

Abstract
Protein-lipid interaction and bilayer regulation of membrane protein functions are largely controlled by the hydrophobic match between the transmembrane (TM) domain of membrane proteins and the surrounding lipid bilayer. To systematically characterize responses of a TM helix and lipid adaptations to a hydrophobic mismatch, we have performed a total of 5.8-μs umbrella sampling simulations and calculated the potentials of mean force (PMFs) as a function of TM helix tilt angle under various mismatch conditions. Single-pass TM peptides called WALPn (n = 16, 19, 23, and 27) were used in two lipid bilayers with different hydrophobic thicknesses to consider hydrophobic mismatch caused by either the TM length or the bilayer thickness. In addition, different flanking residues, such as alanine, lysine, and arginine, instead of tryptophan in WALP23 were used to examine their influence. The PMFs, their decomposition, and trajectory analysis demonstrate that 1), tilting of a single-pass TM helix is the major response to a hydrophobic mismatch; 2), TM helix tilting up to ∼10° is inherent due to the intrinsic entropic contribution arising from helix precession around the membrane normal even under a negative mismatch; 3), the favorable helix-lipid interaction provides additional driving forces for TM helix tilting under a positive mismatch; 4), the minimum-PMF tilt angle is generally located where there is the hydrophobic match and little lipid perturbation; 5), TM helix rotation is dependent on the specific helix-lipid interaction; and 6), anchoring residues at the hydrophilic/hydrophobic interface can be an important determinant of TM helix orientation.
Introduction
The transmembrane (TM) domain of most membrane proteins consists of one or multiple helices, and their interactions with surrounding lipids are important determinants for membrane protein structure and function. Understanding of membrane protein functions thus requires not only the protein structural information, but also information on how the lipid environments affect protein structure and organization. The match between the hydrophobic length of the TM domain and that of the lipid bilayer has been recognized as a central feature in protein-lipid interactions and bilayer regulation of membrane protein functions (1). Responses to an energetically unfavorable hydrophobic mismatch include conformational changes of the TM domain, lipid adaptations by changes in bilayer thickness and lipid chain order, and TM helix association (2). In particular, the changes in TM helix tilt, kink, and rotation angles to relieve any mismatch are often considered key conformational changes implicated in a switch between active and inactive conformations of membrane proteins (3–5). However, structural and dynamic changes of the TM domain in response to a hydrophobic mismatch as well as molecular forces governing such changes remain to be fully understood at the atomic level. In particular, given the abundance of membrane proteins with a single-pass TM helix and their association and conformational changes involved in TM-induced signaling (6,7), it is important and challenging to understand such properties quantitatively.
Mainly due to experimental difficulties associated with membrane proteins of multiple TM helices, our understanding of hydrophobic mismatch effects is mostly based on studies using various single-pass TM helices in model lipid bilayers. The responses to a hydrophobic mismatch have been systematically investigated by changing the hydrophobic length of TM helices or the hydrophobic thickness of lipid bilayers. In particular, designed TM helical peptides, such as WALP and KALP containing poly-leucine/alanine flanked by tryptophan or lysine (Table 1), have been extensively used both in experimental (1,8–13) and computational (14–19) studies to characterize a main response to a hydrophobic mismatch as well as the role of peptide-lipid interactions. In the case of a single-pass TM helix, helix tilting is generally considered the main response to a hydrophobic mismatch with minimum perturbation of lipid bilayers (18,20). Recently, based on the potential-of-mean-force (PMF) calculation as a function of WALP19's tilt angle in a dimyristoylphosphatidyl-choline (DMPC) bilayer, Lee and Im (18) have provided novel insights (to our knowledge) into the driving forces of TM helix tilting in the lipid bilayer: a thermally accessible tilt region of a single-pass TM helix is governed by the intrinsic entropic contribution arising from helix precession (area) around the membrane normal and the helix-lipid interactions that could be TM sequence- and length-specific.
Table 1.
Amino acid sequences of WALPn (n = 16, 19, 23, and 27) and XALP23 (X = A, K, and R) peptides
| Peptide | Sequence∗ | TM hydrophobic length (Å)† |
|---|---|---|
| WALP16 | GWW(LA)5WWA | 15.0 |
| WALP19 | GWW(LA)6LWWA | 19.5 |
| WALP23 | GWW(LA)8LWWA | 25.5 |
| WALP27 | GWW(LA)10LWWA | 31.5 |
| AALP23 | GAA(LA)8LAAA | 31.5 |
| KALP23 | GKK(LA)8LKKA | 25.5 |
| RALP23 | GRR(LA)8LRRA | 25.5 |
The N-terminus of each peptide is blocked by the acetyl group and its C-terminus by the n-methyl amide group.
The lengths are measured in the hydrophobic region (bold) with an assumption of an ideal α-helix.
The aim of this work is to provide in-depth understanding of detailed interplays of specific helix-lipid interactions in TM helix tilting and lipid adaptations under various hydrophobic mismatch conditions. We have performed a total of 5.8-μs umbrella sampling molecular dynamics (MD) simulations (21) to calculate the PMFs of various single-pass TM helices as a function of their tilt and rotation angles (22,23) and analyzed the resulting PMFs by the free energy decomposition technique (18,22).
First, we used WALPn (n = 16, 19, 23, and 27; Table 1) in DMPC bilayers to determine the influence of the TM hydrophobic length on TM helix orientation and lipid adaptations. Second, the results in DMPC were compared with those in palmitoyloleoylphosphatidyl-choline (POPC) bilayers to characterize the influence of the bilayer hydrophobic thickness on TM helix orientation and lipid adaptations. Third and finally, we used alanine, lysine, and arginine as a flanking residue instead of tryptophan in WALP23 in DMPC to examine the influence of various anchoring residues on TM helix orientation and lipid adaptations (12). The PMFs, their decomposition, and trajectory analysis are discussed and generalized in terms of responses of a TM helix and lipid adaptations to various hydrophobic mismatch conditions.
Methods
Defining the tilt and rotation angles
The orientation of a TM helix is defined by its tilt (τ) and rotation (ρ) angles. With the Z axis parallel to the membrane normal, τ is defined as the angle between the helical principal axis and the unit vector along the Z axis (22). To define ρ, both the internal and external references have to be defined (Fig. 1). The internal reference is given by the vector pointing from the helical axis to Cα atom of Leu14 in all WALP peptides except WALP16/19. Because the position of Leu14 in WALP16/19 is too close to the C-terminus, the flexibility of which makes it difficult to define ρ, we instead used Ala7 (WALP16) and Leu10 (WALP19), which are at a similar position to Leu14 on the helical wheel projection. With the unit vector along the Z axis as the external reference, ρ is then defined as the angle between the projections of such reference vectors on the plane made by the second and third helical principal axes. Detailed expressions can be found in our previous works (22,24).
Figure 1.

The definition of helix tilt (τ) and rotation (ρ). The value ρ is defined as the angle between the perpendicular vector (rs) from the helical axis (a) to the selected Cα atom (green circle) and the projection vector (zp) of the Z axis onto the plane (light blue) made by the second and third principal axes. The sign of the rotational angle becomes positive if zp × rs is in the opposite direction to a, or negative otherwise.
Umbrella sampling simulations
The sequence of each peptide studied in this work is given in Table 1. Using the input scripts from the Membrane Builder module (25,26) in CHARMM-GUI (http://www.charmm-gui.org) (27), each peptide with an ideal α-helical conformation (ϕ = −57.8°; ψ = −47.0°) was inserted into a pre-equilibrated lipid bilayer of 72 DMPC (or POPC) and water molecules with τ = 0° and its center at Z = 0. Four chloride ions were added to neutralize the KALP23 and RALP23 systems. To relax the uncorrelated initial systems, 400-ps equilibration was performed with harmonic restraints on heavy atoms. The number of individual components and each system size are listed in Table S1 in the Supporting Material. The snapshots of each system at τ = 0° are shown in Fig. 2.
Figure 2.
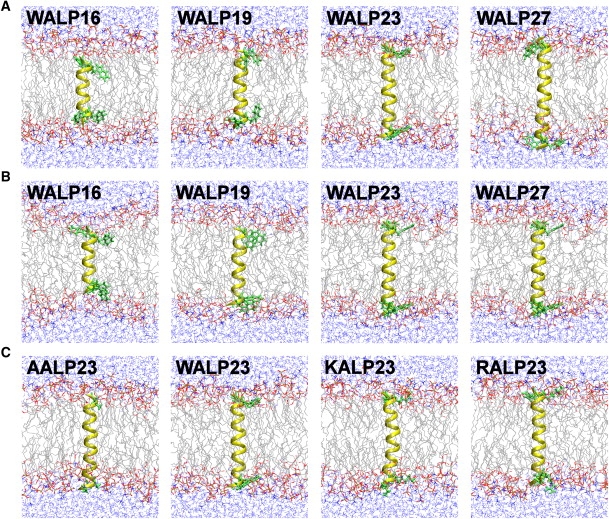
Molecular graphic view of the last snapshot in (A) WALPn/DMPC, (B) WALPn/POPC, and (C) XALP23/DMPC systems at τ = 0° (the helix in yellow, anchoring residues in green, lipid tails in gray, and water molecules in blue).
To perform umbrella sampling MD simulations as a function of TM helix tilt, an initial structure at each window was generated by tilting the helix sequentially from 0° to a specific maximum angle by 1° every 100 ps. The total number of windows and the maximum τ are listed in Table S1. Each window was then subjected to 1-ns equilibration followed by 10- or 12.5-ns production. The force constants of the helix tilt restraint potential (22) were set to 2000 and 6000 kcal/(mol·rad2) (18) for equilibration and production, respectively. All calculations were performed using the biomolecular simulation program CHARMM (28) with the all-atom parameter set PARAM22 for protein (29) including the dihedral cross-term corrections (30) and a modified TIP3 water model (31), as well as recently optimized lipid parameters (32). The cross-sectional areas of DMPC and POPC were set to 60.7 Å2 and 68.3 Å2 at 303.15 K (33), respectively. Following the same protocol in the previous PMF calculation of WALP19 (18), a time-step of 2 fs was used with the SHAKE algorithm (34), and the constant temperature (303.15 K) and pressure (1 atm along the Z-direction) were maintained by the Nosé-Hoover method (35) and the Langevin-piston algorithm (36), respectively, for the NPAT (constant pressure, surface area, and temperature) dynamics. We used the same options for nonbonded interactions in the input scripts provided by the CHARMM-GUI Membrane Builder (25,26).
The PMF, W(τ), as a function of τ was calculated by integrating the reversible work done by the mean force, −〈F(τ)〉τ, along τ;
| (1) |
where U(r) is the potential energy of the system, |J| is the determinant of the Jacobian related to the transformation of the Cartesian coordinate into the generalized coordinate τ, and kB is the Boltzmann constant. To examine the PMF convergence, the trajectory in each window was sequentially divided into every 1-ns duration. The PMFs were then calculated from each subtrajectory. When the PMF was constructed using the last 8-ns trajectory, even the highest standard deviation does not exceed ±2.2 kcal/mol (in the RALP23/DMPC system), illustrating that the calculated PMFs are well converged. The largest standard deviation of the PMFs occurs at either energetically unfavorable small or large tilt angle region (Fig. 3).
Figure 3.

The total PMFs as a function of τ in (A) WALPn/DMPC, (B) WALPn/POPC, and (C) XALP23/DMPC systems.
Results and Discussion
Influence of hydrophobic length of TM helices
Tilting of a TM helix in a bilayer is affected by its TM hydrophobic length (LTM) and the bilayer hydrophobic thickness (Lbilayer) whose difference can cause either positive (LTM > Lbilayer) or negative (LTM < Lbilayer) hydrophobic mismatch. In a DMPC bilayer with Lbilayer ≈ 23 Å (10), WALP16/19 and WALP23/27 at τ = 0° are under negative and positive mismatch conditions, respectively (see LTM in Table 1). Fig. 3 A shows the total PMFs of the WALPn/DMPC systems as a function of τ. The detailed information about the minimum-PMF tilt angle (τmin) and the free energy change from τ = 0° to τmin, ΔW(0→τmin) in each system is summarized in Table 2. As LTM increases, τmin increases, ΔW(0→τmin) decreases, and the thermally accessible tilt region becomes wider, clearly illustrating that it is energetically more favorable for TM helices of longer LTM to have larger τmin in order to maximize the hydrophobic match. In the case of WALP23, its τmin is similar to the average tilt (τ = 33.5°) from the recent multiple MD trajectories (16). Also, although the lipid types are different, the large τmin from the PMF appears to be consistent with the recent fluorescence spectroscopy experiment (20) that shows much larger tilt angle of WALP23 (τ = 24° ± 5°) in a DOPC membrane than the tilt angle (τ = 4.4°∼8.2°) estimated from solid-state 2H-NMR quadrupolar splitting measurements (12,13).
Table 2.
The summary of the PMF in each system
| System: peptide/lipid | Minimum-PMF tilt angle/ ΔW(0→τmin) (kcal/mol) | Thermal-accessible tilt range |
|---|---|---|
| WALP16/DMPC | 12.1°/−3.09 ± 0.27 | 2.6° ∼ 19.4° |
| WALP19/DMPC | 12.1°/−3.30 ± 0.24 | 3.1° ∼ 16.1° |
| WALP23/DMPC | 28.1°/−4.99 ± 0.21 | 14.4° ∼ 39.0° |
| WALP27/DMPC | 43.3°/−7.45 ± 0.51 | 32.3° ∼ 50.7° |
| WALP16/POPC | 6.4°/−3.33 ± 0.21 | 3.3° ∼ 17.4° |
| WALP19/POPC | 12.5°/−3.14 ± 0.29 | 2.7° ∼ 19.9° |
| WALP23/POPC | 14.9°/−3.83 ± 0.28 | 6.7° ∼ 25.6° |
| WALP27/POPC | 38.2°/−4.70 ± 0.49 | 13.7° ∼ 46.3° |
| AALP23/DMPC | 34.2°/−4.60 ± 0.19 | 16.5° ∼ 44.5° |
| KALP23/DMPC | 20.7°/−4.68 ± 0.41 | 13.6° ∼ 29.3° |
| RALP23/DMPC | 15.6°/−3.44 ± 0.53 | 4.0° ∼ 22.4° |
TM helix tilting is governed by the intrinsic entropic contribution (Wentropy) arising from helix precession (area) around the membrane normal and the specific helix-lipid interactions (Wint) (18). To determine detailed interplays of underlying molecular forces in TM helix tilting in the WALPn/DMPC systems, the total PMF of each system was decomposed into Wint and Wentropy based on Eq. 1. It should be noted that Wint also includes the contribution from the helix conformational changes that resulted from the helix-lipid interactions. As shown in Fig. 4, A and B, under the negative mismatch condition such as the WALP16/19 systems, it is evident that tilting up to τmin is driven by Wentropy. After τmin, Wint makes the dominant contribution to the increase of the PMFs. These results clearly explain the microscopic driving forces of previous MD simulation observations that a short TM helix can tilt up to ∼10° in a membrane even under a negative mismatch condition (14). As LTM increases, such as WALP23/27 in Fig. 4, C and D, the Wint contribution to the total PMF becomes more significant to maximize the hydrophobic match and provides additional driving forces for TM helix tilting.
Figure 4.

Decomposition of the total PMF (black) into the helix-lipid interaction (Wint: red) and the entropic contribution (Wentropy: blue) in (A) WALP16, (B) WALP19, (C) WALP23, and (D) WALP27 in DMPC bilayers. The thermally accessible tilt region is indicated by the black-dashed line.
The Wint contribution can be further characterized in terms of lipid adaptations, hydrophobic match, and helix conformational changes as a function of τ. In particular, it has been postulated that the lipids near an integral membrane protein would change their hydrophobic length or that the protein itself undergoes conformational changes to maximize the hydrophobic match (i.e., to minimize the energy penalty of exposing nonpolar residues to aqueous solution) (1). The present umbrella sampling simulation trajectory provides an excellent resource to quantify such changes at the atomic level, which would be difficult to measure in normal MD simulations because of limited sampling along TM helix orientations. In this study, the local lipid adjustment is quantified by
where and are the average hydrophobic thicknesses of contact and bulk lipid bilayers, respectively. Lbilayer is defined as an average distance between the acyl chain C2 carbon atoms in both leaflets (10). A lipid molecule that has any of its heavy atoms within 4 Å from the helix heavy atoms is classified as a contact lipid, otherwise as a bulk one. Therefore, the negative ΔLadaptation indicates a local membrane thinning and the positive ΔLadaptation a local membrane thickening with respect to the bulk lipid bilayer. ΔLadaptation as a function of τ in Fig. 5 A clearly shows that the local lipids respond differently to different hydrophobic mismatch conditions. For WALP16/19 that are under the negative mismatch (Lbilayer > LTM) at τ = 0°, the local membrane thinning (ΔLadaptation < 0) is apparent and ΔLadaptation slightly decreases as τ increases. In contrast, for WALP23/27 that are under the positive mismatch (Lbilayer < LTM) at τ = 0°, the local membrane thickening (ΔLadaptation > 0) occurs at small tilt angles, but becomes reduced as τ increases and disappears at ∼τmin where there is little local lipid perturbation due to the hydrophobic match (see below). After τmin, to maximize hydrophobic match, the contact lipids become thinner (ΔLadaptation < 0) because the effective LTM is reduced as τ increases. Fig. 6 schematically illustrates how the local lipid adaptations occur at τ = 0°, τ ≈ τmin, and τ > τmin, based on Fig. 5 A.
Figure 5.

Local lipid adjustment (ΔLadaptation) as a function of τ in (A) WALPn/DMPC, (B) WALPn/POPC, and (C) XALP23/DMPC systems: , where and are the average hydrophobic thicknesses of contact and bulk lipid bilayers.
Figure 6.

Schematic representation of the local lipid adjustment based on the ΔL change (Fig. 5) under negative (left) and positive (right) hydrophobic mismatch conditions.
To quantify the extent of the hydrophobic match as a function of τ, we calculated the difference between the effective LTM (13) and , i.e.,
where both LTM and were calculated in two different ways: one from the simulation trajectory (solid lines in Fig. 7) and the other from fixed values (LTM in Table 1 and = 23 Å (DMPC) or 26 Å (POPC); dotted lines in Fig. 7). Because LTM remained at nearly the same values as in Table 1 during the simulations, the difference in the two lines represents the influence of the local lipid adaptations on ΔLmismatch. As shown in Fig. 7 A, for WALP16/19, despite the local membrane thinning (Fig. 5 A), the negative hydrophobic mismatch is apparent and slightly increases as τ increases (37). Therefore, it becomes clear that the Wint contribution is not favorable for tilting of WALP16/19 in DMPC (Fig. 4, A and B) because of increased deformation of the lipid bilayer as τ increases. In contrast, for WALP23/27, τ at ΔLmismatch ≈ 0 is well matched with τmin, demonstrating that the local lipid adjustments toward ΔLmismatch ≈ 0 (i.e., ΔLadaptation ≈ 0 in Fig. 5 A) provide the favorable Wint contribution for tilting of WALP23/27 toward τmin (Fig. 4, C and D) and relieve the deformation stress on the lipid bilayer. At τ > τmin, however, bilayer deformation (ΔLadaptation < 0) causes stress on the lipid bilayer, making the Wint contribution unfavorable. While excessive stress on a lipid bilayer at extreme hydrophobic mismatch can induce helix deformations such as helix kink or bending, it also becomes apparent from the thermally accessible tilt regions (Fig. 3 A and Table 2) that the membrane bilayer system can tolerate a certain extent of a hydrophobic mismatch by slight lipid adaptations.
Figure 7.

Extent of the hydrophobic match (ΔLmismatch) as a function of τ in (A) WALPn/DMPC, (B) WALPn/POPC, and (C) XALP23/DMPC systems: , where LTM is the TM hydrophobic length and is the average hydrophobic thicknesses of the contact lipids. Both LTM and were calculated from the simulation trajectory (solid lines) or they were set to fixed values (LTM in Table 1 and = 23 Å (DMPC) or 26 Å (POPC)) (dotted lines).
As shown in Fig. S1 A, the helicity of each helix is well maintained except at energetically unfavorable large τ. The previous study on WALP19 reveals that such helix deformation including helix bending at large τ is attributed to the existence of four Trp anchoring residues at the hydrophobic/hydrophilic interface (18). In other words, it is energetically more favorable for WALP19 to deform the helical conformation slightly at large τ in a DMPC bilayer, than to have Trp side chains inserted into the hydrophobic core (18). Fig. S2 shows the center of mass of each Trp side chain along the Z axis (ZTrp) as a function of τ. Similar to WALP19, ZTrp of WALP16 is around the hydrophobic/hydrophilic interface of the lipid bilayer due to outward-facing of the Trp side chains (see below), despite their relatively short LTM. However, the Trp side chains of WALP23/27 appear to partition into the DMPC hydrophobic core region at large τ. Such difference arises for two reasons. First, because WALP16/19 is under the negative mismatch regardless of τ, the hydrophobic/hydrophilic interfacial matching becomes a dominant factor in optimizing their conformation during helix tilting (in contrast, WALP23/27 gain the favorable Wint contribution by optimizing the hydrophobic match, which becomes more dominant than the interfacial matching, at large τ). Second, the rotation angle of each helix during its tilting, determined by the helix-lipid interactions, also dictates the positioning of the Trp residues.
The direction of helix tilting (i.e., rotation angle, ρ) is also an important determinant of TM helix tilting. In our simulations, because the restraint forces are exerted only on the Cα atoms that define the helical principal axis, the helix can rotate around the helical axis, depending on helix-environment (lipid or water) interactions (23). Fig. 8 shows the change of ρ as a function of τ and also illustrates the tilting direction for each WALPn/DMPC system. In general, ρ shows large fluctuations at τ < 10° (due to ill-definition of ρ and low free energy barriers at small tilt angles), but converged to a specific angle with less fluctuation at ∼τmin. It becomes clear that ZTrp in Fig. S2 is related to the tilting direction. For example, WALP23/27 tilt in a direction between Trp2 and Trp3, so that the Trp residues partition into the membrane hydrophobic region at large τ.
Figure 8.

TM helix rotation angle (ρ) as a function of τ in (A) WALP16, (B) WALP19, (C) WALP23, and (D) WALP27 in DMPC bilayers. The reference atoms, such as Ala7 (for WALP16), Leu10 (for WALP19), and Leu14 (for others), and the tilting direction at τmin, are indicated (solid circles and shaded arrows on the helical view), respectively.
To verify that the helix follows the energetically favorable orientation during the tilting simulations, we have performed additional PMF calculations of WALP23 as a function of τ by restraining ρ at four distinct rotation angles, i.e., ρ = −150°, −60°, 30°, and 120°. As shown in Fig. S3, the tilting energetics is largely dependent on ρ, and the lowest tilting PMF is found with ρ = 120°, which corresponds to ρ at τ = τmin (120.8° ± 18.5°) in Fig. 8 C. This rotation is also well correlated with ρ = 155° (using our definition) estimated from the solid-state 2H-NMR study (12). Clearly, our results demonstrate that each helix prefers to tilt with a specific rotation based on hydrophobic or interfacial matching, and the helix adopts the energetically favorable orientation during the tilting simulations.
Influence of bilayer hydrophobic thickness
In addition to LTM, the TM helix orientation is also affected by Lbilayer. Experimental studies have also used various lipid bilayers with different Lbilayer to induce different hydrophobic mismatch conditions (12,13). To elucidate the effect of Lbilayer on TM helix orientation and lipid adaptations, we used POPC bilayers (Lbilayer = ∼26 Å) (38) for the PMF calculations and compared the results with those in DMPC. In the WALPn/POPC systems, only WALP27 is under the positive mismatch condition at τ = 0°. Similar to the DMPC cases, as shown in Fig. 3 B and Table 2, τmin increases and the thermally accessible tilt region becomes wider as LTM increases. However, each TM helix generally shows smaller τmin and ΔW(0→τmin), and a broader thermally accessible tilt region than in DMPC, illustrating that the TM helices do respond differently in POPC than in DMPC. The PMF decomposition in Fig. S4 clearly shows that tilting up to τmin is mostly driven by Wentropy because all WALP peptides except WALP27 are under the negative mismatch condition in POPC bilayers. The increase in the total PMF after τmin well correlates with Wint, but the Wint contribution appears to be less significant than in DMPC.
Because of the changes in the hydrophobic mismatch condition and thus in the helix-lipid interaction, the local lipid adjustment of the POPC bilayers is different from that of DMPC. As shown in Fig. 5 B, ΔLadaptation in POPC generally shows less change than in DMPC for the same LTM. Compared to DMPC, POPC has the same headgroup but a larger cross-sectional area, as the unsaturated acyl chain makes the hydrophobic packing of lipid tails less tight than found in the fully saturated DMPC acyl chains (39). Its more flexible, dynamic nature is attributed to smaller local lipid adaptations in POPC, which, in addition to its larger Lbilayer, is sufficient to maximize the hydrophobic match even for WALP27 (Fig. 7 B). Hence, the Wint contribution to the total PMF provides less additional driving force for WALP27 tilting than in DMPC.
As shown in Fig. S5, most Trp anchoring residues are positioned around the hydrophobic/hydrophilic interface of the POPC bilayers. Only one Trp residue of WALP16 is below the interface because of its relatively short LTM in POPC with larger Lbilayer. Despite the existence of four Trp anchoring residues at the interface, the WALPn peptides do not show any helical deformation (probably due to the flexible nature of POPC acyl chains; see WALP19 in Fig. S1, A and B, for comparison). ZTrp in Fig. S5 shows only marginal decrease as τ increases, suggesting that the interfacial matching of anchoring residues is dominant throughout TM helix tilting in POPC and thus each helix has broader thermally accessible tilt regions than in DMPC. As shown in Fig. S6, the interfacial matching is closely related to the tilting direction (i.e., rotation angle) having outward-facing of the Trp side chains. Note that the tilting direction is similar for each helix in POPC, but different from that of the WALPn/DMPC systems. Such a difference may arise from the different Lbilayer in DMPC and POPC bilayers.
Influence of anchoring residues
A flanking residue of a single-pass TM helix can also influence helix tilt and rotation as well as lipid adaptations (12,40). To investigate such influences, we used three different anchoring residues (Ala, Lys, and Arg) in addition to Trp in the WALP23/DMPC system for the PMF calculations. Fig. 3 C shows the total PMFs of the XALP23/DMPC (X = A, W, K, and R) systems as a function of τ. Interestingly, even though the XALP23 series have the same LTM except AALP23 (Table 1), their minimum-PMF tilt angles (Table 2) show a direct relationship to the hydrophobicity (41) of its anchoring residues: τmin (RALP23) < τmin (KALP23) < τmin (WALP23) < τmin (AALP23). These results appear to be at odds with the tilt angles estimated from solid-state 2H-NMR experiments (12), showing τ = 4.4° (KALP23) and τ = 5.2° (WALP23). However, it is now apparent that the correct determination of tilt angles using 2H-NMR quadrupolar splitting measurements requires a proper average of rotation angles (16,17,20). For example, as mentioned above, the recent fluorescence spectroscopy experiment (20) shows much larger tilt angle of WALP23 (τ = 24° ± 5°) in a DOPC membrane than the tilt angle (τ = 4.4°∼8.2°) estimated from solid-state 2H-NMR measurements (12,13).
The free energy decomposition (Fig. S7) shows that as the hydrophobicity of anchoring residue increases, there is more Wint contribution to TM helix tilting up to τmin in addition to the intrinsic Wentropy contribution, and the thermally accessible tilt region becomes wider. Such difference is closely related to the local lipid adaptations. As shown in Fig. 5 C, ΔLadaptation disappears around each τmin, and the extent of ΔLadaptation before τmin is inversely proportional to the hydrophobicity of each anchoring residue. In other words, the Wint contribution to TM helix tilting up to τmin arises from the helix-lipid interaction to relieve the hydrophobic mismatch. To avoid exposure of the charged side chains to the hydrophobic membrane interior and to allow them to interact favorably with the aqueous environment (41), as shown in Fig. S8, Lys and Arg anchoring residues prefer to be positioned at 3∼5 Å above the hydrophilic/hydrophobic interface where Trp residues are generally positioned. These snorkeling behaviors (14,42) also affect the local lipid adjustment as shown in Figs. 5 C and 7 C. Consequently, the long charged side chains make a more-dominant interaction with water (Fig. S9), so that they have less τmin.
As shown in Fig. S10, the XALP23/DMPC systems show quite different rotations and thus tilt directions. The large fluctuation in the AALP23 system appears to arise from the flexible dynamics of AALP23 due to a lack of bulky anchoring residue's interaction with membranes. In addition, the large change of ρ and its fluctuation in RALP23 is attributed to the ill-definition of ρ due to slight helix bending at τ > 20° (Fig. S1 C). The tilting direction of KALP23 (−114.3° ± 55.8°) at τ = τmin is well correlated with the rotation angle (ρ = −109°) estimated from the solid-state 2H-NMR measurement (12). These results demonstrate that the interaction of Lys and Arg anchoring residues at the membrane/water interface plays an important role in determining TM helix orientations.
Conclusions
To systematically characterize responses of a single-pass TM helix in terms of its orientation (tilt and rotation) and lipid adaptations under various hydrophobic mismatch conditions, we have performed extensive umbrella sampling MD simulations and calculated the PMFs as a function of TM helix tilt angle (τ) in the WALPn/DMPC (n = 16, 19, 23, and 27), WALPn/POPC, and XALP23/DMPC (X = A, W, K, and R) systems (Table 1 and Table S1). The PMF in each system, its decomposition, and trajectory analysis allow us to generalize such responses and the underlying molecular forces.
-
1.
Tilting of a single-pass TM helix is the major response to a hydrophobic mismatch. Regardless of the negative and positive mismatches, the PMFs in all the systems (Fig. 3 and Table 2) clearly demonstrate that each TM helix prefers to stay around its minimum-PMF tilt angle (τmin) without conformational deformations such as kinks or bending (Fig. S1).
-
2.
Tilting of a single-pass TM helix up to ∼10° is inherent. As shown in Fig. 4, Fig. S4, and Fig. S7, there is the intrinsic entropic contribution (Wentropy) to TM helix tilting in a membrane bilayer, which arises from helix precession around the membrane normal. In other words, the accessible orientational space of the helix is reduced as τ decreases, which causes the entropy cost associated with small tilt angles. In particular, its cost up to ∼10° is high enough to make the TM helix tilt even under negative mismatch conditions, such as WALP16/19 in DMPC bilayers and WALP16/19/23 in POPC bilayers.
-
3.
The favorable helix-lipid interaction provides additional driving forces for TM helix tilting under a positive mismatch. The helix-lipid interactions (Wint) are TM sequence- and length-specific, and indeed vary for different mismatch conditions (Fig. 4, Fig. S4, and Fig. S7). Generally, there is no favorable Wint contribution under negative mismatch conditions because of increased stress on the (already) perturbed lipid bilayer as τ increases (Figs. 5 and 7). However, under positive mismatch conditions (WALP23/27 in DMPC and WALP27 in POPC), molecular interaction forces, in order to decrease such membrane deformation stress at τ < τmin, provide additional driving forces to TM helix tilting to τmin.
-
4.
τmin is generally located where there is the hydrophobic match and little or no lipid perturbation under a positive mismatch, as shown in Figs. 5 and 7. It should be stressed that there are the thermally accessible tilt regions of ∼10°–20° in single-pass TM helices (Fig. 3 A and Table 2) where a certain extent of a hydrophobic mismatch exists. This observation strongly suggests that the membrane system can have some flexibility to tolerate such a mismatch within a certain threshold by slight lipid adaptations.
-
5.
TM helix rotation (ρ) is dependent on the sequence- and length-specific helix-lipid interaction. In other words, ρ is determined by both the anchoring residue type and the mismatch conditions (Fig. 8, Fig. S6, and Fig. S10). As shown in Fig. S3, there is a significant dependence of the tilting energetics on ρ. In general, a single-pass TM helix with bulky anchoring residues can explore various ρ at small τ up to ∼10°, but there are significant energy barriers between different ρ regions after τ ≈ 10°. The precise estimation of thermally accessible rotation angles as a function of τ further requires the two-dimensional PMF calculations as a function of both τ and ρ.
-
6.
Anchoring residues at the hydrophilic/hydrophobic interface can be an important determinant of TM helix orientation. As shown in Fig. S2, Fig. S5, and Fig. S8, it is apparent that anchoring residues, such as Arg, Lys, and Trp, prefer to position at the hydrophilic (lipid headgroup and water)/hydrophobic interface at ∼τmin, regardless of the negative and positive mismatches. In general, there are no other favorable interactions available in the systems under negative mismatch conditions, and thus such interfacial matching appears to be more important. Interestingly, τmin in the XALP23/DMPC systems (Table 2) has a direct relationship to the hydrophobicity of its anchoring residues: τmin (RALP23) < τmin (KALP23) < τmin (WALP23) < τmin (AALP23) (i.e., the more hydrophobic the anchoring residue, the more the TM helix prefers to tilt).
This generalization, based on the extensive PMF calculations and trajectory analysis, provides in-depth insights into the responses of the single-pass TM helix and lipid bilayers to various hydrophobic mismatches. These findings are particularly important because of the abundance of membrane proteins with a single-pass TM helix and their association and conformational changes involved in TM-induced signaling (6,7). Yet, we need further investigation on the influence of the TM helix-helix interaction on the structure and function of these biologically important systems.
Supporting Material
One table and 10 figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00478-9.
Supporting Material
Acknowledgments
We are grateful to Jinhyuk Lee for initial efforts on this work and to Sunhwan Jo and Huan Rui for helpful comments on the manuscript.
This work was supported by National Science Foundation grant No. MCB-0918374. This research was also supported in part by the National Science Foundation (No. OCI-0503992) through TeraGrid resources provided by Purdue University.
References
- 1.Andersen O.S., Koeppe R.E. Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- 2.Killian J.A. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1998;1376:401–415. doi: 10.1016/s0304-4157(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 3.Pelham H.R.B., Munro S. Sorting of membrane proteins in the secretory pathway. Cell. 1993;75:603–605. doi: 10.1016/0092-8674(93)90479-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johannsson A., Smith G.A., Metcalfe J.C. The effect of bilayer thickness on the activity of (Na+ + K+)-ATPase. Biochim. Biophys. Acta Biomembr. 1981;641:416–421. doi: 10.1016/0005-2736(81)90498-3. [DOI] [PubMed] [Google Scholar]
- 5.Froud R.J., Earl C.R.A., Lee A.G. Effects of lipid fatty acyl chain structure on the activity of the (Ca2+ + Mg2+)-ATPase. Biochim. Biophys. Acta Biomembr. 1986;860:354–360. doi: 10.1016/0005-2736(86)90532-8. [DOI] [PubMed] [Google Scholar]
- 6.Arkin I.T., Brünger A.T., Engelman D.M. Are there dominant membrane protein families with a given number of helices? Proteins: Struct. Funct. Gen. 1997;28:465–466. doi: 10.1002/(sici)1097-0134(199708)28:4<465::aid-prot1>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 7.Lichanska A.M., Waters M.J. New insights into growth hormone receptor function and clinical implications. Hormone Res Paed. 2008;69:138–145. doi: 10.1159/000112586. [DOI] [PubMed] [Google Scholar]
- 8.van der Wel P.C.A., Strandberg E., Koeppe R.E. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys. J. 2002;83:1479–1488. doi: 10.1016/S0006-3495(02)73918-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park S.H., Opella S.J. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J. Mol. Biol. 2005;350:310–318. doi: 10.1016/j.jmb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 10.de Planque M.R.R., Killian J.A. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 2003;20:271–284. doi: 10.1080/09687680310001605352. (Review) [DOI] [PubMed] [Google Scholar]
- 11.Killian J.A., Nyholm T.K. Peptides in lipid bilayers: the power of simple models. Curr. Opin. Struct. Biol. 2006;16:473–479. doi: 10.1016/j.sbi.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Ozdirekcan S., Rijkers D.T.S., Killian J.A. Influence of flanking residues on tilt and rotation angles of transmembrane peptides in lipid bilayers. A solid-state 2H NMR study. Biochemistry. 2004;44:1004–1012. doi: 10.1021/bi0481242. [DOI] [PubMed] [Google Scholar]
- 13.Strandberg E., Ozdirekcan S., Killian J.A. Tilt angles of transmembrane model peptides in oriented and non-oriented lipid bilayers as determined by 2H solid-state NMR. Biophys. J. 2004;86:3709–3721. doi: 10.1529/biophysj.103.035402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kandasamy S.K., Larson R.G. Molecular dynamics simulations of model trans-membrane peptides in lipid bilayers: a systematic investigation of hydrophobic mismatch. Biophys. J. 2006;90:2326–2343. doi: 10.1529/biophysj.105.073395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrache H.I., Zuckerman D.M., Woolf T.B. Hydrophobic matching mechanism investigated by molecular dynamics simulations. Langmuir. 2002;18:1340–1351. [Google Scholar]
- 16.Ozdirekcan S., Etchebest C., Fuchs P.F. On the orientation of a designed transmembrane peptide: toward the right tilt angle? J. Am. Chem. Soc. 2007;129:15174–15181. doi: 10.1021/ja073784q. [DOI] [PubMed] [Google Scholar]
- 17.Esteban-Martín S., Salgado J. The dynamic orientation of membrane-bound peptides: bridging simulations and experiments. Biophys. J. 2007;93:4278–4288. doi: 10.1529/biophysj.107.113043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J., Im W. Transmembrane helix tilting: insights from calculating the potential of mean force. Phys. Rev. Lett. 2008;100:018103. doi: 10.1103/PhysRevLett.100.018103. [DOI] [PubMed] [Google Scholar]
- 19.Im W., Brooks C.L. Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2005;102:6771–6776. doi: 10.1073/pnas.0408135102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holt A., Koehorst R.B.M., Killian J.A. Tilt and rotation angles of a transmembrane model peptide as studied by fluorescence spectroscopy. Biophys. J. 2009;97:2258–2266. doi: 10.1016/j.bpj.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torrie G.M., Valleau J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comp. Phys. 1977;23:187–199. [Google Scholar]
- 22.Lee J., Im W. Restraint potential and free energy decomposition formalism for helical tilting. Chem. Phys. Lett. 2007;441:132–135. [Google Scholar]
- 23.Lee J., Im W. Implementation and application of helix-helix distance and crossing angle restraint potentials. J. Comput. Chem. 2007;28:669–680. doi: 10.1002/jcc.20614. [DOI] [PubMed] [Google Scholar]
- 24.Im W., Lee J., Rui H. Novel free energy calculations to explore mechanisms and energetics of membrane protein structure and function. J. Comput. Chem. 2009;30:1622–1633. doi: 10.1002/jcc.21320. [DOI] [PubMed] [Google Scholar]
- 25.Jo S., Lim J.B., Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jo S., Kim T., Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo S., Kim T., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 28.Brooks B.R., Brooks C.L., Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 30.Mackerell A.D., Feig M., Brooks C.L. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 31.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 32.Klauda J.B., Brooks B.R., Pastor R.W. An ab initio study on the torsional surface of alkanes and its effect on molecular simulations of alkanes and a DPPC bilayer. J. Phys. Chem. B. 2005;109:5300–5311. doi: 10.1021/jp0468096. [DOI] [PubMed] [Google Scholar]
- 33.Nagle J.F., Tristram-Nagle S. Structure of lipid bilayers. Biochim. Biophys. Acta Gene Struct. Expression. 2000;1469:159–195. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys. 1977;23:327–341. [Google Scholar]
- 35.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A: At. Mol. Opt. Phys. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 36.Feller S.E., Zhang Y., Brooks B.R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 37.Weiss T.M., van der Wel P.C.A., Huang H.W. Hydrophobic mismatch between helices and lipid bilayers. Biophys. J. 2003;84:379–385. doi: 10.1016/S0006-3495(03)74858-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nezil F.A., Bloom M. Combined influence of cholesterol and synthetic amphiphilic peptides upon bilayer thickness in model membranes. Biophys. J. 1992;61:1176–1183. doi: 10.1016/S0006-3495(92)81926-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Róg T., Murzyn K., Pasenkiewicz-Gierula M. Effects of phospholipid unsaturation on the bilayer nonpolar region: a molecular simulation study. J. Lipid Res. 2004;45:326–336. doi: 10.1194/jlr.M300187-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.de Planque M.R.R., Boots J.-W.P., Killian J.A. The effects of hydrophobic mismatch between phosphatidylcholine bilayers and transmembrane α-helical peptides depend on the nature of interfacially exposed aromatic and charged residues. Biochemistry. 2002;41:8396–8404. doi: 10.1021/bi0257686. [DOI] [PubMed] [Google Scholar]
- 41.Wimley W.C., White S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 42.Strandberg E., Killian J.A. Snorkeling of lysine side chains in transmembrane helices: how easy can it get? FEBS Lett. 2003;544:69–73. doi: 10.1016/s0014-5793(03)00475-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.