

Abstract
Trypanosoma brucei, the causative agent of human African trypanosomiasis, affects tens of thousands of sub-Saharan Africans. As current therapeutics are inadequate due to toxic side effects, drug resistance, and limited effectiveness, novel therapies are urgently needed. UDP-galactose 4′-epimerase (TbGalE), an enzyme of the Leloir pathway of galactose metabolism, is one promising T. brucei drug target. We here use the relaxed complex scheme, an advanced computer-docking methodology that accounts for full protein flexibility, to identify inhibitors of TbGalE. An initial hit rate of 62% was obtained at 100 μM, ultimately leading to the identification of 14 low-micromolar inhibitors. Thirteen of these inhibitors belong to a distinct series with a conserved binding motif that may prove useful in future drug design and optimization.
Introduction
Human African trypanosomiasis (HATa), a condition caused by Tsetse-fly mediated transmission of the unicellular parasite Trypanosoma brucei (T. brucei),(1) affects between 50000 and 70000 people living in sub-Saharan Africa, with an additional 50 million people at risk of infection.(2) The initial stage of the disease, called the hemolymphatic stage, is characterized by mild symptoms including fever, arthralgia, headache, and pruritus. Once the parasite crosses the blood−brain barrier, the neurological symptoms of the chronic stage of the disease are manifest, including severe headaches, nocturnal insomnia, mental and psychological disturbances, coma, and death if untreated.(2)
Pentamidine and suramin, effective against the subspecies T. b. gambiense and T. b. rhodesiense, respectively, are typically administered during the early hemolymphatic stage.3−5 The treatments recommended by the World Health Organization for advanced HAT, however, melarsoprol and eflornithine, are entirely inadequate. Melarsoprol causes encephalopathy in 1 out of every 20 recipients and is subject to growing drug resistance.2,6,7 Eflornithine is ineffective against the T. b. rhodesiense subspecies(8) and can cause fever, seizures, and infections, although combination therapy with nifurtimox ameliorates some of these side effects.(9) To date, no vaccine is available because T. brucei undergoes antigenic variation by altering its surface glycoprotein coat, thereby evading the immune system.(10)
As drugs that target the infections of the developing world are rarely profitable, pharmaceutical companies have largely neglected the development of novel HAT therapeutics. Of the drugs listed above, for example, only one, eflornithine, has been developed since the late 1940s.(11) The trypanocidal effect of eflornithine was discovered only after it failed as an antineoplastic agent,(12) and it is only available today because the compound has also been commercialized as a cosmetic cream for the treatment of hirsutism.
This neglect provides researchers in academia with a unique opportunity to step in and address a largely unmet need. While a few academic institutions do perform high-throughput screens to identify novel inhibitors of pathogenic enzymes, these large-scale projects are often cost prohibitive outside of industry. Fortunately, recent advances in computer-aided drug design have provided academic researchers with powerful tools that in part compensate for insufficient funding.13−15
Motivated by the urgent need for novel HAT therapeutics, computer-aided drug design is here used to identify 14 low-micromolar inhibitors of T. brucei UDP-galactose 4′-epimerase (TbGalE, also known as UDP-glucose 4′-epimerase), a short-chain dehydrogenase/reductase enzyme of the Leloir pathway of galactose metabolism.16,17TbGalE is essential for parasite survival, as null mutants of both the bloodstream and procyclic form of the parasite are not viable.18,19 In T. brucei, galactose is a component of the variant surface glycoproteins used for immune-system evasion,(20) of the transferrin receptor used for iron acquisition,(21) and of the parasite endocytic pathway.(21) Curiously, while T. brucei has a hexose transporter capable of glucose uptake, it is unable to acquire galactose from the host;22,23 intracellular galactose must be synthesized from glucose via TbGalE. Consequently, we are hopeful that the compounds presented here, which inhibit this key step of trypanosomal galactose synthesis, may serve as useful scaffolds for future drug design and optimization.
Results/Discussion
Human African trypanosomiasis threatens 50 million people in sub-Saharan Africa.(2) As current therapies are either too dangerous or too limited, novel drugs are urgently needed. UDP-galactose 4′-epimerase (TbGalE), a protein critical to T. brucei survival, is one potential drug target. We here use computer-aided drug design to identify 14 low-micromolar inhibitors of TbGalE.
Computer Docking and Protein Dynamics
Traditional computer-docking methodologies often fail to identify true-positive inhibitors because the static protein structures typically used do not capture the highly dynamic reality of small-molecule binding.24,25 When a ligand approaches its receptor in vitro or in vivo, it encounters not a static protein structure but rather an ensemble of structures as the protein “breathes” in solution. Upon ligand binding, the population of receptor active-site configurations sampled by the protein may shift to better accommodate the ligand. Additionally, under the influence of a bound ligand, the protein may assume novel active-site configurations not sampled by the apo protein at all.(26)
To better understand TbGalE dynamics, we performed five 20 ns molecular dynamics (MD) simulations. Five short simulations were chosen, as opposed to one long simulation, in order to increase the diversity of protein conformations sampled and to ensure that the conformations sampled were geometrically similar to the known crystal structure. As TbGalE is a homodimer, each simulation provided two monomer trajectories, further increasing the diversity of conformations sampled. Clustering was subsequently used to identify 24 protein structures with active sites representative of the many active-site configurations sampled during the MD simulation. These 24 representative protein structures are said to constitute an ensemble.
The protein conformations of the ensemble proved useful in subsequent computer-docking studies.(27) Recently, a new docking protocol called the relaxed complex scheme (RCS)(25) has been developed that takes into account full protein flexibility. Rather than docking candidate compounds into a static protein receptor, compounds are docked into an ensemble of protein conformations typically extracted from an MD simulation. The compounds are then ranked by an ensemble-based score that accounts for active-site dynamics. The RCS has already been used to successfully identify inhibitors of FKBP,(28) HIV integrase,(29) and T. brucei RNA editing ligase 1.(30)
In the current work, we used AutoDock Vina (Vina)(31) to perform a RCS screen of the NCI Diversity Set II into the 24 ensemble conformations extracted from the MD simulation. Like previous versions of AutoDock, Vina is freely available to the academic community. Additionally, it is 2 orders of magnitude faster than AutoDock 4.0 (AutoDock),(32) the previous version. Vina performs well relative to AutoDock; while AutoDock is slightly better at predicting the energy of binding (standard error of 2.2 kcal mol−1 versus 2.8 kcal mol−1), Vina more accurately reproduces cocrystallized ligand poses.31,32 To our knowledge, Vina has never been used in a RCS screen.
Compounds were docked into both the UDP-glucose and NAD+ binding pockets and were ranked by both an ensemble-average and an ensemble-best scoring scheme (Supporting Information). Twenty-six high-scoring compounds were subsequently tested experimentally.
Experimental Validation Confirms Multiple Hits from the Primary Screen
Of the 26 compounds of the primary screen, 10 showed >50% average inhibition at 100 μM. Interestingly, at this same concentration, six compounds showed greater than 2-fold stimulation, suggesting allosteric cooperativity between the two monomers of the TbGalE homodimer, in harmony with previous studies that demonstrated GalE allostery in Kluyveromyces fragilis and Saccharomyces fragilis.33,34 As we do not expect computer docking to be able to distinguish between an agonist and an antagonist, the effective hit rate of the primary screen was therefore 62% at 100 μM.
The 10 100-μM inhibitors were subsequently tested at 10 μM. Three showed >∼50% average inhibition; compounds 1, 2, and 3 (clorobiocin) had IC50 values of 3.6 ± 0.8, 5.6 ± 0.8, and 5.0 ± 1.2 μM, respectively, and Hill slopes of 1.8 ± 0.6, 1.2 ± 0.3, and 2.1 ± 0.5, respectively (Tables 1 and S1 in Supporting Information). Interestingly, compounds 1 and 2 share a 2′-(phenylcarbamoyl)-[1,1′-biphenyl]-2-carboxylic acid core scaffold.
Table 1. The 14 Low-Micromolar TbGalE inhibitors Indentifieda.
 |
The first column shows compound structures, and the second column shows the compound identification number used throughout the text. The third column shows the IC50 value of each compound as measured in enzymatic assays. The fourth and fifth columns show the EC50 values as measured in whole-cell assays of T. brucei and human MRC5 cells, respectively. The final column shows the predicted LogP value of each compound.
In one recent study, 95% of the inhibitors identified in a high-throughput screen acted through a nonspecific aggregation-based mechanism. This same study suggested that aggregation-based inhibition typically produces steep Hill slopes that are much greater than unity, with average values around 2.2.(35) As the Hill slopes of compounds 2 and 3 (clorobiocin) were significantly greater than unity (Table S1, Supporting Information), TbGalE inhibition was measured at 30 μM inhibitor concentration (∼5 × IC50) in the presence and absence of a detergent that disrupts colloidal aggregates (0.06% n-octylglucopyranoside). No significant differences in inhibition were noted, suggesting that inhibition is specific rather than aggregation-based.(30) Given that the possibility of aggregation was eliminated, the steep Hill slopes of these two compounds provide further evidence for allostery between the two monomers of the TbGalE dimer.36,37
Clorobiocin: An Interesting Inhibitor
One of the hits from the primary virtual screen, compound 3 (clorobiocin), an aminocoumarin derived from several Streptomyces species, has previously been shown to inhibit the growth of Trypanosoma cruzi (T. cruzi), a close relative of T. brucei.(38) As clorobiocin is a known bacterial topoisomerase II inhibitor, some have hypothesized that topoisomerase II may be the T. cruzi protein target as well,(39) although other targets could not be ruled out.(38) The current work suggests that UDP-galactose 4′-epimerase may also be among the proteins targeted by this apparently polypharmacophoric compound.
We note with interest that novobiocin, a compound structurally similar to clorobiocin that likewise inhibits the growth of T. cruzi,(38) did not show greater than 50% TbGalE inhibition at 10 μM despite the fact that our computational model predicted high binding affinity. The crude scoring function employed by Vina, optimized not only for accuracy but also for speed, seems unable to differentiate between the apparently subtle differences in the protein−ligand interactions of clorobiocin and novobiocin.
Predicted Binding Poses of Top Inhibitors
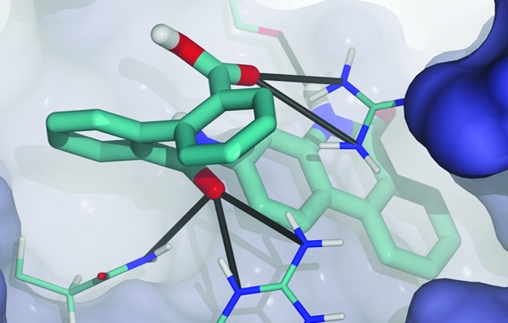
Computer docking suggests that compound 3 (clorobiocin) occupies the NAD+-binding pocket. To further characterize the clorobiocin binding pose, we examined the Vina scores of clorobiocin docked into each of the protein configurations of the ensemble and selected the binding pose/protein configuration associated with the best score for further analysis. The predicted protein−ligand interactions are represented schematically in Figure 1.
Figure 1.

The predicted ligand−protein interactions of clorobiocin bound to TbGalE, shown schematically.
Clorobiocin is predicted to participate in multiple hydrogen bonds with the backbone atoms of amino acids lining the NAD+ binding pocket. One ligand hydroxyl group is predicted to form two hydrogen bonds with the backbone amines of N202 and A203; a second ligand hydroxyl group is predicted to form hydrogen bonds with the backbone amines of V35, G36, and S33. Finally, a ligand secondary amine is predicted to form a hydrogen bond with the backbone carbonyl oxygen atom of A100. We note again, however, that many of these same protein−ligand interactions characterize the predicted binding mode of novobiocin as well, despite the fact that novobiocin binding to TbGalE is weak.
The binding modes of compounds 1 and 2, both predicted to bind in the UDP pocket, were not so paradoxical. These compounds contain similar 2′-(phenylcarbamoyl)-[1,1′-biphenyl]-2-carboxylic acid core scaffolds (Table 1) and similar predicted binding modes (Figure 2). For each of these two ligands, we again examined the Vina scores of the ligand docked into the various protein configurations of the ensemble and selected the binding pose/protein configuration associated with the best score for further analysis (Figure 2). The core scaffold common to compounds 1 and 2 participates in many of the same hydrogen bonds that characterize UDP-glucose binding. R335, N202, and R268 all form hydrogen bonds with the diphosphate moiety of UDP-glucose; they likewise form hydrogen bonds with the carboxylate group and the carbonyl oxygen atom of the core scaffold. Additionally, H221 may also form a hydrogen bond with the carboxylate group of the core scaffold (Figure 2).
Figure 2.

The predicted binding modes of compounds 1 and 2. Some portions of the protein have been removed to facilitate visualization. Hydrogen bonds between the protein and ligand are shown as black lines.
Cation−π interactions seem to play a critical role in the binding of compounds 1 and 2. In these interactions, the positive charge of the cation is electrostatically attracted to the quadrupole moment of an aromatic group.(40) If the predicted binding of the core scaffold of compounds 1 and 2 is correct, both R335 and R268 participate in cation−π interactions with the ligand.
Compounds 1 and 2 have polyaromatic moieties that extend into the pocket normally occupied by the uridine and ribose moieties of UDP-glucose. Much work can yet be done to optimize these fused ring systems in order to improve binding, as hinted at by the hydrogen bond predicted to form between compound 1 and the backbone carbonyl oxygen atom of T220 (Figure 2a). Other potential interacting groups in the uridine and ribose portions of the UDP-glucose binding pocket include the backbone carbonyl oxygen atom of P253 and the backbone amine of F255. It may also be possible to add moieties to the fused ring system that exploit the F255 aromatic side chain, which can participate in π−π and cation−π interactions.
Finally, the nucleophilic side-chain thiol of C266 is attractive from a drug-design perspective; fragments with electronegative moieties could be added to the fused ring systems of compounds 1 and 2 in order to facilitate the formation of a covalent adduct with the C266 thiol group, a strategy similar to that employed by Kerr et al. in designing potent vinyl-sulfone inhibitors of parasitic cysteine proteases, including K777, which, pending FDA approval, will soon enter phase I clinical trials.(41) Additionally, as this cysteine is a glycine in HsGalE, compounds that target C266 may be selective for the trypanosomal form of the enzyme, although caution is advised as nonspecific reactions with protein thiols could lead to toxic side effects.
The Secondary Virtual Screen: A Similarity Search
Encouraged by these initial results, we next searched the entire NCI database for compounds similar to the three low-micromolar inhibitors verified experimentally. Eighty additional compounds were subjected to the same RCS protocol used in the primary virtual screen, and 14 novel compounds were thus identified as potential binders. One of these compounds, kedarcidin, a structural orthologue of clorobiocin, was unavailable from the NCI, and another failed the purity criteria. Thus, 12 compounds were subsequently tested experimentally.
Eleven of the 12 compounds showed >50% average inhibition at 10 μM. Subsequent experimental analysis confirmed that these compounds had IC50 values between 0.9 and 6.8 μM (Table 1). As previously, aggregation effects were excluded, and compound identity and purity were confirmed by accurate mass and LC-MS.
Taken together with the compounds of the initial screen, these inhibitors constitute a novel hit series based on a 2′-(phenylcarbamoyl)-[1,1′-biphenyl]-2-carboxylic acid scaffold. Additionally, a singleton hit (clorobiocin) was also found to be potent, although novobiocin, a related compound that might have otherwise been considered a member of the same hit series, was not.
Whole-Cell Assays
All low-micromolar TbGalE inhibitors identified were tested for their ability to inhibit the growth of cultured T. brucei and human liver MRC5 cells using the established Alamar Blue protocol.42,43 Two compounds containing the 2′-(phenylcarbamoyl)-[1,1′-biphenyl]-2-carboxylic acid core scaffold, compounds 12 and 13, had EC50 values of 24.4 and 28.5 μM against whole-cell T. brucei, respectively. Additionally, the natural product clorobiocin (compound 3) had an EC50 value of 4.4 μM. Only compound 14, a compound with no activity against whole-cell T. brucei, demonstrated inhibition of MRC5 growth (Table 1).
To better understand why most of the TbGalE inhibitors failed to inhibit whole-cell T. brucei, we calculated the LogP value of each (Table 1, Supporting Information). With only one exception, the logP values were all high, either near the upper bound for what is considered “druglike” or, in several cases, well beyond that bound.44,45 As these compounds are very hydrophobic, we postulate that they are retained in the cellular membrane, explaining the reduced efficacy against whole-cell T. brucei. The one compound with a druglike LogP value was compound 3 (clorobiocin), which has a measured whole-cell EC50 of 4.4 μM, suggesting inhibition of intracellular targets including TbGalE and, potentially, topoisomerase II.(39) As an alternate explanation for poor membrane traversal, we note also that the charged carboxylate moiety characteristic of the main hit series, absent in clorobiocin, may likewise block access to intracellular targets.
Designing compounds similar to those based on the 2′-(phenylcarbamoyl)-[1,1′-biphenyl]-2-carboxylic acid core scaffold but with reduced hydrophobicity should not be difficult. We note that these inhibitors have roughly the same IC50 values regardless of the fused ring system that is attached to the core scaffold. This suggests that the fused ring systems likely contribute to the overall binding affinity principally through nonspecific interactions, if at all. Hydrophilic functional groups could likely be added to the fused ring systems without compromising the overall binding affinity, thereby increasing the propensity of these molecules to traverse cellular membranes.
Several of the fused ring systems have functional groups that may be amenable to chemical modification. Hydrophilic moieties could be added to the fused ring systems of compounds 1 and 6 via substitution at the hydroxyl group, for example. Several of the TbGalE inhibitors identified also have aromatic halides that could facilitate fragment addition.
As these compounds are already quite large, however, a better approach may be to entirely substitute the fused ring systems of the compounds tested here with other more hydrophilic molecular fragments. Reactions between the readily available compound [1,1′-biphenyl]-2,2′-dicarboxylic acid (NSC1966) and varied aromatic and even nonaromatic amines could expand the chemical series explored here to include compounds with more favorable partition coefficients.
If the above modifications fail to produce high-affinity inhibitors of cellular growth, the charged carboxylate of the main-series compounds could be methylated to potentially facilitate membrane traversal. Once in the cell, this prodrug could perhaps be demethylated by cellular esterases.(46)
Conclusion
In the current study, we used computer-aided drug design to identify 14 inhibitors of TbGalE, a potential T. brucei drug target. As novel HAT therapeutics are urgently needed, we are hopeful that the hit series described here will serve as a useful scaffold for further drug optimization.
Our study also demonstrates the utility of the RCS. Accounting for receptor flexibility when predicting small-molecule protein inhibition is clearly important, as one of the primary-screen inhibitors would not have been identified had we conducted a virtual screen against the crystal structure alone (Supporting Information). We also show that both the ensemble-average and the ensemble-best docking scores are useful RCS ranking metrics (Supporting Information).
The chemical series of TbGalE inhibitors based on a 2′-carbamoyl-[1,1′-biphenyl]-2-carboxylate core scaffold is promising. Future efforts should focus on improving target and cellular potency. As the fused ring systems seem to contribute to ligand binding only nonspecifically, if at all, they could potential be replaced. The side-chain thiol of C266, which comes within 4 Å of the predicted binding mode of compound 2, could also be exploited for more specific binding. As this cysteine is absent from the active site of the corresponding human protein, inhibitors that exploit C266 may also be more pathogen specific.
Experimental Section
System Preparation
Chains A and B of a 2.0 Å resolution TbGalE crystal structure (PDB ID: 1GY8(16)) were processed for use in a molecular dynamics simulation. All crystallographic waters within 4 Å of the protein were retained, and missing loops were modeled using Discovery Studio 2.1 (Accelrys). Hydrogen atoms were added to the protein residues using the PDB2PQR server;(47) histidine protonation states were manually verified. Additional hydrogen atoms were added to the ligand and cofactor using Discovery Studio 2.1 (Accelrys).
Force-field parameters for NAD+ were obtained from Richard B. Ryce.48,49 Atom-type parameters for uridine diphosphate (UDP) were derived from the ff99 force field based on the uridine monophosphate (RU) residue.(50) The linking ribose oxygen atom of RU was replaced with a hydroxyl group, and the atom types of the UDP β phosphate were chosen to be the same as those of the RU α phosphate. Ab initio partial charges were calculated for the UDP atoms using the GAUSSIAN03 computer program. The Hartree−Fock (HF) method, in conjunction with the 6-31+G* basis set, was used for geometry optimization. The optimized structure was used for subsequent single-point charge calculations performed using multiconformational RESP fitting.(51) The remaining protein residues were parametrized using the ff99 force field.(50)
The LEAP module of AMBER9 was subsequently used to further process the system.(52) The protein was submerged in a TIP3P(53) water box that extended 10 Å beyond the protein in the x, y, and z directions. Eighteen Na+ ions were added to bring the system to electrical neutrality. An additional 13 Na+ and Cl− ions were added to simulate a 20 mM solution.
Molecular Dynamics (MD) Simulations
The system was relaxed via a four-phase minimization protocol. In the first phase, 5000 steps of minimization were applied to the hydrogen atoms alone. In the second phase, 5000 steps of minimization were applied to the hydrogen atoms, the water molecules, and all ions. Ten thousand steps of minimization were then applied to the hydrogen atoms, the water molecules, all ions, and the atoms of the protein backbone. Finally, all atoms were minimized for an additional 25000 steps.
A four-phase equilibration protocol was next used. Starting from the minimized system, four sequential, 250-ps MD simulations at 310 K were executed with harmonic restraining forces of 4.0, 3.0, 2.0, and 1.0 kcal mol−1 Å−2 applied to the atoms of the protein backbone for the first, second, third, and fourth simulation, respectively.
This equilibrated system was used as the starting point for five distinct 20 ns, unrestrained, “productive” MD simulations. Because TbGalE is a homodimer, 200 ns of monomer dynamics were effectively simulated. A 2 fs time step was used. The temperature and pressure of the system were maintained by Langevin dynamics at 310 K and the hybrid Nose−Hoover−Langevin piston method at 1 atm, respectively,(54) with period and decay times of 100 and 50 fs, respectively. Long-range electrostatics were treated using the particle mesh Ewald algorithm.(55) Bonded interactions were calculated at every time step. Nonbonded interactions and full electrostatics were calculated every two time steps. Bonds with hydrogen atoms were constrained using the SHAKEH algorithm.(56) NAMD 2.6 was used to perform all minimizations, equilibrations, and productive simulations.(57)
Conformational Clustering
To identify representative protein conformations sampled during the MD simulation, each of the 10 effective trajectories (five simulations times two monomers per simulation) were concatenated, and the composite trajectory was aligned by minimizing the root-mean-square difference (rmsd) between the active-site atoms of each frame and the corresponding atoms of the first frame. rmsd clustering using the gromos algorithm as implemented in the GROMOS++ analysis software58,59 was used to identify representative structures. A rmsd cutoff of 1.4 Å produced 24 clusters. The centoid member of each cluster was selected; the set of all 24 centroids, representative of all the protein configurations sampled during the MD simulation, is said to constitute an ensemble.
Relaxed Complex Scheme
AutoDock Vina (Vina)(31) was used for all computer dockings, as that program was able to adequately recapture the crystallographic poses of the ligand and cofactor. To account for full protein-receptor flexibility, each compound of the NCI Diversity Set II was docked into the 24 ensemble conformations. Four RCS screens were performed. For three, both the UDP ligand and the NAD cofactor were removed from the receptor, and the compounds of the NCI Diversity Set II were docked into the UDP binding site, into the NAD binding site, and into both contiguous sites simultaneously, respectively. For the fourth, the NAD cofactor was maintained, and the compounds of the NCI Diversity Set II were docked into the UDP binding site.
The compounds were ranked according to two ensemble-based criteria. As each compound was docked into 24 distinct protein conformations, the compounds were first ranked by the average of these 24 docking scores (ensemble-average). Next, the compounds were ranked by the best of these 24 docking scores (ensemble-best) (Supporting Information).
Similarity Searches
An online search utility provided by the NCI (http://129.43.27.140/ncidb2/) was used to search the entire NCI database for compounds similar to those identified in the primary RCS screen. Two methods were used to judge compound similarity. First, a substructure search was performed to identify 19 compounds that contained a 2′-(phenylcarbamoyl)-[1,1′-biphenyl]-2-carboxylic acid substructure. Second, 57 similar compounds were identified using a Tanimoto index(60) with a 90% cutoff. Finally, four additional compounds similar to clorobiocin were identified using a Tanimoto index with an 80% cutoff.
TbGalE Inhibition Assay
All compounds were obtained from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov). Compound identity was independently confirmed by high-resolution mass spectrometry, and LC-MS, which was used to monitor both the negative and positive mode simultaneously in the range 80−1000 m/z, confirmed purity >99% (i.e., all compounds gave a single peak >99% that was of the correct mass).
Recombinant TbGalE containing an N-terminal hexahistidine tag was expressed in Escherichia coli and purified as described previously.(18) The inhibition of TbGalE was measured using high pH anion exchange chromatography (HPAEC) to follow the conversion of UDP-Gal to UDP-Glc by TbGalE.(42) The reaction mixture (1 mM Tris pH 7.6, 100 μM UDP-Gal, 100 μM β-NAD+, 5 μg/mL TbGalE, 1% dimethyl sulfoxide) was incubated at 37 °C for 30 min with or without inhibitor (0.03−100 μM), quenched with 15 μL of 10 mM NaOH, and then subjected to HPAEC chromatography on a CarboPac PA-1 column (Dionex) in 1 mM NaOH with a NH4OAc gradient.(42) The eluant was monitored at 260 nm and peaks assigned by comparison to commercial standards. The IC50 value was calculated using a four-parameter fit of the potency curves derived from three independent experiments.
To exclude the possibility of aggregation effects, the inhibition of the compounds was measured at 30 μM (∼5 × IC50) in the presence and absence of a detergent (0.06% n-octylglucopyranoside), and no difference was observed.
Cytotoxicity Assay
Cytotoxicity assays were preformed against cultured T. brucei and human MRC5 cells as described previously,(42) but further miniaturized to 384 well plates. Briefly, 50 μL of cells (1 × 104 cells/ml) were aliquoted into 384-well plate and incubated with or without inhibitor (100 μM to 30 nM) for a further three days before the number of viable cells was counted using the Alamar blue assay.(43)
Acknowledgments
J.D.D. is funded by a Pharmacology Training Grant through the UCSD School of Medicine. M.D.U. and M.A.J.F. are funded by a Programme Grant from The Wellcome Trust (085622). This work was also carried out with funding from NIH GM31749, NSF MCB-0506593, and MCA93S013 to J.A.M. Additional support from the Howard Hughes Medical Institute, the National Center for Supercomputing Applications, the San Diego Supercomputer Center, the W.M. Keck Foundation, the National Biomedical Computational Resource, and the Center for Theoretical Biological Physics is gratefully acknowledged. All compounds were provided by the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov). We also thank Matthew Durrant for help with virtual screening.
Supporting Information Available
Additional experimental details and discussion, including comments regarding the utility of rescoring AutoDock Vina docked poses with the AutoDock 4.0 scoring function and a comparison of the two ensemble-based ranking criteria described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviation: DTP, Developmental Therapeutics Program; HAT, human African trypanosomiasis; HIV, human immunodeficiency virus; logP, log base ten of the partition coefficient; NAD, nicotinamide adenine dinucleotide; NAMD, nanoscale molecular dynamics; NCI, National Cancer Institute; NIH, National Institutes of Health; NMR, nuclear magnetic resonance; NSF, National Science Foundation; TbGalE, UDP-galactose 4′-epimerase from Trypanosoma brucei; UDP, uridine diphosphate; WHO, World Health Organization.
Supplementary Material
References
- WHO. African trypanosomiasis (sleeping sickness), Fact Sheet 259. http://www.who.int/mediacentre/factsheets/fs259/en/Oct12, 2009.
- Gehrig S.; Efferth T. Development of drug resistance in Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense. Treatment of human African trypanosomiasis with natural products (Review). Int. J. Mol. Med. 2008, 22, 411–419. [PubMed] [Google Scholar]
- Maser P.; Luscher A.; Kaminsky R. Drug transport and drug resistance in African trypanosomes. Drug Resist. Updates 2003, 6, 281–290. [DOI] [PubMed] [Google Scholar]
- Delespaux V.; de Koning H. P. Drugs and drug resistance in African trypanosomiasis. Drug Resist. Updates 2007, 10, 30. [DOI] [PubMed] [Google Scholar]
- Vansterkenburg E. L.; Coppens I.; Wilting J.; Bos O. J.; Fischer M. J.; Janssen L. H.; Opperdoes F. R. The uptake of the trypanocidal drug suramin in combination with low-density lipoproteins by Trypanosoma brucei and its possible mode of action. Acta Trop. 1993, 54, 237–250. [DOI] [PubMed] [Google Scholar]
- Blum J.; Nkunku S.; Burri C. Clinical description of encephalopathic syndromes and risk factors for their occurrence and outcome during melarsoprol treatment of human African trypanosomiasis. Trop. Med. Int. Health 2001, 6, 390–400. [DOI] [PubMed] [Google Scholar]
- Kayser O.; Kiderlen A. F.; Croft S. L. Natural products as antiparasitic drugs. Parasitol. Res. 2003, 90 (Suppl 2), S55–S62. [DOI] [PubMed] [Google Scholar]
- Iten M.; Matovu E.; Brun R.; Kaminsky R. Innate lack of susceptibility of Ugandan Trypanosoma brucei rhodesiense to dl-alpha-difluoromethylornithine (DFMO). Trop. Med. Parasitol. 1995, 46, 190–194. [PubMed] [Google Scholar]
- Priotto G.; Kasparian S.; Mutombo W.; Ngouama D.; Ghorashian S.; Arnold U.; Ghabri S.; Baudin E.; Buard V.; Kazadi-Kyanza S.; Ilunga M.; Mutangala W.; Pohlig G.; Schmid C.; Karunakara U.; Torreele E.; Kande V. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [DOI] [PubMed] [Google Scholar]
- Cross G. A. Antigenic variation in trypanosomes: secrets surface slowly. Bioessays 1996, 18, 283–291. [DOI] [PubMed] [Google Scholar]
- Smith D. H.; Pepin J.; Stich A. H. Human African trypanosomiasis: an emerging public health crisis. Br. Med. Bull. 1998, 54, 341–355. [DOI] [PubMed] [Google Scholar]
- Schechter P. J.; Sjoerdsma A. Difluoromethylornithine in the Treatment of African Trypanosomiasis. Parasitol. Today 1986, 2, 223–224. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajorath J. Integration of virtual and high-throughput screening. Nature Rev. Drug Discovery 2002, 1, 882–894. [DOI] [PubMed] [Google Scholar]
- Abagyan R.; Totrov M. High-throughput docking for lead generation. Curr. Opin. Chem. Biol. 2001, 5, 375–382. [DOI] [PubMed] [Google Scholar]
- Shaw M. P.; Bond C. S.; Roper J. R.; Gourley D. G.; Ferguson M. A.; Hunter W. N. High-resolution crystal structure of Trypanosoma brucei UDP-galactose 4′-epimerase: a potential target for structure-based development of novel trypanocides. Mol. Biochem. Parasitol. 2003, 126, 173–180. [DOI] [PubMed] [Google Scholar]
- Holden H. M.; Rayment I.; Thoden J. B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol. Chem. 2003, 278, 43885–43888. [DOI] [PubMed] [Google Scholar]
- Roper J. R.; Guther M. L.; Milne K. G.; Ferguson M. A. Galactose metabolism is essential for the African sleeping sickness parasite Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 5884–5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper J. R.; Guther M. L.; Macrae J. I.; Prescott A. R.; Hallyburton I.; Acosta-Serrano A.; Ferguson M. A. The suppression of galactose metabolism in procylic form Trypanosoma brucei causes cessation of cell growth and alters procyclin glycoprotein structure and copy number. J. Biol. Chem. 2005, 280, 19728–19736. [DOI] [PubMed] [Google Scholar]
- Mehlert A.; Zitzmann N.; Richardson J. M.; Treumann A.; Ferguson M. A. The glycosylation of the variant surface glycoproteins and procyclic acidic repetitive proteins of Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 91, 145–152. [DOI] [PubMed] [Google Scholar]
- Nolan D. P.; Geuskens M.; Pays E. N-Linked glycans containing linear poly-N-acetyllactosamine as sorting signals in endocytosis in Trypanosoma brucei. Curr. Biol. 1999, 9, 1169–1172. [DOI] [PubMed] [Google Scholar]
- Tetaud E.; Barrett M. P.; Bringaud F.; Baltz T. Kinetoplastid glucose transporters. Biochem. J. 1997, 325 (Pt 3), 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenthal R.; Game S.; Holman G. D. Specificity and kinetics of hexose transport in Trypanosoma brucei. Biochim. Biophys. Acta 1989, 985, 81–89. [DOI] [PubMed] [Google Scholar]
- Dodson G. G.; Lane D. P.; Verma C. S. Molecular simulations of protein dynamics: new windows on mechanisms in biology. EMBO Rep. 2008, 9, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaro R. E.; Baron R.; McCammon J. A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput.-Aided Mol. Des. 2008, 22, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K.; Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: induced-fit versus population-shift mechanisms. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 11182–11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G.; Fechner U. Computer-based de novo design of drug-like molecules. Nature Rev. Drug Discovery 2005, 4, 649. [DOI] [PubMed] [Google Scholar]
- Lin J. H.; Perryman A. L.; Schames J. R.; McCammon J. A. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J. Am. Chem. Soc. 2002, 124, 5632–5633. [DOI] [PubMed] [Google Scholar]
- Schames J. R.; Henchman R. H.; Siegel J. S.; Sotriffer C. A.; Ni H.; McCammon J. A. Discovery of a novel binding trench in HIV integrase. J. Med. Chem. 2004, 47, 1879–1881. [DOI] [PubMed] [Google Scholar]
- Amaro R. E.; Schnaufer A.; Interthal H.; Hol W.; Stuart K. D.; McCammon J. A. Discovery of drug-like inhibitors of an essential RNA-editing ligase in Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17278–17283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- Brahma A.; Banerjee N.; Bhattacharyya D. UDP-galactose 4-epimerase from Kluyveromyces fragilis—catalytic sites of the homodimeric enzyme are functional and regulated. FEBS J. 2009, 276, 6725–6740. [DOI] [PubMed] [Google Scholar]
- Hay M.; Bhaduri A. UDP-Glucose 4-epimerase from Saccharomyces fragilis. Allosteric kinetics with UDP-glucose as substrate. J. Biol. Chem. 1975, 250, 4373–4375. [PubMed] [Google Scholar]
- Feng B. Y.; Simeonov A.; Jadhav A.; Babaoglu K.; Inglese J.; Shoichet B. K.; Austin C. P. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007, 50, 2385–2390. [DOI] [PubMed] [Google Scholar]
- Surig U.; Gaal K.; Kostenis E.; Trankle C.; Mohr K.; Holzgrabe U. Muscarinic allosteric modulators: atypical structure−activity relationships in bispyridinium-type compounds. Arch. Pharm. (Weinheim) 2006, 339, 207–212. [DOI] [PubMed] [Google Scholar]
- Langmead C. J. Screening for positive allosteric modulators: assessment of modulator concentration−response curves as a screening paradigm. J. Biomol. Screening 2007, 12, 668–676. [DOI] [PubMed] [Google Scholar]
- Pate P. G.; Wolfson J. S.; McHugh G. L.; Pan S. C.; Swartz M. N. Novobiocin antagonism of amastigotes of Trypanosoma cruzi growing in cell-free medium. Antimicrob. Agents Chemother. 1986, 29, 426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschmann R. L.; Wolfson J. S.; McHugh G. L.; Dickersin G. R.; Hooper D. C.; Swartz M. N. Novobiocin-induced ultrastructural changes and antagonism of DNA synthesis in Trypanosoma cruzi amastigotes growing in cell-free medium. J. Protozool. 1989, 36, 14–20. [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. Cation−pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [DOI] [PubMed] [Google Scholar]
- Kerr I. D.; Lee J. H.; Farady C. J.; Marion R.; Rickert M.; Sajid M.; Pandey K. C.; Caffrey C. R.; Legac J.; Hansell E.; McKerrow J. H.; Craik C. S.; Rosenthal P. J.; Brinen L. S. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 2009, 284, 25697–25703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak M. D.; Tabudravu J. N.; Msaki A.; Matera K. M.; Brenk R.; Jaspars M.; Ferguson M. A. Identification of novel inhibitors of UDP-Glc 4′-epimerase, a validated drug target for african sleeping sickness. Bioorg. Med. Chem. Lett. 2006, 16, 5744–5747. [DOI] [PubMed] [Google Scholar]
- Raz B.; Iten M.; Grether-Buhler Y.; Kaminsky R.; Brun R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T. b. rhodesiense and T. b. gambiense) in vitro. Acta Trop. 1997, 68, 139–147. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Ghose A. K.; Viswanadhan V. N.; Wendoloski J. J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [DOI] [PubMed] [Google Scholar]
- Testa B. Prodrug research: futile or fertile?. Biochem. Pharmacol. 2004, 68, 2097–2106. [DOI] [PubMed] [Google Scholar]
- Dolinsky T. J.; Czodrowski P.; Li H.; Nielsen J. E.; Jensen J. H.; Klebe G.; Baker N. A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R. C.; de Souza M. M.; Mercer I. P.; Gould I. R.; Klug D. R.. Large and fast relaxations inside a protein: calculation and measurement of reorganization energies in mcohol dehydrogenase. J. Phys. Chem. B 2002, 106, 11658−11665. [Google Scholar]
- Pavelites J. J.; Gao J.; Bash P. A.; Mackerell A. D. Jr. A molecular mechanics force field for NAD+ NADH, and the pyrophosphate groups of nucleotides. J. Comput. Chem. 1998, 18, 221–239. [Google Scholar]
- Ponder J. W.; Case D. A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [DOI] [PubMed] [Google Scholar]
- Bayly C. I., ; Cieplak P., ; Cornell W., ; Kollman P. A., .
- Case D. A.; Cheatham T. E. III; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995, 103, 4613. [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N−log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar]
- Kale L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landon M. R.; Amaro R. E.; Baron R.; Ngan C. H.; Ozonoff D.; McCammon J. A.; Vajda S. Novel druggable hot spots in avian influenza neuraminidase H5N1 revealed by computational solvent mapping of a reduced and representative receptor ensemble. Chem. Biol. Drug Des. 2008, 71, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen M.; Hunenberger P. H.; Bakowies D.; Baron R.; Burgi R.; Geerke D. P.; Heinz T. N.; Kastenholz M. A.; Krautler V.; Oostenbrink C.; Peter C.; Trzesniak D.; van Gunsteren W. F. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 2005, 26, 1719–1751. [DOI] [PubMed] [Google Scholar]
- Tanimoto T.IBM Technical Report Series; 1957.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.