

Abstract
Purpose of the review
The nephrology community lacks a unified view of protein sieving through the glomerular capillary wall (GCW). The GCW consists of three distinct but closely interacting layers: the fenestrated endothelium, with its glycocalyx; the podocytes, with their interdigitated foot processes and slit diaphragms; and the intervening glomerular basement membrane (GBM). Proteinuria is associated with abnormalities in any one layer, suggesting that each contributes to the glomerular filtration barrier (GFB). Proteinuria can also be induced in the context of a normal GCW. Here we review some classic studies as well as some newer concepts and present competing hypotheses about the GFB.
Recent findings
Two almost forgotten concepts have recently emerged. One group has challenged the exquisite selectivity of the GFB to albumin and suggested that proteinuria is the result of abnormal tubular uptake. There has also been a reemphasis on diffusion through the GBM as the driving force behind macromolecular filtration. New evidence suggests that the endothelial glycocalyx is an important charge-selective barrier.
Summary
We suggest viewing the GFB as a dynamic rather than as a rigid barrier, requiring three healthy layers and a hemodynamic steady state. Multiple challenges to studying the endothelium, the tubular handling of albumin, and the role of hemodynamic forces will require new tools, new hypotheses, and open minds.
Keywords: Proteinuria, Glomerular filtration barrier, Permselectivity, Gel permeation, Podocytes
Introduction
The terms glomerular capillary wall (GCW) and glomerular filtration barrier (GFB) were coined during the 1950s to describe the novel structure observed by electron microscopy (EM). The GCW consists of three distinct but interacting layers: fenestrated endothelial cells, podocytes with their foot processes (FPs) and slit diaphragms (SDs), and a shared intervening extracellular matrix called the glomerular basement membrane (GBM) [1]. We now have a better understanding of the complex nature of these three layers and the interactions among them (Figure 1). Either genetic or acquired abnormalities in each of the three layers of the GCW can lead to a defective GFB, proteinuria, and kidney disease. However, it is unclear whether the pathway leading to increased protein concentration in the glomerular filtrate is similar in all cases, or if it differs considerably depending on the primary defect. Importantly, if proteinurias can be segregated based on their causes, can we better predict outcomes or employ better treatments?
Figure 1. View of the glomerular capillary wall by freeze fracture deep-etch scanning electron microscopy.

The GCW consists of the diaphragm-less fenestrated endothelium (Endo), the GBM with its thick central layer (corresponding to the lamina densa by transmission EM), and podocyte FPs with bridging SDs. Note the thin strands connecting podocytes and endothelial cells to the GBM. Image provided by Dr. John Heuser, Washington University School of Medicine.
We would like to direct readers to multiple excellent reviews that discuss in more detail than possible here the many aspects of glomerular filtration and physiology [2–4••,5–7••].
Historical prospective
Karl Ludwig in the 1800s was the first to propose that the glomerulus works as a protein sieve. Multiple studies in the first half of the twentieth century supported his ideas [8]. With the advent of EM and the discovery of the intricate nature of the GCW [1], investigators attempted to localize the GFB within the GCW using different tracers, with variable results [9]. With the arrival of the genomic age, we have gained a much better understanding of the molecular structure of the GCW, but our understanding of the functional aspects of the GFB has not advanced at the same rate. In fact, many of the current controversies surrounding the GFB (size vs. charge selectivity; SD vs. GBM as predominant albumin barrier; glomerular barrier vs. tubular reabsorption as the major “anti-albuminuria” mechanism; determination of sieving coefficients) are not new, as some go back more than 50 years [10,11].
Glomerular sieving coefficient (GSC)
The local sieving coefficient of a molecule is the ratio of its concentration in filtrate to that in plasma at a particular point along a capillary [2], whereas the GSC is the ratio of its concentration in Bowman’s space to that in plasma. Because albumin is the most abundant plasma protein, and considering that albuminuria is viewed by most as one of the most important signs of glomerular disease, determining albumin’s GSC is central to understanding the GFB.
How tight is the GFB?
The glomerulus works as a macromolecular sieve, retarding the passage of plasma proteins and certain exogenous tracers, while allowing relatively free flow of water and small solutes. Early micropuncture studies showed albumin’s concentration in the proximal tubules of normal rats to be 0.7–1 mg/100ml [12], which increased after the induction of proteinuria. Using differential micropuncture techniques to eliminate interstitial contamination, Tojo and Endou determined the albumin concentration in rat glomerular filtrate to be 22.9 μg/ml, with a calculated GSC of 0.00062 [13]. Recently, much higher albumin GSCs of ~.03, about 50 times the long accepted estimates, have been reported by one group of investigators using different techniques [14–21••]. Yet even using the highest GSC estimates, only ~3% of plasma albumin passes through the GFB. Nevertheless, the difference between 3% and the more widely accepted 0.06% is staggering, and it suggests a fundamental flaw in one or perhaps both measurements.
Role of size vs. charge selectivity
The existence of size selectivity is universally accepted. Studies with inert tracers such as Ficoll and dextran indicate that the GSC is inversely related to molecular weight and radius [22–24]. However, the location of the size barrier is ill-defined. The contributions of podocyte SDs, the GBM, and the endothelium are not fully understood.
Charge selectivity was established from classic studies using charged dextrans [25,26]. Negatively charged dextran was more restricted than neutral dextran, which was more restricted than positively charged dextran of similar size. There is a detectable charge selectivity defect associated with proteinuria in animal models and human subjects. Charge selectivity was also suggested to explain the difference between the GSCs of native (anionic) proteins and uncharged Ficoll of similar size [27,28]. However, charge selectivity is not universally accepted. The concept was formulated through the use of charged polysaccharides (dextran and Ficoll) that do not behave like the more rigid spherical proteins [29–31]. Charge selectivity has been challenged by Comper and coworkers to explain the higher than anticipated GSC they have reported.
The location of the charge barrier, if it exists, has not been fully defined. Despite earlier reports, reducing fixed anionic charge sites in the GBM by more than 50% via removal of agrin has no consequences on urinary albumin concentration [32,33]. Cellular glycocalyces (of podocytes and endothelial cells), however, may provide sufficient charge to form an effective barrier (see below) [34], but this is underappreciated, understudied, and requires further morphological analysis.
Where is the barrier?
Data generated over decades suggest that normal macromolecular filtration requires the contribution of all three layers of the GCW: endothelium, GBM, and podocytes [3,7••].
Endothelium
The fenestrated glomerular endothelium may play a direct role in determining protein sieving [35,36]. Although the fenestrae are too large to form any meaningful barrier and lack a diaphragm, endothelial cells have an elaborate luminal surface glycocalyx forming a highly negatively charged coat that covers the fenestrae with plugs (Figure 2), which could be central for charge selectivity [35]. However, the glycocalyx is highly sensitive to the hypoxia encountered during tissue fixation, making it difficult to visualize and contributing to underestimations of its importance [36]. Visualizing the glycocalyx requires fixation under near normoxic conditions or perfusion with positively charged tracers [37•,38]. The extent of the glycocalyx’s interactions with plasma proteins, considering that such interactions may change its restrictive properties, is poorly understood [39].
Figure 2. GCW charges.
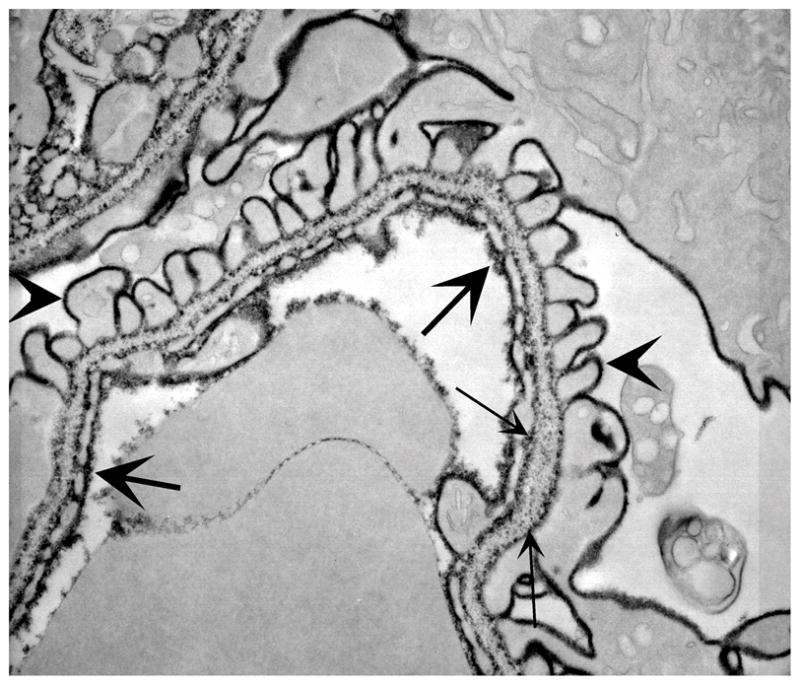
Kidneys were perfused with a high concentration of polyethylenimine (PEI) and post-fixed in phosphotungstic acid and osmium. The extent of the endothelial glycocalyx and fenestral plug is clear (large arrows). Note also the podocyte glycocalyx (arrowheads) and GBM charges (small arrows), which are strongest at the laminae rarae internae and externae.
Pre-eclampsia/eclampsia is the classic proteinuric disease associated with endothelial dysfunction [40] and is now known to be caused by blockade of trophic factors, such as vascular endothelial growth factor (VEGF), that are required for endothelial cell health [41]. In animal models, manipulating VEGF signaling results in an endothelial injury similar to pre-eclampsia, with proteinuria and kidney failure even in the absence of detectable podocyte and GBM injury [42••,43,44]. Diabetic nephropathy is also associated with endothelial dysfunction [45]. Despite advances in our understanding of the mechanisms of glomerular endothelial cell injury, our knowledge of the full extent of the glomerular endothelium’s contribution to permselectivity is limited.
GBM
Classic studies localizing different tracers by electron microscopy indicated an important role for the GBM in glomerular permselectivity [46–48]. The pattern and distribution of ferritin and high molecular weight dextran (62–125 kD) in normal rats was restricted to the subendothelial aspect of the GBM and changed dramatically after puromycin aminonucleoside-induced proteinuria, with deeper penetration of the tracers into the GBM and the appearance of tracer-laden endocytic vesicles within podocytes [47,49]. Studies using endogenous albumin and IgG showed similar distributions, suggesting that the GBM is central to determining GSC [48]. However, many of these studies showed the GBM to be leaky at the site of endothelial cell or podocyte detachment in puromycin aminonucleoside (PAN)-induced proteinuria or after clamping the renal artery and vein [50–52].
It is well known that GBM abnormalities can result in proteinuria. The best example is the absence of laminin β2, which results in congenital nephrotic syndrome in both mice [53,54] and humans [55,56•]. Laminin β2 is normally the only laminin β chain in the GBM, so its absence would be expected to change the properties of the GBM. Lamb2−/−mice are born with mild proteinuria, initially in the absence of significant endothelial or podocyte changes [57], but eventually urinary albumin levels increase greatly, foot processes become effaced, and the mice die at about 1 month of age [54]. The mutant GBM shows increase permeability to ferritin [57]. Pierson syndrome is the corresponding human disease, and the observed pathology includes diffuse mesangial sclerosis [55]. Alport syndrome, a collagen IV disease, is also associated with a disrupted GBM and but initially only mild proteinuria both in humans and in mouse models [58], with increased ferritin permeability observed in mice [59•].
Podocytes and their SDs
The nature of the SD’s molecular structure [60–64] and the link between primary SD abnormalities and development of congenital nephrotic syndrome in humans and in animal models indicates a tight link between SD function and the GFB. Furthermore, podocyte FP effacement and SD abnormalities are the hallmark of proteinuria in nearly all settings.
Rodewald and Karnovsky proposed the current model of the SD as a ladder-like structure with a thick central strand [65]. Studies showed that horseradish peroxidase (40kD) and catalase (210kD) concentrations drop sharply at the level of the SD, suggesting it has barrier properties [52]. The structure was further investigated by electron tomography and shown to consist of multiple layers of nephrin strands connecting adjacent FPs. Pores within this meshwork form elongated channels with a maximum width equals to that of albumin [66]. In the setting of Nphs1 (nephrin) mutation, the slit pores become narrower with only shorter, thinner, less organized strands bridging adjacent FPs and forming relatively larger pores and channels.
However, because the SD is located most distally in the GCW, it is unlikely to function as its most restrictive barrier, even if its pores are smaller than albumin. Proteinuria has been reported in the absence of FP and SD changes [57,67]. Conversely, the degree of such changes in other cases does not correlate with the level of proteinuria [68], and foot process effacement and SD changes are not always associated with proteinuria. Furthermore, albumin and other serum proteins can be podocyte-toxic [69••,70•,71••,72], suggesting that podocytes under normal conditions must be sheltered from high concentrations of plasma proteins.
Mechanisms of protein filtration
Convection refers to drag by fluid flow across the barrier, with the GFB acting as a physical sieve for macromolecules. Convection, however, cannot be the only force driving macromolecular filtration, because changing GFR results in an opposite change in the macromolecular filtration fraction [24,73]. Diffusion has been suggested to explain the lower filtration fraction of macromolecules with increasing GFR. Diffusion also explains the sudden appearance of albumin and IgG in the urinary space after clamping the renal artery, followed by their disappearance after releasing the clamp [48]. Most recently, Oliver Smithies reintroduced this concept in his “Gel Permeation/Diffusion Hypothesis” [6]. According to this hypothesis, diffusion through the GBM, which acts as a modified gel, is the predominant force governing macromolecular movement through the GFB. Diffusion is independent of fluid flow (i.e., GFR), but dependent on the gel’s properties. According to this hypothesis, increased protein concentration in the glomerular filtrate can occur by two different pathways. The first is by an increase in the rate of passage of protein by changes in the gel’s properties (i.e., by alterations to the GBM and perhaps also to the endothelial glycocalyx). The second is by a reduction in the available surface area for filtration, as occurs either with FP effacement or reduced endothelial fenestration. This reduction results in increased hydraulic resistance and reduced single nephron GFR [2,40], while diffusion of plasma proteins remains constant; i.e., approximately the same amount of protein is diluted in a smaller volume. (A normal, properly functioning gel requires stable interactions with the adjacent cells under stable hydrodynamic pressures, which can explain some of the results from studies of isolated GBM [74,75].) The increased protein concentration in the filtrate then overwhelms the tubular resorption pathway, resulting in albuminuria. However, as elegantly simple as Dr. Smithies’ hypothesis appears, there are issues to be resolved. Data in the literature show that the filtered load of albumin is more dependent on GFR [7••] than predicted by the hypothesis, and the hypothesis makes no allowance for the effects of tubule flow rate or residence time [76]. As yet there is no direct experimental evidence published in its support.
In overload proteinuria [69••], the GFB and GSC are normal, but both plasma and glomerular filtrate albumin concentrations are higher, with the latter eventually exceeding the tubular threshold for filtered protein resorption. Furthermore, we postulate that in the appropriate setting, podocyte FP effacement can occur without significant proteinuria as long as the GBM or glycocalyx becomes more restrictive, a situation in which reduced albumin diffusion would compensate for reduced GFR [77••].
Role of tubular absorption
Even considering the most restrictive GSC proposed for albumin in humans (~0.0001), a significant amount of albumin will cross the GFB in the normal kidney. And with the much higher proposed GSC estimates, the amount of filtered albumin would easily exceed the nephrotic range if present in the final urine. Thus, in any scenario there must be significant post-glomerular processing of albumin by tubules.
Tojo and Endou, using a fractional micropuncture technique, found that 94% of filtered albumin was absorbed by proximal tubules [13]. Park and Maack studied the fate of albumin in isolated rabbit proximal tubules [78]. They failed to show any transcellular or paracellular transport of intact albumin. Proximal tubule albumin absorption capacity was estimated to be 3.7 ng/min per mm tubule length, and the rate of albumin removal was proportional to fluid absorption. Most of the absorbed albumin was degraded and transported through the basal surface into the bathing solution.
An earlier micropuncture study comparing glomerular filtrate and final urinary albumin concentrations in normal and nephrotic PAN-treated rats showed that urinary albumin increased by a factor of 43, while glomerular filtrate albumin increased by a factor of only 4.5, suggesting abnormal tubular handling of the filtered albumin [12]. More recently, Comper and colleagues have published multiple papers suggesting that the GFB is more leaky to albumin than generally accepted, by ~50 fold. They hypothesize and have provided evidence that filtered albumin is reclaimed in the S1 segment of proximal tubules, with most of it returned intact to the circulation [15,17–21••,79]. They conclude that proteinuria is a tubular rather than a glomerular disorder. If true, we must change the way we view the GFB and think of even selective proteinuria in the context of tubular abnormalities. However, this concept is not accepted by most in the nephrology community. A new report utilizing a specially labeled albumin that can be visualized by fluorescence only after hydrolysis shows that albumin endocytosed by proximal tubular cells is degraded quickly [80••]. However, there was no indication of the ratio of degraded to intact albumin at the basal pole of the cells.
Concluding remarks
We suggest viewing the GFB as a functional unit consisting of three different elements [3]. We also suggest viewing it as a dynamic sieve, rather than as physically inert. Normal GFB function requires not only three intact GCW layers, but also a hemodynamic steady state in the glomerular capillary and the urinary spaces. A change in any factor (GBM, cell, glycocalyx, local GFR, or plasma albumin concentration) will alter albumin’s concentration in the primary filtrate. When the level of albumin in the filtrate exceeds the tubular absorption threshold, albuminuria will ensue.
We suggest the following approaches for the future:
Analyze in vivo the growing list of proteinuric animal models of different etiologies to better delineate the pathophysiology of proteinuria.
Define the endothelium’s contribution to the GFB, in part by developing the tools necessary to manipulate glomerular endothelial cell gene expression.
Investigate the endothelial glycocalyx with better and reproducible morphological and physiological assessments.
Devise a better “mousetrap” to understand tubular handling of albumin, especially the fate of resorbed albumin: degradation, return to the circulation, or both.
Acknowledgments
Authors funded by National Institutes of Health and American Heart Association
Our research is supported by grants from the National Institutes of Health (R01DK078314 and R21DK074613 to JHM; P30DK079333 to GJ), by an Established Investigator Award from the American Heart Association (JHM), and by an Alaska Kidney Foundation-American Society of Nephrology Research Grant (GJ).
References
- 1.Rhodin J. Electron microscopy of the glomerular capillary wall. Exp Cell Res. 1955;8:572–574. doi: 10.1016/0014-4827(55)90136-1. [DOI] [PubMed] [Google Scholar]
- 2.Deen WM, Lazzara MJ, Myers BD. Structural determinants of glomerular permeability. Am J Physiol Renal Physiol. 2001;281:F579–596. doi: 10.1152/ajprenal.2001.281.4.F579. [DOI] [PubMed] [Google Scholar]
- 3.Deen WM. What determines glomerular capillary permeability? J Clin Invest. 2004;114:1412–1414. doi: 10.1172/JCI23577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••4.Comper WD, Haraldsson B, Deen WM. Resolved: normal glomeruli filter nephrotic levels of albumin. J Am Soc Nephrol. 2008;19:427–432. doi: 10.1681/ASN.2007090997. A point-counterpoint style review of the competing hypotheses of the glomerular vs. tubular origin of proteinuria. [DOI] [PubMed] [Google Scholar]
- 5.Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354:1387–1401. doi: 10.1056/NEJMra052131. [DOI] [PubMed] [Google Scholar]
- 6.Smithies O. Why the kidney glomerulus does not clog: a gel permeation/diffusion hypothesis of renal function. Proc Natl Acad Sci U S A. 2003;100:4108–4113. doi: 10.1073/pnas.0730776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••7.Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. An outstanding extensive review of the physiology of the GFB and the mechanisms of proteinuria. [DOI] [PubMed] [Google Scholar]
- 8.Wearn JT, Richards AN. Observations on the composition of glomerular urine, with particular reference to the problem of reabsorption in renal tubules. Am J Physiol. 1924;71:209–227. [Google Scholar]
- 9.Farquhar MG. Editorial: The primary glomerular filtration barrier--basement membrane or epithelial slits? Kidney Int. 1975;8:197–211. doi: 10.1038/ki.1975.103. [DOI] [PubMed] [Google Scholar]
- 10.Brandt JL, Gruhn JG. Effect of rennin on proteinuria and PAH clearance at low plasma levels. Am J Physiol. 1948;153:458–464. doi: 10.1152/ajplegacy.1948.153.3.458. [DOI] [PubMed] [Google Scholar]
- 11.Rather LJ, Addis T. Renin proteinuria in the rat; evidence that renin does not interfere with the tubular resorption of purified human hemoglobin or bovine albumin. J Exp Med. 1950;91:567–572. doi: 10.1084/jem.91.6.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oken DE, Flamenbaum W. Micropuncture studies of proximal tubule albumin concentrations in normal and nephrotic rats. J Clin Invest. 1971;50:1498–1505. doi: 10.1172/JCI106635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tojo A, Endou H. Intrarenal handling of proteins in rats using fractional micropuncture technique. Am J Physiol. 1992;263:F601–606. doi: 10.1152/ajprenal.1992.263.4.F601. [DOI] [PubMed] [Google Scholar]
- 14.Burne MJ, Osicka TM, Comper WD. Fractional clearance of high molecular weight proteins in conscious rats using a continuous infusion method. Kidney Int. 1999;55:261–270. doi: 10.1046/j.1523-1755.1999.00234.x. [DOI] [PubMed] [Google Scholar]
- 15.Eppel GA, Osicka TM, Pratt LM, et al. The return of glomerular-filtered albumin to the rat renal vein. Kidney Int. 1999;55:1861–1870. doi: 10.1046/j.1523-1755.1999.00424.x. [DOI] [PubMed] [Google Scholar]
- 16.Osicka TM, Hankin AR, Comper WD. Puromycin aminonucleoside nephrosis results in a marked increase in fractional clearance of albumin. Am J Physiol. 1999;277:F139–145. doi: 10.1152/ajprenal.1999.277.1.F139. [DOI] [PubMed] [Google Scholar]
- 17.Eppel GA, Takazoe K, Nikolic-Paterson DJ, et al. Characteristics of albumin processing during renal passage in anti-Thy1 and anti-glomerular basement membrane glomerulonephritis. Am J Kidney Dis. 2000;35:418–426. doi: 10.1016/s0272-6386(00)70194-6. [DOI] [PubMed] [Google Scholar]
- 18.Greive KA, Nikolic-Paterson DJ, Guimaraes MA, et al. Glomerular permselectivity factors are not responsible for the increase in fractional clearance of albumin in rat glomerulonephritis. Am J Pathol. 2001;159:1159–1170. doi: 10.1016/S0002-9440(10)61792-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo LM, Osicka TM, Brammar GC, et al. Renal processing of albumin in diabetes and hypertension in rats: possible role of TGF-beta1. Am J Nephrol. 2003;23:61–70. doi: 10.1159/000068039. [DOI] [PubMed] [Google Scholar]
- 20.Osicka TM, Strong KJ, Nikolic-Paterson DJ, et al. Renal processing of serum proteins in an albumin-deficient environment: an in vivo study of glomerulonephritis in the Nagase analbuminaemic rat. Nephrol Dial Transplant. 2004;19:320–328. doi: 10.1093/ndt/gfg226. [DOI] [PubMed] [Google Scholar]
- ••21.Russo LM, Sandoval RM, McKee M, et al. The normal kidney filters nephrotic levels of albumin retrieved by proximal tubule cells: retrieval is disrupted in nephrotic states. Kidney Int. 2007;71:504–513. doi: 10.1038/sj.ki.5002041. One report in a series of papers suggesting that the normal glomerulus filters a high level of albumin, which is then reclaimed by proximal tubules and returned intact to the circulation. They argue for a tubular origin of proteinuria. [DOI] [PubMed] [Google Scholar]
- 22.Ohlson M, Sorensson J, Haraldsson B. Glomerular size and charge selectivity in the rat as revealed by FITC-ficoll and albumin. Am J Physiol Renal Physiol. 2000;279:F84–91. doi: 10.1152/ajprenal.2000.279.1.F84. [DOI] [PubMed] [Google Scholar]
- 23.Chang RL, Ueki IF, Troy JL, et al. Permselectivity of the glomerular capillary wall to macromolecules. II. Experimental studies in rats using neutral dextran. Biophys J. 1975;15:887–906. doi: 10.1016/S0006-3495(75)85863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang RL, Deen WM, Robertson CR, et al. Permselectivity of of the glomerular capillary wall. Studies of experimental glomerulonephritis in the rat using neutral dextran. J Clin Invest. 1976;57:1272–1286. doi: 10.1172/JCI108395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang RL, Deen WM, Robertson CR, Brenner BM. Permselectivity of the glomerular capillary wall: III. Restricted transport of polyanions. Kidney Int. 1975;8:212–218. doi: 10.1038/ki.1975.104. [DOI] [PubMed] [Google Scholar]
- 26.Bohrer MP, Baylis C, Humes HD, et al. Permselectivity of the glomerular capillary wall. Facilitated filtration of circulating polycations. J Clin Invest. 1978;61:72–78. doi: 10.1172/JCI108927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeansson M, Haraldsson B. Glomerular size and charge selectivity in the mouse after exposure to glucosaminoglycan-degrading enzymes. J Am Soc Nephrol. 2003;14:1756–1765. doi: 10.1097/01.asn.0000072742.02714.6e. [DOI] [PubMed] [Google Scholar]
- 28.Sorensson J, Ohlson M, Haraldsson B. A quantitative analysis of the glomerular charge barrier in the rat. Am J Physiol Renal Physiol. 2001;280:F646–656. doi: 10.1152/ajprenal.2001.280.4.F646. [DOI] [PubMed] [Google Scholar]
- 29.Venturoli D, Rippe B. Ficoll and dextran vs. globular proteins as probes for testing glomerular permselectivity: effects of molecular size, shape, charge, and deformability. Am J Physiol Renal Physiol. 2005;288:F605–613. doi: 10.1152/ajprenal.00171.2004. [DOI] [PubMed] [Google Scholar]
- 30.Asgeirsson D, Venturoli D, Rippe B, Rippe C. Increased glomerular permeability to negatively charged Ficoll relative to neutral Ficoll in rats. Am J Physiol Renal Physiol. 2006;291:F1083–1089. doi: 10.1152/ajprenal.00488.2005. [DOI] [PubMed] [Google Scholar]
- 31.Asgeirsson D, Venturoli D, Fries E, et al. Glomerular sieving of three neutral polysaccharides, polyethylene oxide and bikunin in rat. Effects of molecular size and conformation. Acta Physiol (Oxf) 2007;191:237–246. doi: 10.1111/j.1748-1716.2007.01733.x. [DOI] [PubMed] [Google Scholar]
- 32.Harvey SJ, Jarad G, Cunningham J, et al. Disruption of glomerular basement membrane charge through podocyte-specific mutation of agrin does not alter glomerular permselectivity. Am J Pathol. 2007;171:139–152. doi: 10.2353/ajpath.2007.061116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harvey SJ, Miner JH. Revisiting the glomerular charge barrier in the molecular era. Curr Opin Nephrol Hypertens. 2008;17:393–398. doi: 10.1097/MNH.0b013e32830464de. [DOI] [PubMed] [Google Scholar]
- 34.Galeano B, Klootwijk R, Manoli I, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–1594. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeansson M, Haraldsson B. Morphological and functional evidence for an important role of the endothelial cell glycocalyx in the glomerular barrier. Am J Physiol Renal Physiol. 2006;290:F111–116. doi: 10.1152/ajprenal.00173.2005. [DOI] [PubMed] [Google Scholar]
- 36.Hjalmarsson C, Johansson BR, Haraldsson B. Electron microscopic evaluation of the endothelial surface layer of glomerular capillaries. Microvasc Res. 2004;67:9–17. doi: 10.1016/j.mvr.2003.10.001. [DOI] [PubMed] [Google Scholar]
- •37.Reitsma S, Slaaf DW, Vink H, et al. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454:345–359. doi: 10.1007/s00424-007-0212-8. A paper discussing the endothelilal glycocalyx in general, its importance, and the methods used to visualize it. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andrews PM, Bates SB. Dose-dependent movement of cationic molecules across the glomerular wall. Anat Rec. 1985;212:223–231. doi: 10.1002/ar.1092120302. [DOI] [PubMed] [Google Scholar]
- 39.Schneeberger EE, Hamelin M. Interaction of serum proteins with lung endothelial glycocalyx: its effect on endothelial permeability. Am J Physiol. 1984;247:H206–217. doi: 10.1152/ajpheart.1984.247.2.H206. [DOI] [PubMed] [Google Scholar]
- 40.Lafayette RA, Druzin M, Sibley R, et al. Nature of glomerular dysfunction in pre-eclampsia. Kidney Int. 1998;54:1240–1249. doi: 10.1046/j.1523-1755.1998.00097.x. [DOI] [PubMed] [Google Scholar]
- 41.Maynard S, Epstein FH, Karumanchi SA. Preeclampsia and angiogenic imbalance. Annu Rev Med. 2008;59:61–78. doi: 10.1146/annurev.med.59.110106.214058. [DOI] [PubMed] [Google Scholar]
- ••42.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. This paper shows that anti-tumor therapy in humans with avastin, an anti-VEGF antibody, or podocyte-specific knockout of VEGF in adult mice causes glomerular endothelial cell defects similar to those observed in pre-eclampsia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugimoto H, Hamano Y, Charytan D, et al. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem. 2003;278:12605–12608. doi: 10.1074/jbc.C300012200. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa T, Segal M, Croker B, Johnson RJ. A breakthrough in diabetic nephropathy: the role of endothelial dysfunction. Nephrol Dial Transplant. 2007;22:2775–2777. doi: 10.1093/ndt/gfm380. [DOI] [PubMed] [Google Scholar]
- 46.Caulfield JP, Farquhar MG. The permeability of glomerular capillaries to graded dextrans. Identification of the basement membrane as the primary filtration barrier. J Cell Biol. 1974;63:883–903. doi: 10.1083/jcb.63.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farquhar MG, Palade GE. Segregation of ferritin in glomerular protein absorption droplets. J Biophys Biochem Cytol. 1960;7:297–304. doi: 10.1083/jcb.7.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ryan GB, Hein SJ, Karnovsky MJ. Glomerular permeability to proteins. Effects of hemodynamic factors on the distribution of endogenous immunoglobulin G and exogenous catalase in the rat glomerulus. Lab Invest. 1976;34:415–427. [PubMed] [Google Scholar]
- 49.Caulfield JP, Farquhar MG. The permeability of glomerular capillaries of aminonucleoside nephrotic rats to graded dextrans. J Exp Med. 1975;142:61–83. doi: 10.1084/jem.142.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuhn K, Ryan GB, Hein SJ, et al. An ultrastructural study of the mechanisms of proteinuria in rat nephrotoxic nephritis. Lab Invest. 1977;36:375–387. [PubMed] [Google Scholar]
- 51.Ryan GB, Karnovsky MJ. An ultrastructural study of the mechanisms of proteinuria in aminonucleoside nephrosis. Kidney Int. 1975;8:219–232. doi: 10.1038/ki.1975.105. [DOI] [PubMed] [Google Scholar]
- 52.Venkatachalam MA, Karnovsky MJ, Cotran RS. Glomerular permeability. Ultrastructural studies in experimental nephrosis using horseradish peroxidase as a tracer. J Exp Med. 1969;130:381–399. doi: 10.1084/jem.130.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noakes PG, Miner JH, Gautam M, et al. The renal glomerulus of mice lacking s-laminin/laminin beta 2: nephrosis despite molecular compensation by laminin beta 1. Nat Genet. 1995;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 54.Miner JH, Go G, Cunningham J, et al. Transgenic isolation of skeletal muscle and kidney defects in laminin beta2 mutant mice: implications for Pierson syndrome. Development. 2006;133:967–975. doi: 10.1242/dev.02270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zenker M, Aigner T, Wendler O, et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13:2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- •56.Choi HJ, Lee BH, Kang JH, et al. Variable phenotype of Pierson syndrome. Pediatr Nephrol. 2008;23:995–1000. doi: 10.1007/s00467-008-0748-7. This paper describes patients carrying laminin β2 mutations with very different clinical presentations. [DOI] [PubMed] [Google Scholar]
- 57.Jarad G, Cunningham J, Shaw AS, Miner JH. Proteinuria precedes podocyte abnormalities inLamb2−/− mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest. 2006;116:2272–2279. doi: 10.1172/JCI28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): implications for Alport syndrome. J Cell Biol. 1996;135:1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •59.Abrahamson DR, Isom K, Roach E, et al. Laminin compensation in collagen alpha3(IV) knockout (Alport) glomeruli contributes to permeability defects. J Am Soc Nephrol. 2007;18:2465–2472. doi: 10.1681/ASN.2007030328. A report showing the GBM changes associated with Alport syndrome in mice and the associated increased premeability to ferritin. [DOI] [PubMed] [Google Scholar]
- 60.Kestila M, Lenkkeri U, Mannikko M, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 61.Ruotsalainen V, Ljungberg P, Wartiovaara J, et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A. 1999;96:7962–7967. doi: 10.1073/pnas.96.14.7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Donoviel DB, Freed DD, Vogel H, et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol. 2001;21:4829–4836. doi: 10.1128/MCB.21.14.4829-4836.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 64.Li C, Ruotsalainen V, Tryggvason K, et al. CD2AP is expressed with nephrin in developing podocytes and is found widely in mature kidney and elsewhere. Am J Physiol Renal Physiol. 2000;279:F785–792. doi: 10.1152/ajprenal.2000.279.4.F785. [DOI] [PubMed] [Google Scholar]
- 65.Rodewald R, Karnovsky MJ. Porous substructure of the glomerular slit diaphragm in the rat and mouse. J Cell Biol. 1974;60:423–433. doi: 10.1083/jcb.60.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wartiovaara J, Ofverstedt LG, Khoshnoodi J, et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest. 2004;114:1475–1483. doi: 10.1172/JCI22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karumanchi SA, Epstein FH, Stillman IE. Is loss of podocyte foot processes necessary for the induction of proteinuria? Am J Kidney Dis. 2005;45:436. doi: 10.1053/j.ajkd.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 68.van den Berg JG, van den Bergh Weerman MA, Assmann KJ, et al. Podocyte foot process effacement is not correlated with the level of proteinuria in human glomerulopathies. Kidney Int. 2004;66:1901–1906. doi: 10.1111/j.1523-1755.2004.00964.x. [DOI] [PubMed] [Google Scholar]
- ••69.Yoshida S, Nagase M, Shibata S, Fujita T. Podocyte injury induced by albumin overload in vivo and in vitro: involvement of TGF-beta and p38 MAPK. Nephron Exp Nephrol. 2008;108:e57–68. doi: 10.1159/000124236. This study suggests that albumin overload in vitro and in vivo is podocyte-toxic. Albumin overload was associated with increased podocyte apoptosis and increased TGF-β1 levels and activity. [DOI] [PubMed] [Google Scholar]
- •70.Tojo A, Onozato ML, Kitiyakara C, et al. Glomerular albumin filtration through podocyte cell body in puromycin aminonucleoside nephrotic rat. Med Mol Morphol. 2008;41:92–98. doi: 10.1007/s00795-008-0397-8. This study recapitulated the conclusion of older studies, by showing that in the setting of PAN nephrosis, albumin is taken and transported through podocyte cell body. [DOI] [PubMed] [Google Scholar]
- ••71.Ijpelaar DH, Schulz A, Koop K, et al. Glomerular hypertrophy precedes albuminuria and segmental loss of podoplanin in podocytes in Munich-Wistar-Fromter rats. Am J Physiol Renal Physiol. 2008;294:F758–767. doi: 10.1152/ajprenal.00457.2007. This study showed that in MWF rats, which develop FSGS, albuminuria was preceded by glomerular hypertrophy and is accompanied by signs of podocyte injury and entrapment of albumin in the injured podocytes. The authors did not elaborate whether albumin can induce or contribute to podocyte injury. [DOI] [PubMed] [Google Scholar]
- 72.Morigi M, Buelli S, Angioletti S, et al. In response to protein load podocytes reorganize cytoskeleton and modulate endothelin-1 gene: implication for permselective dysfunction of chronic nephropathies. Am J Pathol. 2005;166:1309–1320. doi: 10.1016/S0002-9440(10)62350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rippe C, Asgeirsson D, Venturoli D, et al. Effects of glomerular filtration rate on Ficoll sieving coefficients (theta) in rats. Kidney Int. 2006;69:1326–1332. doi: 10.1038/sj.ki.5000027. [DOI] [PubMed] [Google Scholar]
- 74.Robinson GB, Walton HA. Glomerular basement membrane as a compressible ultrafilter. Microvasc Res. 1989;38:36–48. doi: 10.1016/0026-2862(89)90015-0. [DOI] [PubMed] [Google Scholar]
- 75.Daniels BS, Hauser EB, Deen WM, Hostetter TH. Glomerular basement membrane: in vitro studies of water and protein permeability. Am J Physiol. 1992;262:F919–926. doi: 10.1152/ajprenal.1992.262.6.F919. [DOI] [PubMed] [Google Scholar]
- 76.Lazzara MJ, Deen WM. Model of albumin reabsorption in the proximal tubule. Am J Physiol Renal Physiol. 2007;292:F430–439. doi: 10.1152/ajprenal.00010.2006. [DOI] [PubMed] [Google Scholar]
- ••77.Chen S, Wassenhove-McCarthy DJ, Yamaguchi Y, et al. Loss of heparan sulfate glycosaminoglycan assembly in podocytes does not lead to proteinuria. Kidney Int. 2008;74:289–299. doi: 10.1038/ki.2008.159. This paper shows that mutation of Ext1, encoding an enzyme required for heparan sulfate glycosaminoglycan biosynthesis, specifically in podocytes results in only mild foot process effacement but no significant albuminuria, despite loss of anionic charge. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park CH, Maack T. Albumin absorption and catabolism by isolated perfused proximal convoluted tubules of the rabbit. J Clin Invest. 1984;73:767–777. doi: 10.1172/JCI111270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Osicka TM, Houlihan CA, Chan JG, et al. Albuminuria in patients with type 1 diabetes is directly linked to changes in the lysosome-mediated degradation of albumin during renal passage. Diabetes. 2000;49:1579–1584. doi: 10.2337/diabetes.49.9.1579. [DOI] [PubMed] [Google Scholar]
- ••80.Slattery C, Lee A, Zhang Y, et al. In vivo visualization of albumin degradation in the proximal tubule. Kidney Int. 2008;74:1480–1486. doi: 10.1038/ki.2008.463. This paper shows that some of the albumin taken up by proximal tubule cells is degraded. [DOI] [PubMed] [Google Scholar]