

Abstract
Classical theory states that ligand binding induces the dimerization of ErbB proteins, leading to their activation. Although we and other investigators have shown the existence of preformed homoclusters of ErbB receptors and analyzed their composition, the stoichiometry of their heteroclusters has not been quantitatively described. Here, we report the development of the fluorescence resonance energy transfer (FRET)-sensitized acceptor bleaching (FSAB) technique to quantitate the ratio of ErbB1 and ErbB2 in their heteroclusters. In FSAB, photolabile acceptors within FRET distance from photostable donors are excited and photobleached by FRET, and the fraction of acceptors that are participating in FRET is determined. In quiescent SKBR-3 breast cancer cells, ∼35% of ErbB1 and ∼10% of ErbB2 have been found in heteroclusters. Epidermal growth factor (ligand of ErbB1) increased the fraction of ErbB2 heteroclustering with ErbB1, whereas the ratio of heteroclustered ErbB1 did not change significantly. The fractions of heteroclustered ErbB1 and ErbB2 were independent of their expression levels, indicating that the formation of these clusters is not driven by the law of mass action. In contrast, the FRET efficiency depended on the donor/acceptor ratio as expected. We present a model in which preformed receptor clusters are rearranged upon ligand stimulation, and report that the composition of these clusters can be quantitatively described by the FSAB technique.
Introduction
The fact that the association of cell surface receptors and their activation are linked was established a long time ago, and it has become a paradigm of receptor biology (1). ErbB proteins are the best characterized of the receptor tyrosine kinases (RTK), but most published results describe their associations in a qualitative way. Four ErbB proteins (ErbB1–4) have been characterized and shown to form an extensive network of homo- and heteroassociations (2). Except for ErbB2, the extracellular domains of ErbB proteins are thought to be in a tethered, inactive conformation that is rearranged upon ligand binding, leading to exposure of the dimerization arm. These events culminate in the formation of ligand-induced homo- and heterodimers and receptor activation (1,3). Dimerization of the intracellular kinase domain is directly involved in its activation, leading to the phosphorylation of several tyrosine residues in the C-terminus and recruitment of SH2 domain-containing proteins (2–4). ErbB2 is at the heart of the association pattern because its extracellular domain constantly assumes a conformation that is capable of forming heterodimers (5). The signaling potency of such heterodimers is significantly enhanced compared to that of homodimers (6). Although information derived from crystal structures of the ErbB2 extracellular domain does not support the formation of ErbB2 homodimers, the existence of overexpression-driven, ligand independent ErbB2 homoassociations is supported by strong experimental evidence (7).
Molecular clusters can be characterized by a multitude of techniques, including crystallography, several molecular biological approaches, the proximity ligation assay, and the VeraTag assay (8,9). Because of its quantitative nature and relative ease of implementation, fluorescence resonance energy transfer (FRET) has become the technique of choice in quantitative studies of receptor clustering (10–12). In hetero-FRET, an excited donor passes its energy to a spectroscopically different acceptor. The fact that the energy transfer process in hetero-FRET is unidirectional limits its sensitivity for distinguishing between clusters with more than two subunits. Although dimers are unquestionably the best-characterized type of receptor clusters, they are obviously not the only kind. Tetramers and even larger clusters of ErbB1 have been detected by fluorescence correlation spectroscopy (13,14). Very large clusters of ErbB2 and ErbB3 involving tens or hundreds of proteins on a scale of tens or hundreds of nanometers have been detected by electron (15) and near-field optical microscopy (16). Homo-FRET involving the interaction between spectroscopically identical fluorophores can also be used to detect the formation of large-scale homoclusters (17). Using flow cytometric homo-FRET measurements, we have shown that the number of monomers in an ErbB1 homocluster increases from ∼4 to ∼10 upon epidermal growth factor (EGF) stimulation. ErbB2 was found to behave in the opposite way, in that it formed extensive, large-scale clusters (∼50–100 proteins/cluster) in the absence of stimulation, which decreased in size after treatment with EGF or neuregulin (18). Although the stoichiometry of large-scale heteroclusters could not be measured, we hypothesized that ligand-bound ErbB1 and ErbB3 recruits ErbB2 to heterodimers, leading to the observed decrease in the cluster size of ErbB2. By using number and brightness (N&B) measurements and two-color fluorescence correlation, investigators may be able to determine the stoichiometry of heteroclusters by measuring the comobility of different receptors (19–21).
In this study, we developed and used a technique originally proposed by Mekler et al. (22,23) to characterize the composition of heteroclusters of ErbB1 and ErbB2 in a quantitative way. Briefly, FRET from a photostable donor to a photolabile acceptor induces acceptor photobleaching. Optimally, only acceptors within FRET distance from donors will undergo photobleaching. Using this approach, which we call FRET-sensitized acceptor bleaching (FSAB), the fraction of acceptors within FRET distance from donors can be quantitatively determined. We demonstrate that FSAB can principally be used to determine the fraction of acceptors within FRET distance of donors by showing that the efficiency of FRET drops to zero long before all acceptor molecules are bleached, using AlexaFluor546-Cy5 as a donor-acceptor pair. In quiescent SKBR-3 breast tumor cells, only ∼10% of ErbB2 heteroassociates with ErbB1, and this fraction is doubled by EGF stimulation. Although a higher fraction of ErbB1 forms heteroclusters with ErbB2, this fraction is not significantly changed by EGF treatment. The FSAB technique is suitable for measuring the heteroclustering of proteins in a quantitative way, and has the potential to provide new insight into the behavior of receptor tyrosine kinases upon activation.
Materials and Methods
Cells and reagents
SKBR-3 breast cancer and A431 epithelial carcinoma cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and grown according to the their specifications. For microscopic experiments, cells were grown in two- or eight-well chambered coverglasses (Nalge Nunc International, Rochester, NY). ErbB1 and ErbB2 were labeled by Mab528 and trastuzumab, respectively. Mab528 was purified from the supernatant of the HB-8509 hybridoma cell line (ATCC) by protein A affinity chromatography. Trastuzumab (Herceptin) was purchased from Roche Ltd. (Budapest, Hungary). Conjugation of antibodies with AlexaFluor (Molecular Probes, Eugene, OR), Cy5 (GE HealthCare, Freiburg, Germany), or Qdot605 (Invitrogen, Karlsruhe, Germany) was carried out according to the manufacturers' specifications. The number of fluorophores on a single antibody was determined by spectrophotometry and was always kept between 1–2 to reduce the possibility of interactions between neighboring Cy5 molecules (24). EGF and CBr4 were purchased from R&D Systems (Minneapolis, MN) and Sigma-Aldrich (Schnelldorf, Germany), respectively.
Stimulation and labeling of cells
SKBR-3 cells grown in chambered coverglass were starved in medium containing 0.1% fetal calf serum for 24 h before experiments were conducted, and stimulated by 100 nM EGF in Hank's buffer supplemented with 1 mg/mL bovine serum albumin for 15 min at 37°C. Control and stimulated cells were labeled by a saturating concentration (10–20 μg/mL) of fluorescent Mab528 and/or trastuzumab in Hank's buffer containing 1 mg/mL bovine serum albumin for 30 min on ice. Unbound antibodies were removed by washing twice in phosphate-buffered saline, and cells were fixed in 1% formaldehyde.
Confocal microscopy and photobleaching
A Zeiss LSM 510 confocal laser scanning microscope (Carl Zeiss AG, Jena, Germany) was used to image the samples. AlexaFluor546 was excited at 543 nm and its emission was detected at 560–615 nm. The fluorescence of Cy5 was excited at 633 nm and detected above 650 nm. When FRET between Alexa546 and Cy5 was measured, a third fluorescence image (FRET channel), excited at 543 nm and recorded above 650 nm, was also measured in addition to the donor (AlexaFluor546) and acceptor (Cy5) channels. Qdot605 was excited at 488 nm and its emission was recorded at 585–615 nm. Fluorescence images were taken as single optical sections using a 63x (NA = 1.4) oil immersion objective focused to the middle of the cell along the Z axis. The pinhole size was adjusted to 2 Airy units. The image size was 512×512 pixels, and the pixel size in the X and Y directions was 400 nm. To induce FRET-sensitized bleaching of Cy5, the sample was illuminated at 543 nm (bleaching beam). Bleaching illumination was interrupted approximately every 30 s, and donor, FRET, and acceptor images were recorded with an attenuated laser beam. The power of the 543 nm laser line was set to 5% and 100% for the imaging and bleaching illuminations, respectively. Photobleaching was carried out in the presence of 8×10−4 M CBr4.
Image analysis
Image processing was carried out with the DipImage toolbox (Delft University of Technology, Delft, The Netherlands) under MATLAB (The MathWorks Inc., Natick, MA). Segmentation of images into membrane and nonmembrane pixels was carried out with the manually seeded watershed algorithm using a custom-written MATLAB program (25,26). Image stacks acquired during photobleaching were corrected for shift using the correctshift command of DipImage. FRET efficiency, unquenched donor and direct acceptor intensities corrected for spectral overspill were calculated in the photobleaching stack in the membrane of selected cells double-labeled by donor- and acceptor-tagged antibody as described previously (27), and the fraction of bleached acceptors (Fbleached in Eq. 5) was determined at the time when the FRET efficiency dropped to zero. The fraction of directly bleached acceptors excited at the excitation wavelength of the donor (BCF in Eq. 5) was determined using a sample labeled with acceptor only.
To calibrate the donor and acceptor fluorescence intensities in terms of the number of molecules, two samples of SKBR-3 cells were separately labeled by either the donor- or acceptor-tagged antibody against the same epitope. The means of the background-corrected fluorescence intensities in the membrane of ∼100 cells were determined separately in the donor-only and acceptor-only labeled samples (〈Id〉 and 〈Ia〉, respectively), and corrected for labeling ratio, i.e., the number of fluorophores per antibody. Since the cells in the two samples contained on average the same number of antibodies per cell, the ratio of the mean fluorescence intensities is the ratio of the fluorescence intensities generated by the same number of fluorophores in the donor and acceptor channels:
| (1) |
where Ld and La are the number of fluorophores on the donor- and acceptor-labeled antibodies, respectively. The fluorescence intensity in the donor image of double-labeled cells was multiplied by Ra/d to ensure that the donor and acceptor fluorescence intensities would be on the same scale. To calibrate these fluorescence intensities in terms of receptor numbers, the number of binding sites of the respective antibody was determined by flow cytometry using Qifikit (DAKO, Hamburg, Germany). Briefly, a series of beads with calibrated numbers of bound primary mouse monoclonal antibodies were labeled with a fluorescent anti-mouse IgG. After correcting for the fluorescence intensity of blank beads, a calibration curve was constructed from which the slope and y-intercept were determined. The cells were labeled by primary mouse monoclonal antibodies against the antigen of interest, followed by secondary labeling by the fluorescent secondary antibody used to label the beads, and flow cytometric determination of their fluorescence intensity. After background correction was performed, the number of antigens on the investigated cell line was determined by linear regression.
The number of molecules was then calculated for the microscopic images according to the following equation:
| (2) |
where Ni and NQifi are the number of molecules in a given cell or pixel and the mean number of molecules in the population of cells determined by Qifikit, respectively, and Ii and 〈I〉 are the fluorescence intensity in a given cell or pixel and the mean fluorescence intensity of the population of cells corrected by Ra/d, respectively.
Theory
Determination of the fraction of bound acceptors
Given a photostable FRET donor and an acceptor that is sensitive to a photochemical reaction, one can determine the FRET efficiency according to the enhanced rate of the photochemical reaction of the acceptor excited at the wavelength of the donor (22). Mekler et al. (23) claimed that even small FRET efficiencies can be accurately determined using this approach, which explains why the technique is called photochemical enhancement of sensitivity (PES). If the photochemical reaction of the acceptor is irreversible photobleaching, then FRET results in the accelerated rate of acceptor photobleaching. To emphasize that our approach is based on the enhanced rate of acceptor photobleaching as a result of FRET, we coined the term “FRET-sensitized acceptor bleaching” (FSAB). Since acceptors that take part in FRET are preferentially bleached, it is possible to calculate the relative contribution of the bleached acceptor subpopulation to the total FRET efficiency by analyzing the relationship between the decays of FRET efficiency and sensitized acceptor emission (23). We were interested in the theoretical endpoint of FSAB, i.e., the point at which all acceptors within FRET distance from donors are bleached. We assumed that two classes of acceptors exist with regard to their association with donors: 1) acceptors bound to donors, i.e., within FRET distance from donors (Abound); and 2) free acceptors (Afree). When all of the bound acceptors are bleached, the FRET efficiency has to drop to zero. By comparing the acceptor intensity (proportional to the number of acceptors) at the time point when FRET drops to zero with the total acceptor intensity before the FSAB process is undertaken, one can determine the fraction of bound acceptors (Abound/A0). Because acceptors are also excited directly at the wavelength of the donor, some free acceptor molecules outside the range of FRET are also bleached. The bleached fraction of Afree (at the time of complete bleaching of Abound, i.e., when FRET decreases to zero) is designated the bleaching correction factor (BCF). Therefore, the fraction of bleached acceptors (Fbleached) and Abound can be calculated according to the following equations:
| (3) |
| (4) |
Solving for Abound yields
| (5) |
Since BCF was found to vary between different antibodies, it had to be determined for every individual labeled batch of any acceptor-conjugated antibody used in the FSAB experiments. To reduce the effects of the possible intensity dependence of Cy5 photobleaching arising from the interactions between neighboring Cy5 molecules (24), BCF was determined on cells with fluorescence intensities similar to those of double-labeled cells used for the FSAB experiments. It is assumed in Eqs. 3–5 that 100% of the bound acceptor population is bleached. However, as discussed later, the bleaching of Cy5, the acceptor chosen for FSAB measurements, may not always be complete. If the bleached fraction of the bound acceptor population is designated by BCFFRET, the fraction of bound acceptors can be calculated according to the following equation:
| (6) |
BCFFRET can be determined with a sample labeled with Cy5-conjugated primary antibody followed by labeling with an AlexaFluor546-tagged secondary antibody.
Results
Selection of an appropriate donor-acceptor pair for FSAB experiments
The prerequisite for successful application of the FSAB technique is a donor-acceptor FRET pair in which the donor is photostable and the acceptor is photolabile. Although Cy5 is known to be relatively photolabile in confocal microscopy, the time required for its complete bleaching is prohibitively long. In accordance with the results of Mekler et al. (22,23), we found that CBr4 significantly accelerated the rate of photobleaching of Cy5 (see Fig. S1 in the Supporting Material). A donor that would be photostable in the presence of CBr4 had to be found. Quantum dots were obvious candidates because of their known photostability, but CBr4 significantly enhanced the rate of their photobleaching (Fig. S1). AlexaFluor546, on the other hand, was photostable in both the absence and presence of CBr4, and therefore we used it as a donor for Cy5 in the FSAB experiments.
We measured the kinetics of Cy5 photobleaching at 633 nm (the excitation wavelength for Cy5) and at 543 nm (the excitation wavelength for AlexaFluor546). Photobleaching of Cy5 at 543 nm was incomplete, i.e., a residual fluorescence intensity amounting to 20–40% of the initial intensity was resistant to bleaching when the intensity was measured at 543 or 633 nm (Fig. S2 A). On the other hand, the photobleaching of Cy5 at 633 nm was complete independent of whether the images were recorded at an excitation wavelength of 543 or 633 nm (Fig. S2 A).
Proof of concept: the FSAB technique reliably detects receptor clusters
We first established theoretically the reliability of the FSAB approach using AlexaFluor546 and Cy5 dyes. We show in the Supporting Material that although the acceptor is photobleached both directly and via the donor, the fraction of bound acceptors can be reliably determined. In addition, donor bleaching does not have a significant effect on the value for the fraction of bound acceptors calculated by FSAB (Fig. S3). Next, we tested the FSAB approach using a positive and a negative control. It was previously shown that the majority of ErbB1 is monomeric, with a minority forming small aggregates in nonstimulated cells. On the other hand, the majority of ErbB2 forms large clusters in quiescent cells (18). We compared the photobleaching kinetics of Cy5 in SKBR-3 cells labeled with a 1:1 mixture of Cy5-conjugated and unlabeled antibodies against ErbB2 or with a 1:1 mixture of AlexaFluor546-tagged and Cy5-tagged anti-ErbB2 antibodies. Unlabeled antibodies were used to reduce the binding of the Cy5-conjugated antibody to levels comparable to those found in donor-acceptor double-labeled samples, to prevent artifacts arising from the possible influence of interactions between Cy5 molecules on the photobleaching kinetics (24). In accordance with the results shown in the previous section, Cy5 was incompletely bleached in cells labeled only with Cy5-tagged antibodies (Fig. 1 A). However, its bleaching was practically complete by the second bleaching step, when the FRET donor was present, and the FRET efficiency dropped to zero at this time point (data not shown). For quantitative evaluation of the fraction of homoclustered ErbB2, we used the fluorescence intensities measured after two bleaching steps because the FRET efficiency dropped to zero at this time point. Substitution into Eq. 5 yielded 83% for the fraction of homoclustered ErbB2 (Fig. 2 A). Since the expression level of ErbB1 is much higher in A431 cells than in SKBR-3 cells, and previous experiments by our group (18) and others (14,28) to measure the homoassociation of ErbB1 were carried out in A431 cells, we used this cell line to compare the results of our previous homo-FRET experiments with those obtained using the FSAB approach. The photobleaching kinetics of Cy5 in cells labeled with a 1:1 mixture of Cy5-conjugated and unlabeled monoclonal antibodies against ErbB1 was comparable to that measured in cells labeled with a 1:1 mixture of AlexaFluor546-tagged and Cy5-tagged anti-ErbB1 antibodies (Fig. 1 A). This observation implies that the majority of ErbB1 is nonclustered. A quantitative comparison of the two curves reveals that ∼13% of ErbB1 forms homoclusters (Fig. 2 A). The measured fraction of homoclustered ErbB1 in EGF-stimulated A431 cells was ∼3-fold higher than in starved cells (Fig. 2 A). However, the fraction of heteroclustered ErbB2 decreased upon EGF stimulation in SKBR-3 cells (Fig. 2 A). The fractions of homoclustered ErbB1 and ErbB2 in starved and EGF-stimulated cells determined by the FSAB technique are in agreement with previous results obtained by homo-FRET (18).
Figure 1.
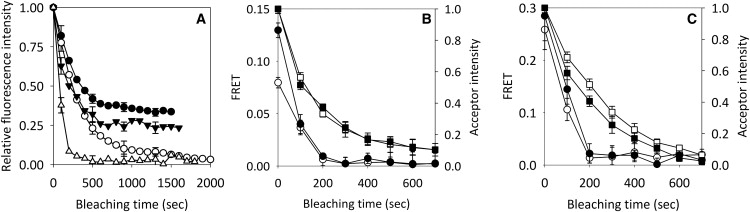
Decrease of FRET efficiency and acceptor fluorescence during FSAB. (A) Determination of the fraction of homoclustered ErbB1 and ErbB2 using FSAB. A431 cells were labeled with Cy5-Mab528 (●) or with a mixture of Cy5-Mab528 and AlexaFluor546-Mab528 (○). SKBR-3 cells were labeled with Cy5-trastuzumab (▾) or with a mixture of Cy5-trastuzumab and AlexaFluor546-trastuzumab (▵), and the samples were photobleached in the presence of CBr4 at 543 nm. The directly excited acceptor fluorescence intensities are shown in the graph. Error bars indicating the mean ± standard error (SE) are shown for every third data point for clarity. (B) Determination of the fraction of heteroclustered ErbB1 in the absence and presence of EGF stimulation. Quiescent and EGF-stimulated SKBR-3 cells were labeled with a mixture of AlexaFluor546-trastuzumab and Cy5-Mab528 against ErbB2 and ErbB1, respectively. Nonsensitized, directly excited acceptor fluorescence and the FRET efficiency were calculated by recording fluorescence images in the donor, FRET, and acceptor channels when the bleaching illumination was interrupted at the excitation wavelength of the donor (□, directly excited acceptor intensity of nonstimulated cells; ○, FRET efficiency of nonstimulated cells; ▪, directly excited acceptor intensity of EGF-stimulated cells; ●, FRET efficiency of EGF-stimulated cells). Photobleaching was carried out in the presence of CBr4. Error bars indicate the mean ± SE of ∼100 cells. (C) Determination of the fraction of heteroclustered ErbB2 in the absence and presence of EGF stimulation. SKBR-3 cells were labeled with a mixture of AlexaFluor546-Mab528 and Cy5-trastuzumab. Otherwise, the experimental conditions and symbol assignments are the same as in B.
Figure 2.
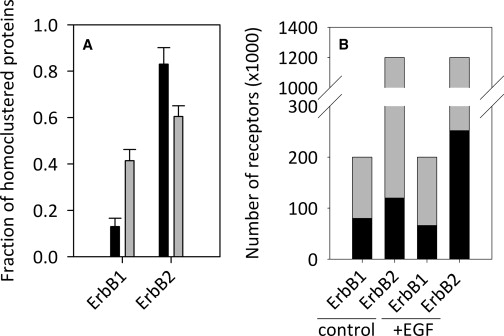
The fraction of homo- and heteroclustered ErbB1 and ErbB2 in quiescent and EGF-stimulated SKBR-3 cells determined by FSAB. (A) Determination of the fraction of homoclustered ErbB1 and ErbB2 in quiescent and stimulated SKBR-3 cells. Starved (black columns) and EGF-stimulated (gray columns) A431 cells were labeled with a mixture of Cy5-tagged and AlexaFluor546-tagged Mab528 (to measure the homoclustering of ErbB1). Starved (black columns) and EGF-stimulated (gray columns) SKBR-3 cells were labeled with Cy5-conjugated and AlexaFluor546-conjugated trastuzumab (to measure the homoclustering of ErbB2), and the fraction of homoclustered acceptor was determined using FSAB. Error bars indicate the mean ± SE of ∼100 cells. (B) Determination of the fraction of heteroclustered ErbB1 and ErbB2 in starved and EGF-stimulated SKBR-3 cells. The fraction of ErbB1 molecules forming heteroclusters with ErbB2 was calculated by labeling SKBR-3 cells with a mixture of AlexaFluor546-trastuzumab and Cy5-Mab528 against ErbB2 and ErbB1, respectively. The fraction of ErbB2 heteroassociating with ErbB1 was determined by labeling cells with AlexaFluor546-Mab528 and Cy5-trastuzumab. The fractions of free/homo- and heteroclustered molecules were converted to absolute numbers after determining the expression levels of ErbB1 and ErbB2. The numbers of heteroclustered and free/homoclustered molecules are shown by the black and gray parts of the bars, respectively, in quiescent and EGF-stimulated cells. The FSAB approach only characterizes whether an acceptor is heteroassociated with a donor; therefore, a free acceptor may indeed be a free (i.e., monomeric) molecule or form homoclusters or heteroclusters with other molecules.
Since the duration of bleaching is long in FSAB experiments (∼200 s), we tested whether protein mobility would affect the results of our experiments. As shown in Fig. S4, the lateral mobility of ErbB1 is abolished in formaldehyde-fixed cells. Therefore, we concluded that only proteins associated with each other at the time of fixation are detected by the FSAB approach, i.e., those acceptors that would transiently associate with donors (if they were free to diffuse) are not counted as bound. Since heteroclusters cannot be analyzed by homo-FRET measurements, we set out to characterize the heteroassociation of ErbB1 and ErbB2 using the FSAB approach. Since SKBR-3 cells express both ErbB1 and ErbB2 at moderate to high levels (∼2×105 ErbB1 and ∼106 ErbB2; values determined by Qifikit), and ErbB2 is only slightly expressed by A431 cells (∼2×106 ErbB1 and ∼2×104 ErbB2), we chose to use SKBR-3 cells for these experiments. Cells were labeled with AlexaFluor546-tagged antibody against ErbB2 and Cy5-tagged antibody against ErbB1, and the directly excited fluorescence intensity of the acceptor and the FRET efficiency were calculated after each bleaching step (Fig. 1 B). In accordance with the aforementioned results, the FRET efficiency dropped to zero after the second bleaching step, indicating that all acceptors within FRET distance of the donors were bleached (Figs. 1 B, 2 B, and 3). However, the fluorescence intensity of the acceptor was only reduced to ∼35% of the initial value, indicating that a significant fraction of the acceptor was not within FRET distance of the donors (Figs. 1 B and 2 B). The labeling ratio (number of fluorophores per antibody) is expected to influence the initial value of FRET, but as long as the starting FRET efficiency is not too small, acceptors within FRET distance from the donors will be bleached. The extent of bleaching of donor fluorescence was much smaller than that of the acceptor or the fractional decrease in the FRET efficiency (Fig. 3). A quantitative evaluation according to Eq. 5 revealed that ∼40% of the acceptor (ErbB1) was heteroclustered with the donor (ErbB2). To obtain reproducible results, we only analyzed cells with bright fluorescence in which FRET decreased monotonously with bleaching and could be reliably determined, and in which the fraction of bleachable acceptors in the double-labeled sample was at least 90%. We concluded that the fraction of heteroclustered proteins can be reliably determined by the FSAB technique, and started a systematic investigation of ErbB1 and ErbB2 in quiescent and stimulated cells.
Figure 3.
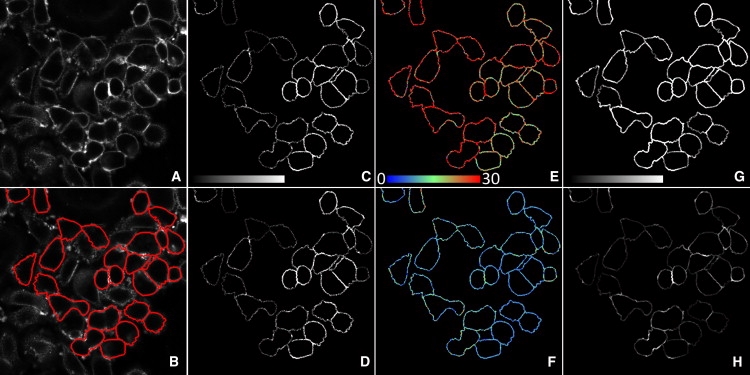
Representative images for calculating the fraction of bound acceptors using the FSAB technique. SKBR-3 cells were labeled with Alexa546-Mab528 and Cy5-trastuzumab, and the fraction of bound acceptors was analyzed by the FSAB approach. (A) The image recorded in the acceptor channel to which the manually seeded watershed segmentation algorithm was applied to segment the image into membrane and nonmembrane pixels. (B) The membrane mask (in red) overlaid on the fluorescence image shown in A. (C and D) Unquenched donor fluorescence intensity calculated for membrane pixels before the beginning of bleaching (C) and after the second bleaching step, corresponding to ∼200 s of photobleaching (D). (E and F) The FRET efficiency calculated before bleaching (E) and after the second bleaching step (F). Pixels are color-coded according to the FRET efficiency, which ranges between 0 and 30%. (G and H) Nonsensitized acceptor emission calculated before bleaching (G) and after the second bleaching step (H). The fluorescence images (C, D, G, and H) were contrast-stretched, and therefore the donor (C and D) and acceptor fluorescence intensities (G and H) are not comparable.
Quantitative analysis of heteroclusters of ErbB1 and ErbB2 in quiescent and EGF-stimulated cells
SKBR-3 breast cancer cells were starved in the presence of 0.1% fetal calf serum for 24 h and labeled with AlexaFluor546-tagged antibody against ErbB1 and Cy5-tagged antibody against ErbB2. The fraction of ErbB2 heteroclustering with ErbB1 in these nonstimulated cells was found to be 10%. After EGF stimulation, ErbB2 was recruited into heteroclusters with ErbB1, which was reflected by an ∼2-fold increase in the fraction of ErbB2 heteroassociating with ErbB1 (Table 1, Figs. 1 C and 2 B). To determine the fraction of ErbB1 that formed heteroclusters with ErbB2, we labeled the former with acceptor-tagged antibodies and the latter with donor-tagged antibodies. The fraction of heteroclustered ErbB1 in quiescent and EGF stimulated cells was 40% and 33%, respectively (Table 1, Figs. 1 B and 2 B). The absolute number of free and bound (i.e., clustered) ErbB1 and ErbB2 was also calculated using the expression levels of the proteins determined by flow cytometry (Table 1 and Table S1). The term “free acceptor” means that the acceptor is not heteroassociated with the donor, but they may form homoclusters or associate with other molecules in the membrane.
Table 1.
The numbers and fractions of free and heteroclustered ErbB1 and ErbB2 in control and EGF-stimulated SKBR-3 cells
| Quiescent (×103) | EGF stimulation (×103) | ||
|---|---|---|---|
| ErbB1 | Free or homoclustered | 120 ± 8 (60 ± 4%) | 134 ± 10 (67 ± 5%) |
| in complex with ErbB2 | 80 ± 8 (40 ± 4%) | 66 ± 10 (33 ± 5%) | |
| ErbB2 | Free or homoclustered | 1080 ± 24 (90 ± 2%) | 948 ± 36 (79 ± 3%) |
| in complex with ErbB1 | 120 ± 24 (10 ± 2%) | 252 ± 36 (21 ± 3%) |
The numbers and fractions of free (or homoclustered) and heteroclustered proteins were determined by FSAB as described in the legend to Fig. 2B. The standard errors of the percentage of heteroclustered molecules determined from ∼100 cells in three independent experiments are also shown. These errors are equal to those of free/homoclustered molecules since the latter was calculated by subtracting the percentage of heteroclustered molecules from 100%.
The fraction of heterocluster-forming ErbB2 was also analyzed on a cell-by-cell basis, and was found to be independent of the expression levels of ErbB1 and ErbB2 (Fig. S5 A). To the contrary, the FRET efficiency was proportional to the acceptor/donor ratio (Fig. S5 B). If free and bound ErbB2 were in equilibrium according to the law of mass action, the fraction of heteroassociating ErbB2 would depend on the expression levels of ErbB1 and ErbB2 (Fig. S5 C). The fact that the fraction of bound ErbB2 was independent of the expression levels of ErbB1 and ErbB2 implies that the formation of these heteroclusters does not follow the law of mass action.
Discussion
In this work, we report the successful implementation of the FSAB technique to measure the heteroclustering of membrane proteins in a quantitative way. The approach is based on preferential, FRET-induced bleaching of photolabile acceptors in the molecular proximity of photostable donors. Although both donor bleaching and direct bleaching of the acceptor take place, they do not compromise the reliability of the approach, and we concluded that AlexaFluor546 and Cy5 are a suitable donor-acceptor pair, since bound acceptors are indeed preferentially bleached (Fig. S3). The photophysics of Cy5 is complicated and involves cis-trans isomerization, reversible photoinduced transition between dark and bright states, triplet and higher excited states, and interaction between neighboring fluorophores (24,29–31). In the absence of oxygen and CBr4, Cy5 emits 100,000–200,000 photons before irreversible photobleaching occurs (30,32). The presence of oxygen, the short lifetime and large molar absorption coefficient, and the consequent rapid cycling between the ground and excited states deteriorates the photostability of Cy5 in confocal microscopy (33). CBr4 substantially decreases the photostability of Cy5 by photoinduced electron transfer (22) without significantly accelerating the rate of AlexaFluor546 bleaching (Fig. S1). Because of their photostability, quantum dots would have been the best choice for the photostable donor, but the rate of their bleaching was significantly increased by CBr4 (Fig. S1). We also considered AlexaFluor555 as a potential donor for Cy5 in the FSAB experiments. Based on its photobleaching quantum yield, AlexaFluor555 is ∼10 times more photostable than AlexaFluor546 (34). In certain cases, AlexaFluor555 may be a suitable dye for FSAB experiments, but its low fluorescence quantum yield and the small R0 for the Alexa-Fluor555-Cy5 pair (R0,A555-Cy5 = 4.9 nm, R0,A546-Cy5 = 6.8 nm) limit its applicability.
Transient, photoinduced dark states of Cy5 can invalidate the interpretation of FRET experiments involving Cy5 as an acceptor (31). However, the bleaching curves of Cy5 reported here represent irreversible photobleaching, since illumination at 543 nm did not recover the fluorescence intensity (data not shown). Contrary to the findings of Eggeling et al. (29), who reported accelerated photobleaching of Cy5 with excitation in the short-wavelength range of the absorption spectrum, we observed partial bleaching of Cy5 with illumination at 543 nm (Fig. S2 A). We do not know the reason for this observation. The ratios of fluorescence intensities excited at 543 and 633 nm were identical before and after photobleaching at 543 nm. In addition, the emission spectra of Cy5 bleached at 543 nm and that of unbleached Cy5 were identical (Fig. S2 B). These findings exclude the possibility that molecules that differed in their ground-state excitation spectrum led to the observation. Either the photobleaching illumination at 543 nm reacts differently compared to the 633 nm light with the homogeneous population of molecules (and does not bleach them completely), or the subpopulation of molecules that display different photosensitivities at 543 and 633 nm have identical excitation spectra. Interaction between Cy5 molecules conjugated to the same IgG is unlikely to be behind the observations, since such interactions were reported to take place only if the number of Cy5 labels per IgG is >2–3 (24). Heterogeneous photobleaching of Cy5 was detected previously and assumed to be caused by heterogeneity of Cy5 generated during protein labeling or by impurities in the dye (30). Although photobleaching of Cy5 carried out at 543 nm was partial, FRET-induced bleaching of Cy5 was complete (Fig. 1). The reason for this finding is also obscure. If the FRET-induced bleaching had been incomplete, it should have been taken into consideration by Eq. 6.
Heteroclusters can be analyzed by several biophysical methods. Fluorescence correlation spectroscopy has been applied in confocal microscopy to study the dynamics of molecules (35). Fluorescence cross-correlation spectroscopy (19,36), cross-correlation raster image spectroscopy (21), and two-color N&B analysis (20) report the stoichiometry of stable molecular heteroassociations. Although homo-FRET is sensitive to the size of protein clusters, it can only be used for the analysis of homoassociations (17,18). Although conventional hetero-FRET measurements do not usually provide information about the stoichiometry of clusters or the fraction of clustered molecules, several FRET-based methods have been put forward to obtain these kinds of information. A systematic analysis of the dependence of FRET efficiency on the local density of acceptors allowed investigators to determine an upper bound for the percentage of clustered GPI-anchored proteins (37). A lower-bound value for the fraction of clustered donor molecules was obtained by comparing the photobleaching kinetics of the donor in the absence and presence of an acceptor (28,38). Dual measurements of the kinetics of donor bleaching in the donor and FRET channels enabled Clayton et al. (38) to calculate the fraction of donors undergoing FRET. Hoppe et al. (39) reported that the stoichiometry of CFP-citrine complexes can be determined provided that the FRET efficiency of a bound donor-acceptor pair is known. FSAB presents an alternative method for determining the fraction of heteroclustered acceptors without requiring sophisticated technologies or complex image processing.
The methods used to characterize molecular associations have different sensitivities for detecting clusters that differ in size and stability (40). Since the FSAB approach is based on hetero-FRET, it has all the inherent advantages and limitations of the hetero-FRET principle. Alterations in FRET efficiency can be caused by changes in the orientation or distance between the donor and the acceptor (27). Due to the flexibility of the chemical linkage between the antibodies and the fluorophores, dynamic averaging of the orientation factor takes place, eliminating the dependence of FRET on orientation. It was also recently shown that although fixation is expected to restrict the rotational mobility of proteins, intermolecular FRET efficiencies are not significantly affected by fixation (41). The sensitivity of the FSAB approach for detecting clusters with different sizes and stabilities also has to be discussed. Steady-state FRET measurements cannot discriminate between dimers (two molecules bound to each other by specific molecular interactions) and the proximity of two molecules as a result of accidental apposition (random association due to high density) or partitioning into the same microdomain (co-confinement). Such discrimination would require an analysis of the dependence of FRET on the donor/acceptor ratio or the acceptor density (42). Consequently, FSAB measurements simply reveal the fraction of acceptors within FRET distance of the donors. In addition to dimers and small-scale clusters, aggregation of proteins on a much larger scale has been identified (16,18). Some acceptors in such clusters may be beyond the FRET distance from donors, especially if they outnumber the donor-labeled protein. Although these large-scale clusters are dynamic (43), because of the restricted lateral mobility of proteins in fixed cells, these distant acceptors are not reached by donors. Consequently, the contribution of large-scale clusters to the fraction of bleached acceptors is underestimated in FSAB measurements.
We applied the FSAB technique to investigate heteroclustering of ErbB1 and ErbB2. Almost half of ErbB1 is heteroclustered with ErbB2 in quiescent cells, whereas only ∼10% of ErbB2 is in heteroclusters with ErbB1 (Table 1 and Table S1). The fact that the majority of ErbB2 is not heteroclustered with ErbB1 reflects the much greater numbers of ErbB2 expressed by SKBR-3 cells and the strong tendency of ErbB2 to form homoclusters (Fig. 4 B) (18). After EGF treatment, the fraction of heteroclustered ErbB1 did not change significantly, because EGF induces the formation of both ErbB1 homodimers and ErbB1-2 heterodimers. ErbB2 behaved in a different way. EGF induced an increase in the fraction of ErbB2 in heteroclusters with ErbB1. We previously reported that the size of ErbB2 homoclusters decreases after EGF stimulation (18). We assumed that this phenomenon is caused by ErbB2 being removed from its homoclusters due to recruitment into ErbB1-2 heteroclusters. Our current findings corroborate this hypothesis, as summarized in Fig. 4 B. According to the model, large-scale homoclusters of ErbB2 are partially disrupted after EGF stimulation. ErbB2 homoclusters contain inactive ErbB2, providing a pool for recruitment into heteroclusters with other ErbB proteins. Xiao et al. (44) reached a similar conclusion in a recent study. They found that the lateral diffusion coefficient and the size of the confinement zone of ErbB2 increased upon heregulin stimulation. Although they concluded that the observed changes were caused by cytoskeletal interactions, a decrease in the size of ErbB2 homoclusters is also expected to lead to the same effects.
Figure 4.
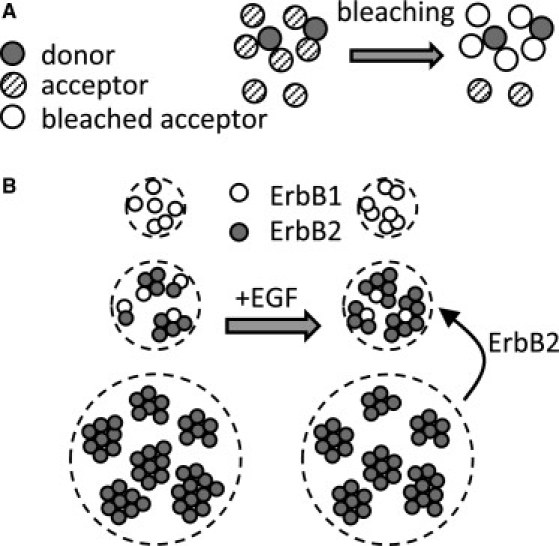
Clustering of membrane proteins revealed by FSAB. (A) Heteroclusters contain a mixture of donor- and acceptor-tagged antibodies. Multiple acceptors surrounding a donor molecule take part in FSAB interactions. All such acceptors in the immediate vicinity of donors will be photobleached. (B) A mixture of homo- and heteroclusters and free (unclustered) receptors exists in unstimulated SKBR-3 cells. The majority of ErbB1 is not involved in heteroclusters with ErbB2. According to our previous work (18), most of these ErbB1 molecules are monomeric. The overwhelming majority of ErbB2 is outside ErbB1-2 heteroclusters and forms large-scale homoclusters (18). Upon EGF stimulation, ErbB2 is recruited from ErbB2 homoclusters to form heteroclusters with ErbB1, leading to a decrease in the homocluster size of ErbB2 and an increase in the fraction of heteroclustered ErbB2. At the same time, the size of the ErbB1 homoclusters increases because EGF induces the formation of ErbB1 homoaggregates in addition to ErbB1-2 heteroclusters. Due to the opposing effects of homo- and heterodimerization, the fraction of heteroclustered ErbB1 does not substantially change upon EGF stimulation. The percentages of homo- and heteroclustered ErbB proteins are expected to be cell-type-dependent, but the general tendency depicted in the figure is likely to be valid for cells overexpressing ErbB2 with a moderate expression level of ErbB1.
We found the fraction of heteroclustered ErbB2 to be independent of the expression levels of ErbB1 and ErbB2 when analyzed at the level of single cells (Fig. S5). This observation implies that the formation of clusters detected by FSAB is not governed by the law of mass action. We believe that the composition of these clusters is established during their export by the vesicular transport system to the cell membrane, and that the density of proteins in the membrane of these vesicles is more or less constant and independent of the number of proteins expressed in the cell membrane.
In conclusion, we have demonstrated that the FSAB technique can be applied to determine the fraction of heteroclustered molecules in a quantitative way, and to investigate the composition of ErbB1-2 heteroclusters. Our interpretation of the experimental findings is in agreement with our previous results and suggests that the size of large-scale ErbB2 clusters decreases upon EGF stimulation. FSAB and other quantitative methods are required to shed light on the intricate details of the first steps of activation of the ErbB receptor tyrosine kinases.
Supporting Material
One table and five figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00428-5.
Supporting Material
Acknowledgments
We thank György Vámosi for helpful discussions about the FSAB principle.
This work was supported by research grants from the Hungarian Scientific Research Fund (K72677, K68763, K62648), the National Office for Research and Technology (NKTH EA-0888-005/2010), the European Commission (LSHC-CT-2005-018914), and the New Hungary Development Plan, cofinanced by the European Social Fund and the European Regional Development Fund (TÁMOP-4.2.2-08/1-2008-0019).
References
- 1.Lemmon M.A. Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 2009;315:638–648. doi: 10.1016/j.yexcr.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yarden Y., Sliwkowski M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 3.Bose R., Zhang X. The ErbB kinase domain: structural perspectives into kinase activation and inhibition. Exp. Cell Res. 2009;315:649–658. doi: 10.1016/j.yexcr.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 5.Cho H.S., Mason K., Leahy D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 6.Klapper L.N., Glathe S., Yarden Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA. 1999;96:4995–5000. doi: 10.1073/pnas.96.9.4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worthylake R., Opresko L.K., Wiley H.S. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J. Biol. Chem. 1999;274:8865–8874. doi: 10.1074/jbc.274.13.8865. [DOI] [PubMed] [Google Scholar]
- 8.Shi Y., Huang W., Winslow J. A novel proximity assay for the detection of proteins and protein complexes: quantitation of HER1 and HER2 total protein expression and homodimerization in formalin-fixed, paraffin-embedded cell lines and breast cancer tissue. Diagn. Mol. Pathol. 2009;18:11–21. doi: 10.1097/PDM.0b013e31818cbdb2. [DOI] [PubMed] [Google Scholar]
- 9.Söderberg O., Gullberg M., Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 10.Jares-Erijman E.A., Jovin T.M. FRET imaging. Nat. Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 11.Spriet C., Trinel D., Heliot L. Enhanced FRET contrast in lifetime imaging. Cytometry A. 2008;73:745–753. doi: 10.1002/cyto.a.20581. [DOI] [PubMed] [Google Scholar]
- 12.Nagy P., Szöllősi J. Proximity or no proximity: that is the question—but the answer is more complex. Cytometry A. 2009;75:813–815. doi: 10.1002/cyto.a.20782. [DOI] [PubMed] [Google Scholar]
- 13.Clayton A.H., Walker F., Burgess A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor—a multidimensional microscopy analysis. J. Biol. Chem. 2005;280:30392–30399. doi: 10.1074/jbc.M504770200. [DOI] [PubMed] [Google Scholar]
- 14.Clayton A.H., Tavarnesi M.L., Johns T.G. Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry. 2007;46:4589–4597. doi: 10.1021/bi700002b. [DOI] [PubMed] [Google Scholar]
- 15.Yang S., Raymond-Stintz M.A., Wilson B.S. Mapping ErbB receptors on breast cancer cell membranes during signal transduction. J. Cell Sci. 2007;120:2763–2773. doi: 10.1242/jcs.007658. [DOI] [PubMed] [Google Scholar]
- 16.Nagy P., Jenei A., Jovin T.M. Activation-dependent clustering of the erbB2 receptor tyrosine kinase detected by scanning near-field optical microscopy. J. Cell Sci. 1999;112:1733–1741. doi: 10.1242/jcs.112.11.1733. [DOI] [PubMed] [Google Scholar]
- 17.Yeow E.K., Clayton A.H. Enumeration of oligomerization states of membrane proteins in living cells by homo-FRET spectroscopy and microscopy: theory and application. Biophys. J. 2007;92:3098–3104. doi: 10.1529/biophysj.106.099424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szabó A., Horváth G., Nagy P. Quantitative characterization of the large-scale association of ErbB1 and ErbB2 by flow cytometric homo-FRET measurements. Biophys. J. 2008;95:2086–2096. doi: 10.1529/biophysj.108.133371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacia K., Kim S.A., Schwille P. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods. 2006;3:83–89. doi: 10.1038/nmeth822. [DOI] [PubMed] [Google Scholar]
- 20.Digman M.A., Wiseman P.W., Gratton E. Stoichiometry of molecular complexes at adhesions in living cells. Proc. Natl. Acad. Sci. USA. 2009;106:2170–2175. doi: 10.1073/pnas.0806036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Digman M.A., Wiseman P.W., Gratton E. Detecting protein complexes in living cells from laser scanning confocal image sequences by the cross correlation raster image spectroscopy method. Biophys. J. 2009;96:707–716. doi: 10.1016/j.bpj.2008.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mekler V.M. A photochemical technique to enhance sensitivity of detection of fluorescence resonance energy transfer. Photochem. Photobiol. 1994;59:615–620. [Google Scholar]
- 23.Mekler V.M., Averbakh A.Z., Kharitonova O.V. Fluorescence energy transfer-sensitized photobleaching of a fluorescent label as a tool to study donor-acceptor distance distributions and dynamics in protein assemblies: studies of a complex of biotinylated IgM with streptavidin and aggregates of concanavalin A. J. Photochem. Photobiol. B. 1997;40:278–287. doi: 10.1016/s1011-1344(97)00070-5. [DOI] [PubMed] [Google Scholar]
- 24.Gruber H.J., Hahn C.D., Knaus H.G. Anomalous fluorescence enhancement of Cy3 and cy3.5 versus anomalous fluorescence loss of Cy5 and Cy7 upon covalent linking to IgG and noncovalent binding to avidin. Bioconjug. Chem. 2000;11:696–704. doi: 10.1021/bc000015m. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez R.C., Woods R.E., Eddins S.L. Segmentation using the watershed algorithm. In: Gonzalez R.C., Woods R.E., Eddins S.L., editors. Digital Image Processing Using MATLAB. Pearson Prentice Hall; Upper Saddle River, NJ: 2004. pp. 417–425. [Google Scholar]
- 26.Pályi-Krekk Z., Barok M., Nagy P. EGFR and ErbB2 are functionally coupled to CD44 and regulate shedding, internalization and motogenic effect of CD44. Cancer Lett. 2008;263:231–242. doi: 10.1016/j.canlet.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Nagy P., Vereb G., Szöllősi J. Measuring FRET in flow cytometry and microscopy. In: Robinson J.P., editor. Curr. Protoc. Cytom. John Wiley & Sons; 2006. Unit12 18. [DOI] [PubMed] [Google Scholar]
- 28.Gadella T.W., Jovin T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995;129:1543–1558. doi: 10.1083/jcb.129.6.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eggeling C., Widengren J., Seidel C.A. Analysis of photobleaching in single-molecule multicolor excitation and Förster resonance energy transfer measurements. J. Phys. Chem. A. 2006;110:2979–2995. doi: 10.1021/jp054581w. [DOI] [PubMed] [Google Scholar]
- 30.Füreder-Kitzmüller E., Hesse J., Schütz G.J. Non-exponential bleaching of single bioconjugated Cy5 molecules. Chem. Phys. Lett. 2005;404:13–18. [Google Scholar]
- 31.Heilemann M., Margeat E., Tinnefeld P. Carbocyanine dyes as efficient reversible single-molecule optical switch. J. Am. Chem. Soc. 2005;127:3801–3806. doi: 10.1021/ja044686x. [DOI] [PubMed] [Google Scholar]
- 32.Eggeling C., Widengren J., Seidel C.A.M. Photostability of fluorescent dyes for single-molecule spectroscopy. In: Rettig W., Strehmel B., Schrader S., Seifert H., editors. Applied Fluorescence in Chemistry, Biology and Medicine. Springer-Verlag; Berlin: 1999. pp. 193–240. [Google Scholar]
- 33.Vogelsang J., Kasper R., Tinnefeld P. A reducing and oxidizing system minimizes photobleaching and blinking of fluorescent dyes. Angew. Chem. Int. Ed. Engl. 2008;47:5465–5469. doi: 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- 34.Gould T.J., Bewersdorf J., Hess S.T. A quantitative comparison of the photophysical properties of selected quantum dots and organic fluorophores. Z. Phys. Chem. 2008;222:833–849. [Google Scholar]
- 35.Brock R., Hink M.A., Jovin T.M. Fluorescence correlation microscopy of cells in the presence of autofluorescence. Biophys. J. 1998;75:2547–2557. doi: 10.1016/S0006-3495(98)77699-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoevesandt O., Brock R. One-step analysis of protein complexes in microliters of cell lysate using indirect immunolabeling & fluorescence cross-correlation spectroscopy. Nat. Protoc. 2006;1:223–229. doi: 10.1038/nprot.2006.34. [DOI] [PubMed] [Google Scholar]
- 37.Kenworthy A.K., Petranova N., Edidin M. High-resolution FRET microscopy of cholera toxin B-subunit and GPI-anchored proteins in cell plasma membranes. Mol. Biol. Cell. 2000;11:1645–1655. doi: 10.1091/mbc.11.5.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clayton A.H., Klonis N., Nice E.C. Dual-channel photobleaching FRET microscopy for improved resolution of protein association states in living cells. Eur. Biophys. J. 2005;34:82–90. doi: 10.1007/s00249-004-0427-y. [DOI] [PubMed] [Google Scholar]
- 39.Hoppe A., Christensen K., Swanson J.A. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 2002;83:3652–3664. doi: 10.1016/S0006-3495(02)75365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lidke D.S., Wilson B.S. Caught in the act: quantifying protein behaviour in living cells. Trends Cell Biol. 2009;19:566–574. doi: 10.1016/j.tcb.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anikovsky M., Dale L., Petersen N. Resonance energy transfer in cells: a new look at fixation effect and receptor aggregation on cell membrane. Biophys. J. 2008;95:1349–1359. doi: 10.1529/biophysj.107.124313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kenworthy A.K., Edidin M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 A using imaging fluorescence resonance energy transfer. J. Cell Biol. 1998;142:69–84. doi: 10.1083/jcb.142.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagy P., Mátyus L., Damjanovich S. Cell fusion experiments reveal distinctly different association characteristics of cell-surface receptors. J. Cell Sci. 2001;114:4063–4071. doi: 10.1242/jcs.114.22.4063. [DOI] [PubMed] [Google Scholar]
- 44.Xiao Z., Ma X., Fang X. Single-molecule study of lateral mobility of epidermal growth factor receptor 2/HER2 on activation. J. Phys. Chem. B. 2008;112:4140–4145. doi: 10.1021/jp710302j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.