

Abstract
Matrix metalloproteinase-13 (collagenase-3), a member of the family of matrix metalloproteinases (MMPs), plays a major pathological role in the cartilage destruction of arthritis. A dramatic up-regulation of MMP-13 by inflammatory cytokines such as interleukin (IL)-1β or by fibronectin fragments has been observed in chondrocytes. In this study, we investigated the inhibitory effects of insulin-like growth factor-1 (IGF-1) and osteogenic protein-1 (OP-1) on the expression of MMP-13, which was induced by fibronectin fragment or IL-1β in human immortalized or human primary chondrocytes. IGF-1 and OP-1 each significantly reduced the basal level as well as fibronectin fragment- or IL-1β-stimulated transcription of the MMP-13 gene in a dose-dependent fashion with the corresponding decreases in the protein level of MMP-13. The most prominent suppressive effect was observed by the combination of IGF-1 and OP-1, which decreased the basal promoter activity by 60% and almost completely abrogated the fibronectin fragment-stimulated MMP-13 promoter activity. OP-1 was found to enhance mRNA levels of IGF-1 and the IGF-1 receptor, the latter of which appeared to be responsible for the combined effect of IGF-1 and OP-1. The suppressive effect of IGF-1 and OP-1 on MMP-13 expression was due in part to down-regulation of the expression of pro-inflammatory cytokines and the activity of their intermediate molecules, including NF-κB and AP-1 factors. We propose that IGF-1 and OP-1 could be key physiological regulators of MMP-13 gene expression and that the combination of IGF-1 and OP-1 may be useful in controlling the excess catabolic activity in arthritis.
Cartilage homeostasis is a well synchronized balance between anabolic and catabolic processes. During the development of osteoarthritis, this balance is disrupted resulting in progressive degradation of the articular cartilage (1). Studies have demonstrated that members of the matrix metalloproteinase (MMP)1 family are the major pathophysiological mediators of the cartilage destruction process in osteoarthritis (2). Recently, in vitro, clinical, and transgenic studies have provided evidence that chondrocyte MMP-13 (collagenase-3) is a leading candidate enzyme mediating the degradation of type II collagen in osteoarthritis (2–4). A dramatic up-regulation of MMP-13 gene expression in response to inflammatory cytokines, such as interleukin-1β (IL-1β) (5), or by fibronectin fragment (Fn-f) (6) has been observed in chondrocytes. However, there is limited knowledge of the cellular mechanisms that regulate MMP-13 gene expression in chondrocytes.
Accumulating evidence demonstrates that the bone morphogenetic proteins (BMPs), a subfamily of the transforming growth factor-β superfamily, are inhibitors of MMP-13 expression in human fetal chondrocytes (7) or rat osteoblast-enriched cells (8, 9). Upon ligand binding, the specific serine/threonine kinase activity of BMP receptors (type I or II) transduce signals (10) by allowing the association of Smad1 and Smad4 in the cytoplasm followed by the translocation of the Smad1/4 complexes into the nucleus (11). Smads up- or down-regulate the transcription of a subset of target genes that includes Runx2 (12). BMP-7, also known as the osteogenic protein-1 (OP-1), has been found to induce new cartilage and bone formation in vitro and in vivo (13–15) and regulate cell proliferation and differentiation (16, 17). OP-1 blocks cartilage damage caused by Fn-f and may promote repair by enhancing proteoglycan and collagen synthesis (18). More recently, it has been demonstrated that OP-1 is expressed in human articular chondrocytes and in osteoarthritis cartilage (19). Taken together, these findings imply potential involvement of endogenous OP-1 in cartilage repair mechanisms, either by enhancing matrix synthesis (18) or by repressing the pro-inflammatory expression of cytokines, such as IL-6 and IL-1β, as well as the chemokines IL-8 and monocyte chemotatic protein-1 (20). It has been proposed that the effects of OP-1 in chondrocytes are mediated through type I serine/threonine kinase receptors (activin receptor-like kinase), ALK/BMPR-IB (21).
Synergistic stimulation of cell differentiation and proliferation by insulin-like growth factor (IGF-1) and OP-1 has been demonstrated in rat osteoblastic cells (22) and in human primary chondrocytes.2 Yeh and colleagues (17, 23) have presented data indicating that IGF-1 gene expression may be up-regulated by OP-1 at both transcriptional and post-transcriptional level in osteoblastic cells. IGF-1 is a polypeptide that exerts anabolic/anticatabolic effects on cartilage. The over-expression of human IGF-1 promoted new tissue formation in an ex vivo model of articular chondrocyte transplantation (24). The anabolic effects of IGF-1 appeared to be mediated by stimulating proteoglycan synthesis in articular chondrocytes (24, 25) or by down-regulating the transcriptional induction of MMP enzymes (e.g. MMP-1, MMP-3, MMP-8, and MMP-13) and inflammatory cytokines (26, 27).
In the present study, we assessed the inhibitory effects of IGF-1 and OP-1 on the basal as well as Fn-f- and IL-1β-induced expression of MMP-13. We demonstrated that the combination of IGF-1 and OP-1 was effective at inhibiting both the Fn-f- and IL-1β-stimulated MMP-13 gene expression and defined the molecular mechanisms underlying the observed inhibitory effects.
EXPERIMENTAL PROCEDURES
Cell Culture
Human chondrocytes were isolated by enzymatic digestion of normal ankle articular cartilage obtained from tissue donors through the Gift of Hope Organ and Tissue Donor Network (formerly the Regional Organ Bank of Illinois) as previously described (6). The immortalized human chondrocyte C-28I2 cells were kindly provided by Dr. Mary Goldring (New England Baptist Bone and Joint Institute, Harvard Medical School, Boston, MA). The primary and immortalized chondrocytes both were maintained in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium containing 10% fetal bovine serum and antibiotics (Invitrogen).
Plasmid Construction and Transfection
A series of human MMP-13 promoter deletional DNA fragments, −1600MMP-13, −736MMP-13, −370MMP-13, and −186MMP-13, were subcloned into a firefly luciferase promoterless vector, pGL2-Enhancer (Promega) at the sites of SacI and BglII. The human MMP-1 promoter region (−562/+1) was sub-cloned into the same vector at the sites of XhoI and HindIII. For transfection, the cells were plated 24 h prior to transfection at a density of 1 × 106 cells/well in 6-well plates and transiently transfected with 2 μg of promoter-reporter constructs or 500 ng of cDNA expression vectors using FuGENE 6 transfection reagent (Roche Applied Science) following the manufacturer’s instructions. The promoterless vector, pGL2-Enhancer was used as a control, and a Renilla luciferase construct was included as an internal control for transfection efficiency. After 24 h incubation, the cells were rinsed in phosphate-buffered saline and changed to serum-free condition for 20–24 h followed by treatment with the 110-kDa Fn-f (1 μM) (Chemicon, Temecula, CA), IL-1β (25 ng/ml) (R&D System, Minneapolis, MN), IGF-1 (100 ng/ml) (a gift from Chiron Cooperation, Emeryville, CA), or OP-1 (100 ng/ml) (provided by Stryker Biotech, Hopkinton, MA). Inhibition of IL-1β was performed using 100 ng/ml IL-1Ra (Anakinra; a gift from Amgen, Thousand Oakes, CA). A neutralizing antibody to human IGF-1 (100 μg/ml) (R&D Systems, Minneapolis, MN) or IGF-1 receptor monoclonal antibody (100 μg/ml) (Oncogene, San Diego, CA) and IgG (100 μg/ml) for negative control (Sigma) were added directly to the medium. The cells were harvested after 24 h, and luciferase activity was assayed using a dual luciferase reporter assay system (Promega). All of the transfection experiments were repeated at least three times in duplicate.
An IGF-1 receptor cDNA construct, CVNIGF-1R, and its dominant negative, hIGF-1RDN, were provided by Dr. R. Baserga (Thomas Jefferson University, Philadelphia, PA). The human IGF-1 promoter reporter construct, IGF-1Luc, was provided by Dr. Peter S. Rotwein (Oregon Health Sciences University, Portland, OR). The constitutive active AP-1 factors, c-Jun and c-Fos cDNA constructs, LNCXc-Fos, and LNCXc-Jun were provided by Dr. Stephen Murphy (Mayo Clinic, Rochester, MN). NF-κB dominant negative, pCMV-IkBκM, and cis-reporting systems of pNF-κB-Luc, pAP-1-Luc, were purchased from Clontech (Palo Alto, CA) and Stratagene (La Jolla, CA).
RNA and Genomic DNA Extraction
Total cellular RNA and genomic DNA were isolated using the RNeasy® Mini kit and DNeasy® Tissue kit (Qiagen), respectively, according to the manufacturer’s instructions. All of the samples were stored at −80 °C until analyzed.
Reverse Transcription and Semi-quantitative Polymerase Chain Reaction
Reverse transcription (RT) was carried out with 2 μg of total cellular RNA using the ThermoScript™ RT-PCR System (Invitrogen) for first strand cDNA synthesis in 50 μl of reaction volume. The sequences for the primers and the conditions for their use are summarized in Table I. For all experiments, the conditions were determined to be in the linear range for the PCR amplification as described previously (29). Briefly, all of the samples were subjected to RT at the same time, and subsequently, all of the samples of cDNA were amplified by PCR at the same time to avoid any potential experiment to experiment variation in efficiency. Each RT sample was assessed for GAPDH cDNA. The levels of GAPDH mRNA did not vary with time after the addition of Fn-f, cytokine, or growth factors following 24 PCR cycles. Genomic DNA was included for the PCR to ensure that there was no genomic DNA contamination in the total RNA samples. The cDNA was amplified by PCR using 24 cycles of 95 °C for 30 s, 58~69 °C for 30 s, and 72 °C for 30 s in the presence of Taq polymerase (Invitrogen), 50 pmol of sense and antisense primers. PCR primers and annealing temperature used is shown in Table I. PCR products were resolved on 1.5% agarose gels and visualized by staining with ethidium bromide and UV transillumination. Integrated density values for the genes in question were normalized to the GAPDH values to yield a semi-quantitative assessment.
Table I.
PCR primers and annealing temperature used
| Gene | Primersa | Annealing temperature | RT-PCR product |
|---|---|---|---|
| °C | bp | ||
| MMP-13 | f:GGC TCC GAG AAA TGC AGT CTT TCT T r:ATC AAA TGG GTA GAA GTC GCC ATG C |
64 | 337 |
| IL-1β | f:GAG CTC GCC AGT GAA ATG ATG GC r:CAA GCT TTT TTG CTG TGA GTC CCG |
58 | 387 |
| IL-6 | f:GGA TGC TTC CAA TCT GGA TTC AAT GAG r:CGC AGA ATG AGA TGA GTT GTC ATG TCC |
66 | 302 |
| IL-8 | f:CGT GGC TCT CTT GGC AGC CTT CCT GAT r:TCA AAA ACT TCT CCA CAA CCC TCT GCA |
68 | 270 |
| IGF-I | f:CAC TGT CAC TGC TAA ATT CAG AGC AGA T r:AGC CCC TGT CTC CAC ACA CGA ACT GAA G |
66 | 378 |
| IGF-1 receptor | f:CGA GTA CTT GCT GCT GTT CCG AGT r:CAC AGC GCC AGC CCT CAA ACC TGT |
69 | 583 |
| GAPDH | f:GGT ATC GTG GAA GGA CTC AT r:ACC ACC TGG TGC TCA GTG TA |
60 | 341 |
f, forward; r, reverse.
MMP-13 Analysis by Western Blot
Conditioned media from control and treated chondrocytes were concentrated (10:1) using Centricon YM 10 filters (Millipore, Bedford, MA) and centrifugation. The soluble protein concentration was determined with BCA reagent (Pierce), and the samples containing equal amounts of total protein were separated by SDS-PAGE followed by transferring to nitrocellulose for immunoblotting. The detection of MMP-13 was performed by using antibody to MMP-13 and ECL system (Amersham Biosciences). MMP-13 antibody L2916 was generously provided by Dr. Gillian Murphy (Norwich, UK).
RESULTS
Characterization of Basal Promoter Activity of the 5′-Flanking Region of the Human MMP-13 Gene in Chondrocytes
The MMP-13 promoter contains multiple putative recognition sites for distinct classes of transcription factors. To evaluate the basal promoter activity of the MMP-13 gene, we prepared a series of 5′ deletion constructs of MMP-13 and a MMP-1 promoter-luciferase reporter construct (for the purpose of comparison) (Fig. 1). The 5′ promoter region deletion constructs of MMP-13 and −562MMP-1 were transiently transfected into immortalized human chondrocytes. The full-length of MMP-13 promoter construct, −1600MMP-13, showed the strongest basal promoter activity. The shortest promoter construct, −186MMP-13, containing the essential promoter region, such as TATA box, AP-1, Ets/PEA3, and Runx2 sites, demonstrated ~60% of the basal promoter activity shown in −1600MMP-13. The −562MMP-1 promoter construct showed activity similar to that of −370MMP-13. These results suggested that not only the proximal core factors, such as AP-1 or Ets, but also the multiple putative recognition motifs for transcription factors present in the 5′-flanking region are necessary to exert the maximum basal promoter activity of the MMP-13 gene in chondrocytes.
Fig. 1. Structural features of 5′-flanking sequences (nucleotides −1600 to +1) of the MMP-13 gene and the characterization of basal promoter activity.

A, positions of putative transcription factor binding sites on the MMP-13 promoter. Core transcriptional regulatory sites including a TATA box are filled in black. The TATA box is located 22 bp upstream from the transcription initiation site (TIS). B, promoter activities of the MMP-13 and −562MMP-1 promoter-luciferase deletion constructs. Immortalized human chondrocytes were seeded 24 h before the transfection in growth culture medium followed by transient transfection with the promoter deletion constructs. 24 h post-transfection, the cells were harvested and subjected to luciferase assay. The length of the tested promoter fragments is indicated. The numbers indicate the relative positions with respect to the transcription start site (TIS). Luciferase activity is shown as n-fold value compared with cells transfected with the promoterless pGL2-Enhancer vector (Basic, which was assigned an activity value of 1.0). The data represent the means of three independent experiments in duplicate, with at least two different plasmid preparations.
IGF-1 and OP-1 Down-regulate Chondrocyte-associated Basal MMP-13 Gene Expression
Transcriptional down-regulation of MMP-13 expression by IGF-1 has been previously reported in rat bone cell cultures (26) and in bovine nasal cartilage (27). We investigated whether the treatment of IGF-1, OP-1, or the combination of IGF-1 and OP-1 is capable of down-regulating the basal level of the MMP-13 expression in human articular chondrocytes. As shown in Fig. 2A, the full-length of MMP-13 promoter construct, −1600MMP-13, demonstrated the strongest response to IGF-1 or OP-1 with an ~30% reduction in the promoter activity. Further deletions (−736, −370, and −186MMP-13) resulted in the gradual decrease of the inhibitory effect of IGF-1 and OP-1 by down-regulating the promoter activities approximately from 20% to 15% (Fig. 2A). These results suggest that not a single motif but multiple regulatory motifs within the MMP-13 promoter are responsible for the maximum inhibitory effect of IGF-1 and/or OP-1. The combination of IGF-1 and OP-1 further reduced the basal promoter activity of MMP-13 (60% reduction) compared with control. The inhibitory effects of IGF-1, OP-1, and especially the combination of the two growth factors on MMP-13 transcription were further confirmed by Western blot analysis using primary human chondrocytes (Fig. 2, B and C).
Fig. 2. Inhibitory effects of IGF-1 and OP-1 on MMP-13 and MMP-1 gene expression.

A, a series of human MMP-13 promoter deletion constructs and the MMP-1 (-562) promoter construct were transiently transfected into the immortalized human chondrocytes, C-28I2. 24 h post-transfection, the cells were changed to serum-free medium and subsequently treated with IGF-1 (100 ng/ml), OP-1 (100 ng/ml), or combination of IGF-1 and OP-1 followed by further incubation for another 24 h. Then, the cells were harvested and subjected to luciferase activity assay. B, immunoblotting analysis of endogenous MMP-13 protein secreted by human primary chondrocytes cultured for 24 h in serum-free medium in the presence or absence of IGF-1 (100 ng/ml), OP-1 (100 ng/ml), or the combination of IGF-1 and OP-1, each 100 ng/ml, respectively. C, relative amount of the MMP-13 protein level from densitometric scanning of immunoblots (n = 3) depicted as relative intensity to control which was assigned as 1.0. This histogram represents the means and S.E. of three independent experiments. D, the full-length MMP-13 promoter-luciferase construct, −1600MMP-13, was transiently transfected into immortalized human chondrocytes, which were then treated with increasing amounts of IGF-1 or OP-1. Each growth factor was tested alone or in combination with a fixed amount of the other growth factor.
The inhibitory effect of IGF-1 and OP-1 on MMP-13 promoter activity was concentration-dependent. The maximum inhibitory activity of IGF-1 or OP-1 was observed at the concentration of 100 ng/ml, whereas the treatment with more than 200 ng/ml of either IGF-1 or OP-1 alone showed no further dose-dependent inhibitory effect on MMP-13 expression (Fig. 2D). Individual treatment of IGF-1 or OP-1 at the concentration of 10 ng/ml showed no effect on the MMP-13 promoter activity. However, it was noted that when a fixed amount of IGF-1 or OP-1 (each 100 ng/ml) was preincubated, the concentration of 10 ng/ml of the other growth factor was able to further down-regulate MMP-13 promoter activity. These results suggest that one growth factor enhances the cellular sensitivity for the other growth factor.
IGF-1 and OP-1 Inhibit IL-1β- and Fn-f-elicited MMP-13 Expression
Up-regulation of MMP-13 expression in response to Fn-f or IL-1β has been observed in chondrocytes. Therefore, we examined whether IGF-1 and OP-1 had suppressive effects on Fn-f- or IL-1β-stimulated MMP-13 expression by transient transfection assays. IGF-1 and OP-1 alone and in combination showed a stronger inhibitory effect on Fn-f-induced MMP-13 promoter activity (Fig. 3A) than the activity induced by IL-1β (Fig. 3B). The combination of IGF-1 and OP-1 almost completely inhibited Fn-f-stimulated activity and partially inhibited IL-1β-stimulated activity of MMP-13 promoter expression. Similar results were obtained in cells transfected with the −562MMP-1 promoter construct. Taken together, we conclude that IGF-1 and OP-1 exert a combinatory effect in the suppression of basal as well as Fn-f- or IL-1β-stimulated MMP-13 gene expression in human chondrocytes.
Fig. 3. Inhibitory effects of IGF-1 and OP-1 on Fn-f- and IL-1β-elicited MMP-13 and MMP-1 promoter activity.

Transiently transfected immortalized human chondrocytes with a full-length MMP-13 and −562MMP-1 promoter-luciferase constructs were serum-starved for 20–24 h followed by treatment with Fn-f (A, 1 μM) or IL-1β(B, 25 ng/ml) in the presence or absence of IGF-1 (100 ng/ml) or OP-1 (100 ng/ml). After 24 h of incubation, luciferase activity was measured and shown as n-fold value compared with cells transfected with the promoterless pGL2-Enhancer vector (Basic, which was assigned an activity value of 1.0). The data represent the means of three independent experiments in duplicate, with at least two different plasmid preparations.
The Combined Effect of IGF-1 and OP-1 on MMP-13 Expression Is Due to Enhancement of the IGF-1 Autocrine System by OP-1
Individual treatment with the increased amounts of either IGF-1 (2 μg) or OP-1 (2 μg) did not show any further inhibitory effect on the promoter activity of MMP-13 (Fig. 2D). In addition, the preincubated fixed amount (100 ng/ml) of IGF-1 or OP-1 allowed the cells to respond as to low a concentration as 10 ng/ml of IGF-1 or OP-1 by increasing the cellular sensitivity. Therefore, studies were initiated to understand the molecular mechanisms by which the combination of two growth factors, IGF-1 and OP-1, exerts the most prominent effect on the expression of MMP-13 gene in chondrocytes. The elevated mRNA levels of rat IGF-1 and IGF-1-binding proteins but not IGF-1 receptors by human OP-1 have been reported in rat osteoblast cells (23). Thus, we postulated that IGF-1 and OP-1 may influence each other, stimulating the cellular autocrine system for either IGF-1 or OP-1. To study the influence of OP-1 on the expression of the IGF-1 gene, a human IGF-1 promoter construct containing a luciferase reporter gene was utilized. Transient transfection of the IGF-1 promoter-luciferase construct followed by the treatment with OP-1 increased the IGF-1 promoter activity ~100% in immortalized chondrocytes and 60% in primary human chondrocytes above controls (Fig. 4A). The induction of IGF-1 expression by OP-1 was further supported by semi-quantitative RT-PCR assay. The addition of OP-1 increased the mRNA levels of not only IGF-1 but also the IGF-1 receptor in immortalized chondrocytes (Fig. 4B). Treatment with IGF-1 for 36 h, on the other hand, showed no significant effect on the mRNA or protein levels of OP-1 or OP-1 receptors including ActR-I, BMPR-1A, BMPR-1B, and BMPRII (data not shown). Taken together, these results suggest that OP-1 may stimulate the IGF-1 autocrine system at the level of transcription in human chondrocytes.
Fig. 4. Stimulatory effect of OP-1 on the IGF-1 autocrine system.

A, the human IGF-1 promoter-luciferase construct was transiently transfected into immortalized human chondrocytes (open bars) or into primary human chondrocytes (filled bars). The cells were serum-starved (24 h) and then treated with OP-1 (100 ng/ml) followed by incubation for another 24 h. Luciferase activity is shown as n-fold value compared with cells transfected with the promoterless pGL2-Enhancer vector (Basic, which was assigned an activity value of 1.0). The data represent the means of three independent experiments in duplicate, with at least two different plasmid preparations. B, RT-PCR analysis by using total RNA extracted from immortalized chondrocytes in the presence and absence of OP-1 (100 ng/ml). Lanes M, 100 bp DNA size marker; lanes 1, control; lanes 2, incubated with OP-1 for 36 h. The expected RT-PCR produce sizes are shown in Table I.
To determine whether the stimulated autocrine system of IGF-1 caused the suppression of the MMP-13 gene, we tested the effect of a blocking monoclonal antibody to the IGF-1 receptor and an IGF-1 neutralizing antibody on the inhibitory activity of IGF-1. Treatment with nonspecific IgG (100 μg/ml) was included as a negative control. Indeed, the treatment with either the antibody to IGF-1 receptor or the IGF-1 neutralizing antibody completely abolished the IGF-1 effect on MMP-13 promoter activity (Fig. 5A). The treatment with control IgG did not alter the MMP-13 promoter activity. Next, we examined the effect of the same antibodies on the inhibitory activity of OP-1. Importantly, we found that the same antibodies also blocked the inhibitory activity of OP-1 on MMP-13 gene expression. Similar results were obtained by transient transfection studies performed by using human IGF-1 receptor dominant negative (hIGF-1RDN), which blocks IGF-1-induced cellular signaling pathway. The forced expression of the hIGF-1RDN almost completely abolished cellular responses to not only IGF-1 but also OP-1 (Fig. 5B). The empty vector that was used for negative control did not show any significant effect on the promoter activity. These results suggest that OP-1 was acting through stimulation of the IGF-1 system to down-regulate MMP-13 gene expression. However, it was worth noting that the antibodies to IGF-1, IGF-1 receptor, or the co-expression of hIGF-1RDN only partially abolished the combinatory effects of IGF-1 and OP-1 on MMP-13 expression (Fig. 5).
Fig. 5. Inhibition of the IGF-1 autocrine system reduces the inhibitory effects of IGF-1 and OP-1 on MMP-13 expression.

A, the full-length MMP-13 promoter-luciferase construct was transiently transfected into immortalized human chondrocytes. The transfected cells were treated with IGF-1, OP-1, or the combination of IGF-1 and OP-1 in the presence or absence of antibody to IGF-1 (IGF-1Ab) or IGF-1 receptor (IGF-1R-Ab) at a concentration of 100 μg/ml. In parallel, treatment with IgG (100 μg/ml) was carried out as a negative control. B, the full-length MMP-13 promoter-luciferase construct was co-transfected with 0.5 μg of human IGF-1 receptor dominant negative cDNA construct (hIGF-1RDN) in immortalized human chondrocytes. Co-expression with empty vector was performed as a negative control.
The fact that increasing the amount of IGF-1 above 100 ng/ml had no further inhibitory effect on MMP-13 expression, along with the fact that OP-1 increases the IGF-1 receptor expression, suggested that the IGF-1 receptor could be a limiting factor in this system. If the cellular level of IGF-1 receptor is the limiting factor in the response to IGF-1, we may observe dose-dependent down-regulation of MMP-13 gene expression by forced expression of the IGF-1 receptor. Transient transfection experiments were performed to express human IGF-1 receptor (pCVNIGF-1R) followed by the treatment of increasing IGF-1 up to 2 μg/ml in chondrocytes. We observed an enhanced cellular response to IGF-1 (100 ng/ml) by the forced expression of IGF-1 receptor (Fig. 6A). Higher concentration of IGF-1 (2 μg/ml) with the forced expression of IGF-1 receptor did not show a further enhanced cellular response. No significant enhancement of the IGF-1 effect on MMP-13 promoter activity was observed when the OP-1 receptor (BMPR-1B) was expressed followed by the addition of IGF-1. On the other hand, the forced expression of either IGF-1 receptor or OP-1 receptor enhanced the inhibitory effect of OP-1 on MMP-13 promoter activity (Fig. 6B). Taken together, we concluded that OP-1-mediated up-regulation of IGF-1 and the IGF-1 receptor, thereby stimulating the IGF-1 autocrine system, was responsible in part for the combinatory effect of IGF-1 and OP-1 on MMP-13 expression in human chondrocytes.
Fig. 6. Forced expression of the IGF-1 receptor enhances the effect of IGF-1 and OP-1 on MMP-13 expression.
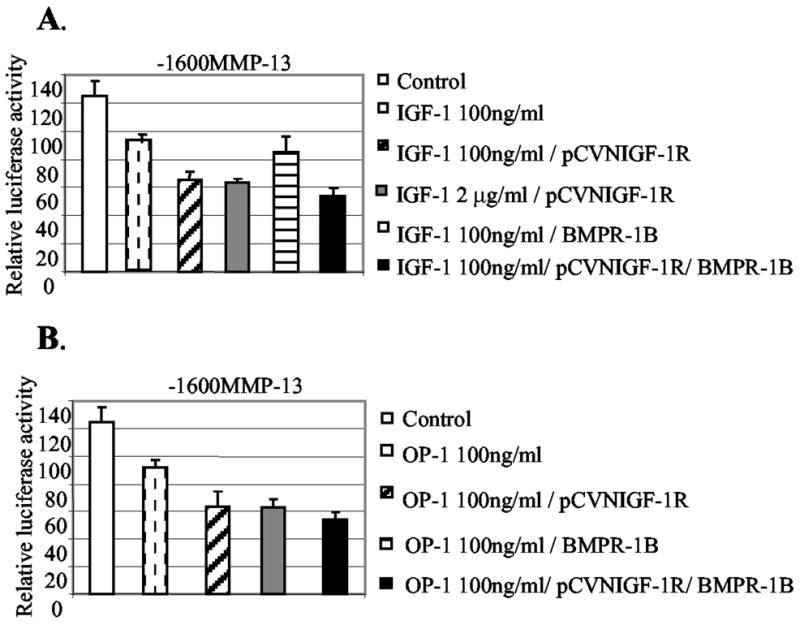
A, the human IGF-1 receptor expression vector (pCVNIGF-1R) and/or OP-1 receptor expression vector (BMPR-1B) were transiently co-expressed (0.5 μg) with the full-length promoter-reporter construct, −1600MMP-13, in immortalized human chondrocytes in the presence of IGF-1 at the concentrations 100 ng/ml or 2 μg/ml. B, promoter construct, −1600MMP-13, was transiently transfected in the presence of OP-1 with or without co-expression of the OP-1 receptor (BMPR-1B), IGF-1 receptor (pCVNIGF-1R), or the combination of these two receptor expression vectors. Luciferase activity is shown as n-fold value compared with cells transfected with the promoterless pGL2-Enhancer vector, which was assigned as activity value of 1.0. The data represent the means of two independent experiments in duplicate, with at least two different plasmid preparations for the transient transfection into immortalized human chondrocytes.
IGF-1 and OP-1 Mediate Repression of Inflammatory Cytokines in Human Chondrocytes
It has been reported that IGF-1 and OP-1 suppress inflammatory cytokine expression in primary human proximal tubule epithelial cells or in skin (20, 30). The pro-inflammatory cytokines are also strong inducers for MMPs (31–33). Therefore, we postulated that the potent inhibitory effects of IGF-1 and OP-1 on MMP-13 expression may in part result from the suppression of inflammatory cytokine expression and/or interference with the inflammatory cytokine-associated intermediate gene product signaling pathways in human chondrocytes. IL-1β or Fn-f stimulation significantly increased the mRNA level of MMP-13 as well as inflammatory cytokines in immortalized chondrocytes (Fig. 7A). These included IL-1β, IL-6, and IL-8. Upon the addition of the combination of IGF-1 and OP-1, we observed decreased expression of endogenous inflammatory cytokines that were induced by Fn-f or IL-1β (Fig. 7, B and C). Several studies have demonstrated that the inhibition of inflammatory cytokine-stimulated AP-1 and NF-κB transcription factors down-regulates MMP-13 gene expression in chondrosarcoma cells or human chondrocytes (32, 33), showing the direct/indirect effects of these transcriptional factors in modulating MMP-13 expression. Thus, we tested whether the blocking of NF-κB or overexpression of AP-1 factors, transcription factors required for the induction of MMP-13 by IL-1 (34), can modulate the IGF-1 and OP-1 effect on MMP-13 expression. Co-transfection of mutant IκB cDNA construct, pCMV-IkBαM, which can block NF-κB signaling, dramatically diminished the basal level as well as the IL-1β- or Fn-f-induced MMP-13 promoter activity (Fig. 7D). The inhibition of NF-κB signaling pathway could cause apoptosis in chondrocytes (35). Therefore, we performed DNA fragmentation analysis to make sure that the reduced promoter activity of MMP-13 was not due to cell death. We observed no sign of apoptosis from the cells transfected with pCMV-IkBαM (data not shown). In addition, the co-expression of AP-1 factors, such as c-Fos and c-Jun, abrogated the IGF-1-and OP-1-mediated suppression of MMP-13 promoter activity (Fig. 7D).
Fig. 7. IGF-1 and OP-1 reduce inflammatory cytokine expression in chondrocytes.

A, immortalized human chondrocytes were serum-starved for 24 h followed by incubation with IL-1β (25 ng/ml) or Fn-f (1 μM) for 36 h. Each sample was harvested for total RNA extraction and subjected to semi-quantitative RT-PCR. Lanes M, 100-bp DNA size marker; lanes G, genomic DNA; lanes 1, control; lanes 2, treatment with IL-1β; lanes 3, treatment with Fn-f. Expected RT-PCR product sizes are shown in Table I. Genomic DNA for MMP-13 amplification (1.1 kb) was included as a control. The densitometric band intensity of PCR products was depicted after normalization by GAPDH. B and C, immortalized human chondrocytes were serum-starved for 24 h and incubated with Fn-f (B, 1 μM) or IL-1β (C, 25 ng/ml) in the presence or absence of the combination of IGF-1 and OP-1 at the concentration of 100 ng/ml, respectively. After incubation for 36 h, each sample was harvested for total RNA extraction and subjected to semi-quantitative RT-PCR. Lanes M, 100 bp DNA size marker; lanes 1, control; lanes 2, treatments with the combination of IGF-1 and OP-1 at the concentration of 100 ng/ml. D, full-length promoter construct, −1600MMP-13, was transiently co-transfected with a mutant IκB cDNA construct, IkBαM, which can block the NF-κB signaling pathway, or with AP-1 factors (c-Fos/c-Jun). The transiently transfected cells were treated with IL-1β (25 ng/ml) or Fn-f (1 μM) in the presence or absence of combination of OP-1(100 ng/ml) and IGF-1(100 ng/ml). The relative luciferase activity is shown as n-fold value compared with cells transfected with promoterless pGL2-Enhancer vector, which was assigned an activity value of 1.0.
In parallel, transient transfection with the NF-κB-responsive control plasmid, pNF-κB-Luc or AP-1 control plasmid, pAP1-Luc, which contains three tandem repeats of NF-κB or AP-1 consensus sequences, respectively, was performed as a control for the responsiveness to IL-1β, mutant IκB (pCMV-IκBαM), or the AP-1 expression vectors. Without stimulation, the basal expression pattern of these control plasmids was not significantly modulated in response to IGF-1, OP-1, or the combination of the two growth factors. However, when chondrocytes were stimulated with Fn-f or IL-1β, the combination of IGF-1 and OP-1 significantly reduced IL-1β- or Fn-f-elicited activity of pNF-κB-Luc and pAP-1-Luc (Fig. 8). The co-expression of pCMV-IkBαM, which blocks the NF-κB signaling pathway, completely abolished the IL-1β-stimulated and, to a lesser extent, Fn-f-stimulated promoter activity of pNF-κB-Luc, whereas the co-expression of AP-1 factors (c-Fos/c-Jun) dramatically increased the promoter activity of pAP1-Luc (16-fold). In addition, the co-expression of AP-1 factors abrogated the suppressive effect of IGF-1 and OP-1 on the promoter activity of pAP1-Luc. Taken together, these results suggest that NF-κB and AP-1 factors are downstream regulators involved in the stimulation of MMP-13 expression by Fn-f or IL-1β in human chondrocytes. Therefore, the down-regulation of inflammatory cytokine expression and/or their intermediate signaling molecules may be part of the mechanism by which IGF-1 and OP-1 exert their suppressive effects on MMP-13 expression in human chondrocytes.
Fig. 8. The combination of IGF-1 and OP-1 down-regulates the activity of the cis-acting control plasmids, pNF-κB-Luc, and pAP-1-Luc.

The cis-acting control plasmids, pNF-κB-Luc (A) or pAP-1-Luc (B), which contain three tandem repeats of NF-κB or AP-1 consensus sequences, respectively, were transiently transfected into immortalized human chondrocytes in the presence or absence of factors as indicated. The luciferase activity was calculated by comparing with the luciferase activity of control plasmid, pNF-κB-Luc or pAP-1-Luc (assigned as 100). The data represent the means of two independent experiments in duplicate, with at least two different plasmid preparations for the transient transfection into immortalized human chondrocytes.
The Effects of the IL-1Ra on Fn-f- or IL-1β-stimulated MMP-13 Expression in Human Chondrocytes
There have been controversial reports on whether the Fn-f-induced MMP-13 expression is IL-1-dependent. Several studies have shown that Fn-f increase MMP expression via an IL-1 auto-crine loop (36, 37). On the other hand, stimulated expression of MMPs mediated by an IL-1-independent mechanism has been reported (38). Previously, we reported that the IL-1 signal transduction pathway may at least in part control Fn-f-induced MMP-13 expression (6), suggesting the cross-talk between Fn-f and IL-1 signaling molecules leading to the stimulation of MMP-13 expression. Therefore, we determined the effect of IL-1Ra on Fn-f-induced MMP-13 gene expression compared with that of IL-1β in the presence or absence of IGF-1 and OP-1. IL-1Ra almost completely abolished IL-1β-induced MMP-13 promoter activity, whereas it decreased Fn-f-induced MMP-13 expression ~25% in immortalized human chondrocytes (Fig. 9A). When combined with IGF-1 and OP-1, IL-1Ra further reduced Fn-f- or IL-1β-induced MMP-13 promoter activity to the basal levels. In parallel experiments, similar results were obtained in cells transfected with the MMP-1 promoter construct (data not shown). Our transient transfection results were further confirmed by semi-quantitative RT-PCR (Fig. 9B). Consistent with the transient transfection results, the addition of IL-1Ra significantly reduced IL-1β-induced MMP-13 mRNA, and to a lesser extent, Fn-f-induced MMP-13 mRNA levels. IL-1Ra combined with IGF-1 and OP-1 further down-regulated MMP-13 mRNA levels, completely abolishing the induction of MMP-13 expression medicated by Fn-f or IL-1β. Taken together with our previous findings in which IL-1Ra significantly reduced IL-1β-induced MMP-13 protein level and, to a lesser extent, Fn-f-induced MMP-13 protein expression by human primary chondrocytes (6), the results suggest that the Fn-f-induction of MMP-13 occurs through two mechanisms, via IL-1-dependent and IL-1-independent pathways.
Fig. 9. Inhibitory effects of IL-1Ra together with IGF-1 and OP-1 on IL-1β- or Fn-f-elicited MMP-13 or MMP-1 expression.

A, full-length MMP-13 promoter-reporter construct 1600MMP-13 was transiently transfected into immortalized human chondrocytes. These transfected cells were serum-starved for 24 h followed by incubation with IL-1β (25 ng/ml) or Fn-f (1 μM) in the presence or absence of the combination of IGF-1 and OP-1 or IL-1Ra at the concentration of 100 ng/ml, respectively. The cells were harvested, and luciferase activity was measured as a relative activity to the promoterless plasmid, pGL2-Enancer vector (Basic). B, modulation of MMP-13 mRNA levels was analyzed by semi-quantitative RT-PCR. IL-1β-stimulated (25 ng/ml) or Fn-f-stimulated (1 μM) cells were treated with IL-1Ra (100 ng/ml) alone or the combinations of IL-1Ra with IGF-1 (100 ng/ml) and OP-1 (100 ng/ml) for 36 h. The cells were harvested, and the total RNA was extracted. RT-PCR analysis was performed. Lane G, genomic DNA; lane M, 100 bp DNA marker; lane 1, control; lane 2, treatment with IL-1β; lane 3, treatment with IL-1β + IL-1Ra; lane 4, treatment with Fn-f; lane 5, treatment with Fn-f + IL-1Ra; lane 6, treatment with IL-1β + IL-1Ra + IGF-1 + OP-1; lane 7, treatment with Fn-f + IL-1Ra + IGF-1 + OP-1.
DISCUSSION
In the present study we investigated the inhibitory effects of IGF-1 and OP-1 on MMP-13 expression in human chondrocytes. We found that the suppressive effect of IGF-1 and OP-1 on the MMP-13 promoter activity was dose-dependent at the transcriptional level with a corresponding decrease in the level of MMP-13 protein. We report that IGF-1 and OP-1 each reduced not only the basal chondrocyte-related expression of MMP-13 and MMP-1 but also the induced expression of these two genes following stimulation by IL-1β or Fn-f. The most prominent suppression of basal and induced MMP-13 expression was observed following treatment with IGF-1 and OP-1 together, presumably because the dual treatment allows a more complete blockade of MMP expression. In addition, we observed that the combined suppressive effect of IGF-1 and OP-1 was more pronounced on Fn-f-induced MMP-13 promoter activity than that induced by IL-1β. On the other hand, IL-1Ra more efficiently inhibited IL-1β-induced MMP-13 promoter activity than that induced by Fn-f. These results support our previous finding that the signaling from Fn-f that regulates MMP-13 expression can be transduced by both IL-1-dependent and IL-1-independent pathways (6). Combining IL-1Ra with IGF-1 and OP-1 exerts a slightly stronger suppressive effect on both the Fn-f- and IL-1β-induced MMP-13 expression, as reflected by a down-regulation of promoter activity and mRNA similar to the basal control level. Also, from the fact that IL-1Ra has less of a suppressive effect on the Fn-f-induced MMP-13 activity than on the IL-1β induced MMP-13 expression, we conclude that the combination of IGF-1 and OP-1 together with IL-1Ra represents an effective strategy for controlling Fn-f-induced MMP-13 gene expression.
Our data indicate that the combined effect of IGF-1 and OP-1 on MMP-13 expression is mediated, at least in part by (a) the OP-1-elicited stimulation of the IGF-1 cellular autocrine system in human chondrocytes and (b) the ability of IGF-1 and OP-1 to reduce the expression of inflammatory cytokines and the activity of the inflammatory cytokine-associated intermediate gene products. This concept is based on several findings. First, recombinant human OP-1 increases the transcription of IGF-1 and the IGF-1 receptor, whereas no significant induction of OP-1 or OP-1 receptor mRNA synthesis was observed by IGF-1. Second, an excess amount of antibodies to IGF-1 and the IGF-1 receptor or forced expression of the human IGF-1 receptor dominant negative completely abolishes the inhibitory effect of IGF-1 as well as OP-1 on MMP-13 promoter activity. Third, forced expression of the human IGF-1 receptor enhances the inhibitory effect of IGF-1 and OP-1; yet forced expression of an OP-1 receptor (BMPR-1B) shows no significant enhancement of the IGF-1 effect on MMP-13 promoter activity. Further investigation would be necessary for other OP-1 receptor components such as BMPR-1A or BMPR-II. The enhanced OP-1 effect upon elevating IGF-1 receptor protein levels by forced expression may be due to the stimulated IGF-1 autocrine system by OP-1, which reflects cellular conditions similar to adding the combination of IGF-1 and OP-1. Finally, down-regulated expression of inflammatory cytokines or the reduced activation of the inflammatory cytokine-associated gene products by the combination of IGF-1 and OP-1 may directly/indirectly reinforce the combined effect of IGF-1 and OP-1.
Our observations also suggest that a complicated process through cross-talk between the signaling molecules of IGF-1 and OP-1 in chondrocytes may be involved in the combined effects of IGF-1 and OP-1. For example, an excess amount of antibodies to IGF-1 and the IGF-1 receptor completely abrogates the individual inhibitory effect of IGF-1 and OP-1 but only partially blocks the combined effect of IGF-1 and OP-1. Furthermore, the preincubation of cells with IGF-1 (100 ng/ml) followed by a gradual increase in the amount of OP-1 or vice versa sensitizes cells to OP-1 or IGF-1. Preincubated chondrocytes by either IGF-1 or OP-1 were able to respond to concentrations of as low as 10 ng/ml of IGF-1 or OP-1. Furthermore, preliminary data revealed that IGF-1 treatment significantly down-regulates Smad7 gene expression, causing interference with the OP-1 signaling pathway.3 This observation suggests that IGF-1 may also in turn stimulate the OP-1 autocrine system through indirect mechanisms in human chondrocytes. Defining the molecular basis for the cross-talk between the IGF-1 and OP-1 signaling pathways should be undertaken as future studies for better understanding of the inhibitory mechanism of IGF-1 and OP-1 on MMP-13 gene expression in chondrocytes.
Transient transfection of a series of deletion promoter constructs of MMP-13 into immortalized or human primary chondrocytes showed the highest luciferase activity driven by the full-length MMP-13 promoter, −1600MMP-13. The −1600MMP-13 promoter encodes multiple putative recognition motifs for transcription factors, including the core binding sites for AP-1 and Ets, as well as two copies of Runx2 (Cbfa I), HRE, NF-κB, and several Sox family members. With further deletion of the 5′-flanking region, we observed decreased MMP-13 promoter activity. This observation suggested that although the proximal core binding sites are critical for basal promoter activity (39), the multiple putative recognition sites in the distal 5′-flanking region of MMP-13 gene may be necessary to mediate maximal MMP-13 promoter activity in human chondrocytes.
Our data also revealed that the full-length of MMP-13 promoter construct, −1600MMP-13, was most responsive to the treatment of IGF-1 and OP-1. The response was gradually decreased as the promoter deletion progressed. The shortest promoter construct, −186MMP-13, showed the minimal response to the growth factors. These results suggested that the inhibitory effects of the growth factors may not be mediated by a single response element but are rather mediated through multiple cis-acting elements present in the distal 5′-flanking promoter region of the MMP-13 gene. Although the DNA-protein interaction of AP-1 factors in the regulation of MMP-13 gene is well known (39–41), the molecular mechanism of NF-κB, including the cis-acting element on MMP-13 promoter region, is still not clear. Our gene analysis identified the putative NF-κB motif in the proximal 5′-flanking MMP-13 promoter region between −736 and −1600, and we propose that this region would be a potential candidate to study for the NF-κB-mediated MMP-13 gene expression.
Mengshol et al. (31) reported that transiently transfected MMP-13 promoters were not IL-1-inducible. In our system, however, using either primary or immortalized human chondrocytes, promoter activity of MMP-13 was responsive to treatment with IL-1β. The IL-1β-stimulated MMP-13 promoter activity was abolished upon the addition of IL-1Ra, which is known to antagonize activity of the IL-1 receptor. These results suggest that the MMP-13 promoter construct is responsive to treatment with IL-1β. The difference in our results from the previous report may be due to either the cell system used for the experiments or from the way in which the promoter-reporter constructs were generated. We utilized a firefly luciferase reporter vector, pGL2-Enhancer, which is promoterless but contains an enhancer region reflecting much higher sensitivity than the pGL3-Basic that was used by the other group.
In summary, the present study demonstrates the inhibitory effects of IGF-1 and OP-1 on basal as well as on the Fn-f- and IL-1β-induced expression of the MMP-13 gene in immortalized human chondrocytes in a concentration-dependent manner. The combination of these two growth factors shows prominent inhibition of MMP-13 expression at the transcriptional level with corresponding decreases in MMP-13 protein. The molecular mechanisms by which OP-1 and IGF-1 exert their combined effects on MMP-13 appeared to be in part through OP-1-mediated stimulation of the IGF-1 autocrine system and the suppression of pro-inflammatory cytokines by IGF-1 and OP-1 in human chondrocytes. Our findings suggest that IGF-1 and OP-1 could be important physiological regulators of MMP-13 expression, and the combination of IGF-1 and OP-1 may be useful in controlling the catabolic activity in arthritis.
Acknowledgments
We thank the Gift of Hope Organ and Tissue Donor Network and Dr. Arkady Margulis for providing human donor tissues. We thank Stryker Biotech for providing OP-1, Amgen for IL-1Ra, Chiron for IGF-1 and Stephan Soeder for semi-quantitative RT-PCR analysis. We also thank Drs. Mary Goldring, Peter S. Rotwein, Renato Baserga, Stephen Murphy, and Gillian Murphy for providing valuable reagents.
Footnotes
The abbreviations used are: MMP, matrix metalloproteinase; IL, interleukin; Fn-f, fibronectin fragment; IGF-1, insulin-like growth factor-1; OP-1, osteogenic protein-1; BMP, bone morphogenetic protein; IL-1Ra, IL-1 receptor antagonist; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; RT, reverse transcription.
Loeser, R. F., Pacione, C. A., and Chubinskaya, S. (2003) Arthritis Rheum., in press.
H.-J. Im, C. Pacione, S. Chubinskaya, and R. F. Loeser, unpublished observation.
This work was supported by National Institutes of Health Grants AG16697 (to R. F. L.), AG47654 (to S. C.), and AR49003 (to R. F. L.).
References
- 1.Goldring MB. Arthritis Rheum. 2000;43:1916–1926. doi: 10.1002/1529-0131(200009)43:9<1916::AID-ANR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 2.Billinghurst RC, Dahlberg L, Ionescu M, Reiner A, Bourne R, Rorabeck C, Mitchell P, Hambor J, Diekmann O, Tschesche H. J Clin Invest. 1997;99:1534–1545. doi: 10.1172/JCI119316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, Geoghegan KF, Hambor JE. J Clin Invest. 1996;97:761–768. doi: 10.1172/JCI118475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahlberg L, Billinghurst RC, Manner P, Nelson F, Webb G, Ionescu M, Reiner A, Tanzer M, Zukor D, Chen J. Arthritis Rheum. 2000;43:673–682. doi: 10.1002/1529-0131(200003)43:3<673::AID-ANR25>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Shlopov BV, Gumanovskaya ML, Hasty KA. Arthritis Rheum. 2000;43:195–205. doi: 10.1002/1529-0131(200001)43:1<195::AID-ANR24>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 6.Forsyth CB, Pulai J, Loeser RF. Arthritis Rheum. 2002;46:2368–2376. doi: 10.1002/art.10502. [DOI] [PubMed] [Google Scholar]
- 7.Johansson N, Saarialho-Kere U, Airola K, Herva R, Nissinen L, Westermarck J, Vuorio E, Heino J, Kahari VM. Dev Dyn. 1997;208:387–397. doi: 10.1002/(SICI)1097-0177(199703)208:3<387::AID-AJA9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 8.Varghese S, Canalis E. Endocrinology. 1997;138:1035–1040. doi: 10.1210/endo.138.3.4978. [DOI] [PubMed] [Google Scholar]
- 9.Gazzero E, Rydziel S, Canalis E. Endocrinology. 1999;140:562–567. doi: 10.1210/endo.140.2.6493. [DOI] [PubMed] [Google Scholar]
- 10.Yamashita H, Ten Dijke P, Heldin CH, Miyazono K. Bone. 1996;19:569–574. doi: 10.1016/s8756-3282(96)00259-1. [DOI] [PubMed] [Google Scholar]
- 11.Lagna G, Hata A, Hemmati-Brivanolou A, Massague J. Nature. 1996;383:832–836. doi: 10.1038/383832a0. [DOI] [PubMed] [Google Scholar]
- 12.Ito Y, Zhang YW. J Bone Miner Metabol. 2001;19:188–194. doi: 10.1007/s007740170041. [DOI] [PubMed] [Google Scholar]
- 13.Sampath TK, Maliakal JC, Hauschka PV, Jones WK, Sasaki H, Tucker RF, White KH, Coughlin JE, Tucker MM, Pang RHL, Corbett C, Ozkaynak E, Oppermann H, Rueger DC. J Biol Chem. 1992;267:20352–20362. [PubMed] [Google Scholar]
- 14.Reddi AH, Cunningham NS. J Bone Miner Res. 1993;8:499–502. doi: 10.1002/jbmr.5650081313. [DOI] [PubMed] [Google Scholar]
- 15.Wozney JM, Rosen V. Clin Orthop Relat Res. 1998;346:26–37. [PubMed] [Google Scholar]
- 16.Vukicevic S, Lluyten FP, Reddi AH. Proc Natl Acad Sci U S A. 1989;86:8793–8797. doi: 10.1073/pnas.86.22.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeh LCC, Adamo ML, Kitten AM, Olson MS, Lee JC. Endocrinology. 1996;137:1921–1931. doi: 10.1210/endo.137.5.8612532. [DOI] [PubMed] [Google Scholar]
- 18.Koepp HE, Sampath KT, Kuettner KE, Homandberg GA. Inflamm Res. 1999;48:199–204. doi: 10.1007/s000110050446. [DOI] [PubMed] [Google Scholar]
- 19.Chubinskaya S, Merrihew C, Cs-Szabo G, Mollenhauer J, McCartney J, Rueger DC, Kuettner KE. J Histochem Cytochem. 2000;48:239–250. doi: 10.1177/002215540004800209. [DOI] [PubMed] [Google Scholar]
- 20.Gould SE, Day M, Jones SS, Dorai H. Kidney Int. 2002;61:51–60. doi: 10.1046/j.1523-1755.2002.00103.x. [DOI] [PubMed] [Google Scholar]
- 21.Haaijman A, Burger EH, Goei SW, Nelles L, Ten Dijke P, Huylebroeck D, Bronckers AL. Growth Factors. 2000;17:177–192. doi: 10.3109/08977190009001067. [DOI] [PubMed] [Google Scholar]
- 22.Yeh LCC, Adamo ML, Olson MS, Lee JC. Endocrinology. 1997;138:4181–4190. doi: 10.1210/endo.138.10.5465. [DOI] [PubMed] [Google Scholar]
- 23.Yeh LCC, Adamo ML, Duan C, Lee JC. J Cell Physiol. 1998;175:78–88. doi: 10.1002/(SICI)1097-4652(199804)175:1<78::AID-JCP9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Madry H, Zurakowski D, Trippel SB. Gene Ther. 2001;8:1443–1449. doi: 10.1038/sj.gt.3301535. [DOI] [PubMed] [Google Scholar]
- 25.Fortier LA, Mohammed HO, Lus G, Nixon AJ. J Bone Joint Surg Br. 2002;84:276–288. doi: 10.1302/0301-620x.84b2.11167. [DOI] [PubMed] [Google Scholar]
- 26.Delany AM, Rydziel S, Canalis E. Endocrinology. 1996;137:4665–4670. doi: 10.1210/endo.137.11.8895331. [DOI] [PubMed] [Google Scholar]
- 27.Hui W, Rowan AD, Cawston T. Ann Rheum Dis. 2001;60:254–261. doi: 10.1136/ard.60.3.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deleted in proof
- 29.Tsuzaki M, Guyton G, Garrett W, Archambault JM, Herzog W, Almekinders L, Bynum D, Yang X, Banes AJ. J Orthop Res. 2003;21:256–264. doi: 10.1016/S0736-0266(02)00141-9. [DOI] [PubMed] [Google Scholar]
- 30.Spies M, Nesic O, Barrow RE, Perez-Polo JR, Herndon DN. Gene Ther. 2001;8:1409–1415. doi: 10.1038/sj.gt.3301543. [DOI] [PubMed] [Google Scholar]
- 31.Mengshol JA, Vincenti MP, Brinckerhoff CE. Nucleic Acids Res. 2001;29:4361–4372. doi: 10.1093/nar/29.21.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liacini A, Sylvester J, Li WQ, Zafarullah M. Matrix Biol. 2002;21:251–262. doi: 10.1016/s0945-053x(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 33.Bondeson J, Brennan F, Foxwell B, Feldmann M. J Rheumatol. 2000;27:2078–2089. [PubMed] [Google Scholar]
- 34.Leeman MF, Curran S, Murray GI. Crit Rev Biochem Mol Biol. 2002;37:149–166. doi: 10.1080/10409230290771483. [DOI] [PubMed] [Google Scholar]
- 35.Kuhn K, Lotz M. Arthritis Rheum. 2001;44:1644–1653. doi: 10.1002/1529-0131(200107)44:7<1644::AID-ART287>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 36.Yasuda T, Poole RA. Arthritis Rheum. 2002;46:138–148. doi: 10.1002/1529-0131(200201)46:1<138::AID-ART10051>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 37.Homandberg GA, Hui F, Wen C, Purple C, Bewsey K, Koepp H, Huch K, Harris A. Biochem J. 1997;321:751–757. doi: 10.1042/bj3210751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanton H, Ung L, Fosang AJ. Biochem J. 2002;364:181–190. doi: 10.1042/bj3640181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tardif G, Pelletier JP, Dupuis M, Hambor JE, Johanne MP. Biochem J. 1997;323:13–16. doi: 10.1042/bj3230013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buttice G, Quinones S, Kurkinen M. Nucleic Acids Res. 1991;19:3723–3731. doi: 10.1093/nar/19.13.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D’Alonzo RC, Selvamurugan N, Karsenty G, Partridge NC. J Biol Chem. 2002;277:816–822. doi: 10.1074/jbc.M107082200. [DOI] [PubMed] [Google Scholar]