1141–1147. [DOI] [PubMed] [Google Scholar]
- Dubarry, N., Pasta, F. & Lane, D. (2006). ParABS systems of the four replicons of Burkholderia cenocepacia: new chromosome centromeres confer partition specificity. J Bacteriol 188, 1489–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easter, J., Jr & Gober, J. W. (2002). ParB-stimulated nucleotide exchange regulates a switch in functionally distinct ParA activities. Mol Cell 10, 427–434. [DOI] [PubMed] [Google Scholar]
- Ebersbach, G. & Gerdes, K. (2004). Bacterial mitosis: partitioning protein ParA oscillates in spiral-shaped structures and positions plasmids at mid-cell. Mol Microbiol 52, 385–398. [DOI] [PubMed] [Google Scholar]
- Ebersbach, G., Briegel, A., Jensen, G. J. & Jacobs-Wagner, C. (2008). A self-associating protein critical for chromosome attachment, division, and polar organization in Caulobacter. Cell 134, 956–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed, A. K., Hothersall, J. & Thomas, C. M. (2001). Quorum-sensing-dependent regulation of biosynthesis of the polyketide antibiotic mupirocin in Pseudomonas fluorescens NCIMB 10586. Microbiology 147, 2127–2139. [DOI] [PubMed] [Google Scholar]
- Erdmann, N., Petroff, T. & Funnell, B. E. (1999). Intracellular localization of P1 ParB protein depends on ParA and parS. Proc Natl Acad Sci U S A 96, 14905–14910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figge, R. M., Easter, J., Jr & Gober, J. W. (2003). Productive interaction between the chromosome partitioning proteins, ParA and ParB, is required for the progression of the cell cycle in Caulobacter crescentus. Mol Microbiol 47, 1225–1237. [DOI] [PubMed] [Google Scholar]
- Funnell, B. E. (1991). The P1 plasmid partition complex at parS. J Biol Chem 266, 14328–14337. [PubMed] [Google Scholar]
- Gal-Mor, O., Borovok, I., Av-gay, Y., Cohen, G. & Aharonowitz, Y. (1998). Gene organization in the trxA/B-oriC region of the Streptomyces coelicolor chromosome and comparison with other eubacteria. Gene 217, 83–90. [DOI] [PubMed] [Google Scholar]
- Glaser, P., Sharpe, M. E., Raether, B., Perego, M., Ohlsen, K. & Errington, J. (1997). Dynamic, mitotic-like behavior of a bacterial protein required for accurate chromosome partitioning. Genes Dev 11, 1160–1168. [DOI] [PubMed] [Google Scholar]
- Godfrin-Estevenon, A.-M., Pasta, F. & Lane, D. (2002). The parAB gene products of Pseudomonas putida exhibit partition activity in both P. putida and Escherichia coli. Mol Microbiol 43, 39–49. [DOI] [PubMed] [Google Scholar]
- Gordon, G. S., Sitnikov, D., Webb, C. D., Teleman, A., Straight, A., Losick, R., Murray, A. W. & Wright, A. (1997). Chromosome and low-copy-plasmid segregation in E. coli: visual evidence for distinct mechanisms. Cell 90, 1113–1121. [DOI] [PubMed] [Google Scholar]
- Irani, V. R. & Rowe, J. J. (1997). Enhancement of transformation in Pseudomonas aeruginosa PAO1 by Mg2+ and heat. Biotechniques 22, 54–56. [DOI] [PubMed] [Google Scholar]
- Ireton, K., Gunther, N. W., IV & Grossman, A. D. (1994). spo0J is required for normal chromosome segregation as well as the initiation of sporulation in Bacillus subtilis. J Bacteriol 176, 5320–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagura-Burdzy, G., Ibbotson, J. P. & Thomas, C. M. (1991). The korF region of broad-host-range plasmid RK2 encodes two polypeptides with transcriptional repressor activity. J Bacteriol 173, 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagura-Burdzy, G., Macartney, D. P., Zatyka, M., Cunliffe, L., Cooke, D., Huggins, C., Westblade, L., Khanim, F. & Thomas, C. M. (1999). Repression at a distance by the global regulator KorB of promiscuous IncP plasmids. Mol Microbiol 32, 519–532. [DOI] [PubMed] [Google Scholar]
- Jakimowicz, D., Brzostek, A., Rumijowska-Galewicz, A., Żydek, P., Dolzblasz, A., Smulczyk-Krawczyszyn, A., Zimniak, T., Wojtasz, Ł., Zawilak-Pawlik, A. & other authors (2007a). Characterization of mycobacterial chromosome segregation protein ParB and identification of its target in Mycobacterium smegmatis. Microbiology 153, 4050–4060. [DOI] [PubMed] [Google Scholar]
- Jakimowicz, D., Żydek, P., Kois, A., Zakrzewska-Czerwińska, J. & Chater, K. F. (2007b). Alignment of multiple chromosomes along helical ParA scaffolding in sporulating Streptomyces hyphae. Mol Microbiol 65, 625–641. [DOI] [PubMed] [Google Scholar]
- Jensen, R. B. & Gerdes, K. (1997). Partitioning of plasmid R1. The ParM protein exhibits ATPase activity and interacts with the centromere-like ParR-parC complex. J Mol Biol 269, 505–513. [DOI] [PubMed] [Google Scholar]
- Kahn, M. R., Kolter, R., Thomas, C. M., Figurski, D., Meyer, R., Remault, E. & Helinski, D. R. (1979). Plasmid cloning vehicles derived from plasmids ColE1, F, R6K, and RK2. Methods Enzymol 68, 268–280. [DOI] [PubMed] [Google Scholar]
- Kim, S.-K. & Shim, J. (1999). Interaction between F plasmid partition proteins SopA and SopB. Biochem Biophys Res Commun 263, 113–117. [DOI] [PubMed] [Google Scholar]
- Kim, H.-J., Calcut, M. J., Schmidt, F. J. & Chater, K. F. (2000). Partitioning of the linear chromosome during sporulation of Streptomyces coelicolor A3(2) involves an oriC-linked parAB locus. J Bacteriol 182, 1313–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler, T., Curty, L. K., Barja, F., van Delden, C. & Pechere, J. C. (2000). Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signalling and requires flagella and pili. J Bacteriol 182, 5990–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasocki, K., Bartosik, A. A., Mierzejewska, J., Thomas, C. M. & Jagura-Burdzy, G. (2007). Deletion of the parA (soj) homologue in Pseudomonas aeruginosa causes ParB instability and affects growth rate, chromosome segregation, and motility. J Bacteriol 189, 5762–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, P. S. & Grossman, A. D. (2006). The chromosome partitioning proteins Soj (ParA) and Spo0J (ParB) contribute to accurate chromosome partitioning, separation of replicated sister origins, and regulation of replication initiation in Bacillus subtilis. Mol Microbiol 60, 853–869. [DOI] [PubMed] [Google Scholar]
- Lee, P. S., Lin, D. C.-H., Moriya, S. & Grossman, A. D. (2003). Effects of the chromosome partitioning protein Spo0J (ParB) on oriC positioning and replication initiation in Bacillus subtilis. J Bacteriol 185, 1326–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M.-J., Liu, C.-H., Wang, S.-Y., Huang, C.-T. & Huang, N. (2006). Characterization of the Soj/Spo0J chromosome segregation proteins and identification of putative parS sequences in Helicobacter pylori. Biochem Biophys Res Commun 342, 744–750. [DOI] [PubMed] [Google Scholar]
- Leonard, T. A., Butler, P. J. & Lowe, J. (2005). Bacterial chromosome segregation: structure and DNA binding of the Soj dimer – a conserved biological switch. EMBO J 24, 270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, P. J. & Errington, J. (1997). Direct evidence for active segregation of oriC regions of the Bacillus subtilis chromosome and co-localization with the Spo0J partitioning protein. Mol Microbiol 25, 945–954. [DOI] [PubMed] [Google Scholar]
- Lewis, R. A., Bignell, C. R., Zeng, W., Jones, A. C. & Thomas, C. M. (2002). Chromosome loss from par mutants of Pseudomonas putida depends on growth medium and phase of growth. Microbiology 148, 537–548. [DOI] [PubMed] [Google Scholar]
- Lin, D. C.-H. & Grossman, A. D. (1998). Identification and characterization of a bacterial chromosome partitioning site. Cell 92, 675–685. [DOI] [PubMed] [Google Scholar]
- Lukaszewicz, M., Kostelidou, K., Bartosik, A. A., Cooke, G. D., Thomas, C. M. & Jagura-Burdzy, G. (2002). Functional dissection of the ParB homologue (KorB) from IncP-1 plasmid RK2. Nucleic Acids Res 30, 1046–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston, A. L. & Errington, J. (1999). Dynamic movement of the ParA-like Soj protein of B. subtilis and its dual role in nucleoid organization and developmental regulation. Mol Cell 4, 673–682. [DOI] [PubMed] [Google Scholar]
- Mohl, D. A. & Gober, J. W. (1997). Cell-cycle dependent polar localization of chromosome partitioning proteins in Caulobacter crescentus. Cell 88, 675–684. [DOI] [PubMed] [Google Scholar]
- Mohl, D. A., Easter, J., Jr & Gober, J. W. (2001). The chromosome partitioning protein, ParB, is required for cytokinesis in Caulobacter crescentus. Mol Microbiol 42, 741–755. [DOI] [PubMed] [Google Scholar]
- Mullis, K., Faloona, F., Scharf, S., Saiki, R., Horn, G. & Erlich, H. (1986). Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51, 263–273. [DOI] [PubMed] [Google Scholar]
- Murray, H., Ferreira, H. & Errington, J. (2006). The bacterial chromosome segregation protein Spo0J spreads along DNA from parS nucleation sites. Mol Microbiol 61, 1352–1361. [DOI] [PubMed] [Google Scholar]
- Niki, H. & Hiraga, S. (1997). Subcellular distribution of actively partitioning F plasmid during the cell division cycle in E. coli. Cell 90, 951–957. [DOI] [PubMed] [Google Scholar]
- Ogasawara, N. & Yoshikawa, H. (1992). Genes and their organization in the replication origin region of the bacterial chromosome. Mol Microbiol 6, 629–634. [DOI] [PubMed] [Google Scholar]
- Quisel, J. D. & Grossman, A. D. (2000). Control of sporulation gene expression in Bacillus subtilis by the chromosome partitioning proteins Soj (ParA) and Spo0J (ParB). J Bacteriol 182, 3446–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid, M. H. & Kornberg, A. (2000). Inorganic polyphosphate is needed for swimming, swarming, and twitching motilities of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 97, 4885–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin, N. V., Rech, J. & Lane, D. (2003). Mapping of functional domains in F plasmid partition proteins reveals a bipartite SopB-recognition domain in SopA. J Mol Biol 329, 875–889. [DOI] [PubMed] [Google Scholar]
- Reece, K. S. & Phillips, G. J. (1995). New plasmids carrying antibiotic-resistance cassettes. Gene 165, 141–142. [DOI] [PubMed] [Google Scholar]
- Saint-Dic, D., Frushour, B. P., Kehrl, J. H. & Kahng, L. S. (2006). A parA homolog selectively influences positioning of the large chromosome origin in Vibrio cholerae. J Bacteriol 188, 5626–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Molecular Cloning: a Laboratory Manual, 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Sharpe, M. E. & Errington, J. (1996). The Bacillus subtilis soj-spo0J locus is required for a centromere-like function involved in prespore chromosome partitioning. Mol Microbiol 21, 501–509. [DOI] [PubMed] [Google Scholar]
- Simon, R., O'Connell, M., Labes, M. & Puhler, A. (1986). Plasmid vectors for the genetic analysis and manipulation of rhizobia and other gram-negative bacteria. Methods Enzymol 118, 640–659. [DOI] [PubMed] [Google Scholar]
- Spratt, B. G., Hedge, P. J., de Heesen, S., Edelman, A. & Broome-Smith, J. K. (1986). Kanamycin-resistant vectors that are analogues of plasmids pUC8, pUC9, EMBL8 and EMBL9. Gene 41, 337–342. [DOI] [PubMed] [Google Scholar]
- Surtees, J. A. & Funnell, B. E. (1999). P1 ParB domain structure includes two independent multimerization domains. J Bacteriol 181, 5898–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaichi, Y. & Niki, H. (2000). Active segregation by the Bacillus subtilis partitioning system in Escherichia coli. Proc Natl Acad Sci U S A 97, 14656–14661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron, C., Vieira, J. & Messing, J. (1985). Improved M13 phage cloning vectors and host strains: nucleotide sequence of the M13mp18 and pUC19 vectors. Gene 33, 103–119. [DOI] [PubMed] [Google Scholar]