

Abstract
Cancer risk assessment utilizing rodents requires extrapolation across five orders of magnitude to estimate the Virtually Safe Dose (VSD). Regulatory agencies rely upon the Linear Extrapolated Dose (LED) except when sufficient information on mechanism of action justifies alternative models. Rainbow trout (Oncorhynchus mykiss) has been utilized at Oregon State University as a model for human cancer for forty years. Low cost and high capacity, made possible by our unique facility, along with low spontaneous background and high sensitivity, allow design and conduct of statistically challenging studies not possible in rodents. Utilization of custom microarrays demonstrates similarities in gene expression in trout and human hepatocellular carcinoma (HCC). We have completed one study employing over 42,000 trout with dibenzo[a,l]pyrene (DBP) and determined the dose resulting in 1 additional cancer in 5,000 animals, a 50-fold enhancement over the mouse ED01 study. Liver tumor incidence at low dose deviated significantly from linearity (concave down), whereas, DBP-DNA adductions deviated slightly (convex up). A second study is underway with aflatoxin B1 (AFB1). Results to date indicate AFB1 at low dose, in contrast to DBP, elicits a linear dose-response function on the log-log scale which falls below the LED with a slope slightly greater than 1.0. Such studies demonstrate the statistical power of the trout cancer model and strengthen the case for incorporation of these data-sets into risk assessment for these environmental human carcinogens.
Keywords: Cancer Risk; Dibenzo[a,l]pyrene; Polycyclic Aromatic Hydrocarbons; Aflatoxin B1; Low Dose Cancer Risk; Hepatocellular Carcinoma
Introduction
Regulatory agencies charged with keeping the public safe from exposure to environmental carcinogens often set their target at a dose producing 1 additional cancer in 106 (Meijers et al., 1997). Typically we have to rely on rodent tumor data which usually generates a dose statistically resulting in an incidence of 10−1 (USEPA, 1996). The largest study ever performed in rodents, the ED01 study, utilized approximately 25,000 mice and the liver and bladder carcinogen, 2-acetylaminofluorene (Cairns, 1979; Gaylor, 1979). The results, although still being discussed with respect to interpretation (Anonymous, 1981a; 1981b; Haseman, 2003; Kodell et al., 1983; Lensing and Kodell, 1995; Travis et al., 1996; Waddell, 2003), showed a linear response in liver, but a sub-linear response in bladder. This study was successful in reducing the uncertainty to four orders of magnitude rather than five. The second largest study in rodents used dimethyl- and diethyl-nitrosamine in approximately 4,000 animals (Peto et al., 1991a; 1991b). Their data appear to support the LED model, but again requires extrapolation across 4–5 orders of magnitude.
The rainbow trout has a number of advantages as a model for low dose carcinogen testing including a high sensitivity to a number of human carcinogens (reviewed in Bailey et al., 1996; Walter et al., 2008) and a low spontaneous tumor incidence (typically 0.1% over forty years of monitoring at our facility). The issue of spontaneous incidence is critical when it comes to the design of large ultra-low dose studies (Figure 1). A mouse with only a 1% background incidence would require over 56,000 mice in the control and lowest dose groups. Such a study is, of course, likely to never be undertaken due to the tremendous cost. In the trout model rare carcinogens may be administered by microinjection at the embryo or sac-fry stage (Bailey et al., 1997) or by embryo immersion. Our studies have employed dietary exposures utilizing a purified diet, the Oregon Test Diet (OTD), developed by our laboratory (Lee et al., 1991). Large scale trout studies would not be possible were it not for the unique Sinnhuber Aquatic Research Laboratory (SARL) facility. This 15,000 square foot building houses over 350 tanks (3–4 feet in diameter, Figure 2) with a capacity to raise about 40,000 trout to 1 year of age (the length of our cancer studies). The facility is a fully functional hatchery and histopathology laboratory. The source of the water is from wells near the Willamette River. Water is subjected to charcoal filtration and UV sterilization to eliminate trace chemical contaminants and potential pathogens, respectively.
Figure 1.

Estimated number of animals required in each group to provide 50% power to demonstrate a statistically significant difference (p-value of 0.1) for one excess tumor in 1000 animals based on background tumor incidence. Calculations were performed according to LaMorte WW: Sample size calculations in research. Boston University Medical Center (http://www.uml.edu/ora/institutionalcompliance/IACUC/LaMorte%20Sample%20Size%20Calc%20Paper.pdf).
Figure 2.

Tanks (top) housing rainbow trout (bottom) used in the cancer studies at the SARL.
Ultra-low Dose Cancer Study with the Polycyclic Aromatic Hydrocarbon (PAH), Dibenzo[a,l]pyrene (DBP)
The first ED001 was directed by Dr. George Bailey and utilized 42,000 trout and eight doses of DBP: 0, 0.45, 1.27, 3.57, 10.1, 28.4, 80 and 225 ppm. A polycyclic aromatic hydrocarbon (PAH) was selected due to the re-emergence of this class of environmental pollutants as a human health risk. Due to increased use of fossil fuels for energy production, PAHs are increasing in the environment. China and the U.S. generate 70 and 50%, respectively, of their electricity from the burning of coal (Xu et al., 2006; Zhang and Smith, 2007). In China, two new coal-powered electricity plants, of a size capable of powering a city the size of Dallas, come on line every week. China is also the second largest auto market and growing rapidly (Tao et al., 2006). Gasoline and diesel exhaust generate PAHs. PAHs formed in China reach the U.S. following atmospheric transport (Primbs et al., 2007) and can account for 25% of the PAHs on any given day in the Los Angeles Basin (Reisen and Arey, 2005). The increased coal burning in China, often without strict emission controls, has been associated with recent increases in lung diseases in that country (Mumford et al., 1995; Watts, 2006; Yu et al., 2006). However, even though PAHs are released from burning of organic material, most of the human exposure to these pollutants is dietary (Luch, 2005).
DBP was administered to the trout for four weeks beginning at about four months of age (average weight about 2 g). PAHs in animals, including trout, are bioactivated by three potential mechanisms. The first involves trans-dihydrodiol formation through epoxygenation catalyzed primarily by cytochrome P450s (CYPs) in the 1 family (CYP1A3 in trout), followed by epoxide hydrolase action. The dihydrodiol is then epoxygenated a second time, again primarily by CYPs in the 1 family to form four potential dihydrodiol-epoxide enantiomers. Benzo[a]pyrene (BaP), the most studied PAH, is a “bay-region” PAH and the ultimate mutagenic and carcinogenic metabolite is the (−)7S-trans-7,8-dihydrobenzo[a]pyrene-7,8-diol-anti-9,10-epoxide ((−)-anti-BPDE). DBP is a more potent carcinogen than BAP in most animal models (Ralston et al., 1994; Ruan et al., 2002) owing to its “fjord” structure. CYP-dependent epoxygenation followed by hydrolysis again results in formation of a dihydrodiol (Figure 3A). The second pathway of PAHs bioactivation is through one electron reactions to reactive radical cations (Figure 3A) (Cavalieri and Rogan, 1990). The third mechanism requires the action of the enzyme aldo-keto reductase which converts the dihydrodiol to a catechol (Jiang et al., 2005). As with other catechols, a quinone can be produced and redox cycling between the catechol, hydroquinone and quinone generates reactive oxygen species which may contribute to PAH carcinogenesis (Figure 3B).
Figure 3.

(A) Bioactivation of DBP through CYP dependent formation of DBPDE or aldo-ketoreductase (AKR) formation of the catechol followed by (B) redox cycling.
Following carcinogen treatment, the trout were switched to OTD for the remainder of the study. At 11–12 months of age, the trout were euthanized with buffered MS-222 (all protocols approved by the Oregon State University IACUC). The study was performed in quartiles due to the length of time required to perform gross necropsy on 10,000 animals. The trout tanks, within and among treatment groups, were randomized prior to necropsy to ensure that growth during the 4–6 week sampling period did not influence the results. We had previously shown that hepatocarcinogenesis in this trout model was correlated to body weight. The results from this study are presented in much greater detail elsewhere (Williams et al., 2003; Bailey et al., in review). In summary, the liver tumor incidence significantly deviated from the LED with decreasing dose (Figure 4). The estimated dose of DBP producing one cancer in 106 was about 1000-fold higher using the actual data (probit fit) than what would have been calculated from an LED from tumor incidences with 10% as the lower bound (the typical situation with rodent studies). This large difference in VSD could have a tremendous impact on regulatory control of PAHs. Interestingly one of the most common biomarkers used in such studies, DNA adduction, displayed a slightly exponential (convex upward) behavior down to the limit of detection (1.27 ppm, the second highest dose), not a downward-curving function like tumor incidence. The use of liver DBP-DNA adduction as a biomarker indicative of hepatocarcinogenesis risk would thus have been misleading and, like the LED extrapolation of tumor incidence, would have markedly over-estimated the VSD.
Figure 4.
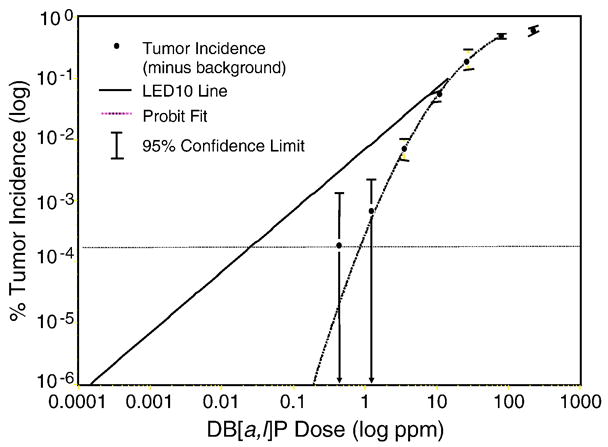
ED001 Liver tumor incidence with DBP. Each point represents the means (bars 95% confidence interval) of the four quartiles. The solid line is calculated from the LED at tumor incidences of 10% and higher. The small arrows indicate the estimated dose using the two extrapolation methods (LED versus probit fit) that would result in a 10−6 incidence.
The second trout ED001 study undertaken utilized the important human dietary carcinogen, aflatoxin B1 (AFB1). AFB1, a mycotoxin produced by Aspergillus flavus and Aspergillus parasiticus, is hepatotoxic, producing a syndrome known as aflatoxicosis in addition to hepatocarcinogenesis (Eaton and Gallagher, 1994; Wild, 2007; Williams et al., 2004). Outbreaks of aflatoxicosis in human populations in Kenya and other parts of the world (also known as kwashiorkor) continue to produce disease and mortalities, as have outbreaks of aflatoxin-associated juandice in human populations (Wild, 2007; Williams et al., 2004). Epidemiology studies support the conclusion that AFB1 is hepatocarcinogenic in humans (Chen et al., 1996; Wang et al., 1996) and the IARC classified AFB1 and aflatoxin mixtures as Group 1 carcinogens in humans in 1992 (Dominguez-Malagon and Gaytan-Graham, 2001).
Primary hepatocellular carcinoma (HCC) is a common malignant tumor worldwide (over 1 million new cases annually, fifth most common worldwide) with the third highest overall mortality rate (Bosch et al., 2005). HCC, although currently relatively rare in the U.S., exhibits the fastest increase among solid tumors (Dominguez-Malagon and Gaytan-Graham, 2001). HCC is epidemic in other regions of the world including Africa and Southeast Asia where climate and post-harvest storage of foods is conducive to growth of Aspergillus. In portions of China, primary liver cancer is the number one cause of all deaths in males (Dominguez-Malagon and Gaytan-Graham, 2001). The high incidences of HBV and HCV infection in these populations and the high dietary intake of AFB1 appear to function synergistically to produce these high incidences (Kew, 2003; Kuang et al., 2004). The risk from AFB1 may be increasing in the future owing to such factors as climate change (Cotty and Maime-Garcia, 2007) and the increased use of corn (a favorite substrate for Aspergillus) in ethanol production (Wu and Munkvold, 2008).
AFB1 is metabolically activated by CYPs in the 1A and 3A sub-families to AFB1-8,9-exo-epoxide (Eaton and Gallagher, 1994; Guengerich et al., 1996) which, like a PAH-epoxide, can be converted to the diol spontaneously or by epoxide hydrolase or it can conjugated by glutathione S-transferases (Tiemersma et al., 2001). If not metabolized by these pathways, the epoxide can bind to DNA, predominantly producing the trans-8,9-dihydro-(N7-guanyl)-9-hydroxy-AFB1 adduct. This N7-guanine adduct undergoes depurination or rearrangement to a more stable ring-opened formamidopyrimidine (FAPY) adduct (Alekseyev et al., 2004; Smela et al., 2002) (Figure 5). It is remarkable to note the agreement between rat and trout with respect to plots of AFB1-DNA adducts and hepatocarcinogenesis; these data basically fall on the same line (Bechtel, 1989). Based on LED of human epidemiological data from the U.S., an exposure of 0.46 ng/Kg/day would result in a risk of 10−6 (Eaton and Gallagher, 1994). The LED from rat tumor data gives a VSD (10−6) of 0.0016 ng/Kg/day which would predict a cancer rate of 98/100,000 for individuals living in the Southeastern U.S. (average daily AFB1 intake of 110 ng/kg) (Eaton and Gallaher, 1994). This is 20-fold higher than the primary liver cancer incidence in the U.S. from all sources.
Figure 5.

Metabolic pathways for bioactivation and detoxication of AFB1.
The sensitivity of the trout to AFB1-dependent hepatocarcinogenesis has been utilized for over 40 years at Oregon State University and a large database acquired on tumor response as a function of dose, route of administration, time to tumor, age, growth rate, etc. (reviewed by Bailey et al., 1996). In addition, a complete histopathology of AFB1-dependent hepatocarcinogenesis has been described for this model (Hendricks et al., 1984). The metabolism of AFB1 in trout models human liver at least as well as rat and better than mouse (Bailey et al., 1996; Williams and Buhler, 1983). The primary DNA adduct is the same in rodents, trout and humans. Trout have a slower rate of bulky adduct global repair than rodents, but again, the correlation of this adduct with tumor incidence, over a wide AFB1 dose range, falls upon the same plot for rat and trout (Bechtel, 1989).
We have completed two of the four quartiles of the AFB1 ED001 study with trout (about 20,000 animals). The tumor response appears to be linear at low dose and falls on a line below the LED on a log-log scale, with a slope slightly greater than 1.0 (Figure 6). These studies should be considered preliminary but demonstrate that different carcinogens may exhibit markedly differerent dose-response patterns at ultra-low dose. Based on just the first two quartiles, the 10−6 VSD would be about 0.1 ppb.
Figure 6.

Preliminary Tumor Incidence for Quartiles 1 and 2. Background incidence (0.1%) has been subtracted.
In addition to tumor incidence, we have been examining potential immunohistochemical biomarkers and indicators of mechanism. From the results In Figure 7, it is apparent that apoptosis (TUNEL) is not a useful low dose biomarker, whereas, cell proliferation (BrdU) does show a dose-response relationship in the low tumor incidence range.
Figure 7.
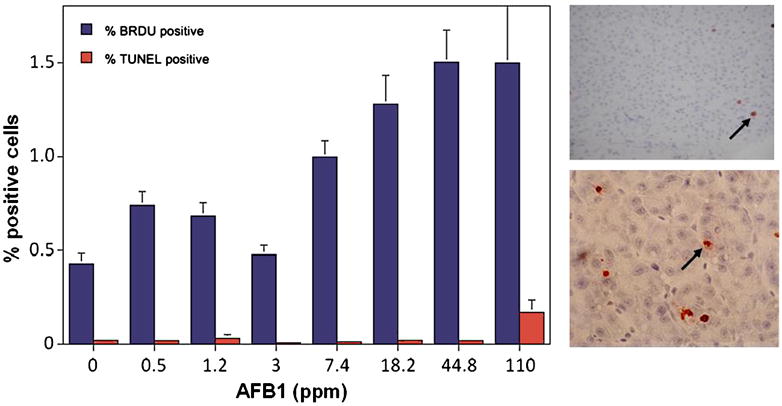
Cell proliferation and apoptosis in trout liver as a function of AFB1 dose.
We have also made use of the custom cDNA GRASP array (Schalburg et al., 2008) and a custom oligonucleotide trout array of our own design to compare gene expression in trout HCC versus adjacent normal appearing liver from the same animal or from livers of control animals (Tilton et al., 2005; 2007; 2008). When AFB1-induced trout HCC is compared to adjacent tissue (Table 1) a number of genes involved in cell proliferation are up-regulated, along with those involved in extracellular matrix and vascularization and immunoregulation. The pathways impacted are similar to those of human HCC (Tilton et al., 2005). A direct comparison is difficult as the trout genome has not been sequenced and annotation is problematic. However, a cursory examination of the genes up- and down-regulated indicate that trout HCC is an aggressive tumor and, if given more time, would likely metastisize.
Table 1.
Select genes differentially regulated in AFB1-initiated HCC compared to normal adjacent tissue
| Gene name (accession number) | Fold change (P-value)a |
|---|---|
| Cell proliferation (signal transduction, transcription factors) | |
| Transmembrane 4 superfamily (AF281357) | 5.44 (0.008) |
| Transcription factor JunB (Q800B3,) | 3.52 (0.005) |
| Calmodulin (X61432) | 3.11 (0.035) |
| Estrogen receptor beta (AJ289883) | 2.94 (<0.001) |
| Protein/Ion stability and transport | |
| Cathepsin L-like cysteine peptidase (AY332270) | 3.82 (0.027) |
| Hepcidin (AF281354) | 8.33 (<0.001) |
| Extracellular matrix and vascularization | |
| Plasminogen activator, urokinase receptor | 13.26 (0.041) |
| Collagen alpha 2(VIII) chain (AK074129) | 6.90 (0.006) |
| Putative interlectin, fibrinogen (AF281350) | 7.22 (0.071) |
| Hemagglutinin aggregation factor (M96983) | 0.32 (<0.001) |
| Redox regulation | |
| Catalase (AAF89686) | 0.30 (0.003) |
| Thioredoxin (AAH49031) | 4.83 (0.002) |
| Glutathione peroxidase 4 (AAO86704) | 2.59 (<0.001) |
| Drug, lipid, glucose and retinol metabolism/homeostasis | |
| Retinol binding protein (AF257326) | 0.30 (0.007) |
| Cytochrome P450 1A (AF059711) | 0.33 (0.049) |
| Aldo-keto reductase 1D1 (Z28339) | 0.19 (<0.001) |
| Insulin-like growth factor II (M95184) | 0.34 (0.002) |
| Glucose-6-phosphatase (AAH54228) | 0.28 (0.004) |
| Potential immunoregulators and acute phase response proteins | |
| Chemotaxin (AF271114) | 16.9 (0.041) |
| Apolipoprotein A1 (AB046208) | 6.96 (0.039) |
| MHC class IA heavy chain precursor | 3.86 (0.012) |
The fold change is a background corrected, LOWLESS-normalized signal ratio of HCC to normal tissue. Stringent criteria were used to filter for genes regulated at least 1.8-fold consistently in all features from biological replicates and had a P value <0.05 by Welch’s t-test. Data is adapted from Tilton et al, (2005) which gives further details on analysis of microarray data and gene validation.
In summary, we have exploited the advantages of the rainbow trout model and the unique facilities available to us to conduct the largest tumor studies performed in any animal model. The first study, which has been completed, was with the potent PAH, DBP, and the ongoing study is utilizing the potent dietary inducer of human HCC, AFB1. Analysis of the dose-response for DBP is striking in that it significantly deviates from linearity at low dose to the degree that the 10−6 VSD is 1000-fold higher than would be calculated from the LED. AFB1, on the other hand, appears linear on the log-log scale at low dose, with a slope slightly steeper than that for the LED extrapolation line. It must be kept in mind that, for the AFB1 studies, these are preliminary results. A significant difference between our trout model and lifetime rodent exposure studies is the relatively short (typically 4 weeks) of dietary carcinogen exposure in trout followed by an 8–9 month grow out period. Trout have an indefinite lifespan, making lifetime exposures impractical. At the conclusion of our studies we rarely see metastasis, however, the tumors can be quite large. Studies of the HCC transcriptome, compared to normal adjacent tissue, suggest that these tumors possess a very aggressive phenotype and would metastasize if the study were extended (Tilton, 2005). It is important to note that these two human carcinogens exhibit very different properties at low dose, an important consideration for human risk assessment. It is our hope that governmental agencies make use of the results from this powerful and unique model. We believe we have established that in many ways the trout readily complements rodents for the study of some carcinogens and there is certainly no mammalian model capable of analyzing tumor responses at the doses and incidences possible with the trout.
Acknowledgments
The authors would like to thank Dr. Robert Tanguay, Mr. Eric Johnson, Ms. Tammie McQuistan, Dr. Christiana Löhr, Ms. Kay Fischer, Ms. Tanya Percifield, Ms. Cari Buchner, Mr. Greg Gonnerman and Ms. Shiela Cleveland for their contributions to this work. This study was supported by ES00210 and ES013534 from the National Institutes of Health.
Footnotes
This paper is derived from a presentation given at the 4th Aquatic Animal Models of Human Disease Conference: hosted by Duke University’s Nicholas School of the Environment and Earth Sciences, and Duke’s Comprehensive Cancer Center, Durham, NC, USA, January 31 - February 3, 2008.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
David E. Williams, Department of Environmental and Molecular Toxicology, The Environmental Health Sciences Center, Oregon State University.
Gayle Orner, Department of Environmental and Molecular Toxicology, The Environmental Health Sciences Center, Oregon State University.
Kristin D. Willard, Department of Environmental and Molecular Toxicology, The Environmental Health Sciences Center, Oregon State University
Susan Tilton, Department of Environmental and Molecular Toxicology, The Environmental Health Sciences Center, Oregon State University.
Jerry D. Hendricks, Department of Environmental and Molecular Toxicology, The Environmental Health Sciences Center, Oregon State University
Clifford Pereira, Department of Statistics and The Linus Pauling Institute, Oregon State University.
Abby D. Benninghoff, Department of Environmental and Molecular Toxicology, The Environmental Health Sciences Center, Oregon State University
George S. Bailey, Department of Environmental and Molecular Toxicology, The Environmental Health Sciences Center, Oregon State University
References
- Alekseyev YO, Hamm ML, Essigmann JM. Aflatoxin B1 formamidopyrimidine adducts are preferentially repaired by the nucleotide excision repair pathway in vivo. Carcinogenesis. 2004;25:1045–1051. doi: 10.1093/carcin/bgh098. [DOI] [PubMed] [Google Scholar]
- Anonymous. Re-examination of the ED-01 study-risk assessment using time. Fund Appl Pharmacol. 1981a;1:88–123. [PubMed] [Google Scholar]
- Anonymous. Re-examination of the ED01 study. Review of statistics: the need for realistic statistical models for risk assessment. Fund Appl Pharmacol. 1981b;1:124–126. [PubMed] [Google Scholar]
- Bailey GS, Dashwood R, Loveland PM, Pereira C, Hendricks JD. Molecular dosimetry in fish: quantitative target organ DNA adduction and hepatocarcinogenicity for four aflatoxins by two exposure routes in rainbow trout. Mut Res. 1997;339:233–244. doi: 10.1016/s0027-5107(97)00258-3. [DOI] [PubMed] [Google Scholar]
- Bailey GS, Pereira CB, Harttig U, Baird WM, Spitsbergen JM, Hendricks JD, Orner GA, Williams DE, Swenberg JS, Reddy AP. Non-linear cancer response at ultra-low dose: A 42,000-animal ED0002 study. 200x doi: 10.1021/tx9000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey GS, Williams DE, Hendricks JD. Fish models for environmental carcinogenesis: the rainbow trout. Environm Hlth Perspect. 1996;104(Suppl):5–19. doi: 10.1289/ehp.96104s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel DH. Molecular dosimetry of hepatic aflatoxin B1-DNA adducts: linear correlation with hepatic cancer risk. Reg Toxicol Pharmacol. 1989;10:74–81. doi: 10.1016/0273-2300(89)90014-7. [DOI] [PubMed] [Google Scholar]
- Bosch FX, Ribes J, Cléries R, Díaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis. 2005;9:191–211. doi: 10.1016/j.cld.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Cairns T. The ED01 study: Introduction, objectives, and experimental design. J Environ Pathol Toxicol. 1979;3:1–8. [PubMed] [Google Scholar]
- Cavalieri EL, Rogan EG. Radical cations in aromatic hydrocarbon carcinogenesis. Free Radic Res Commun. 1990;11:77–87. doi: 10.3109/10715769009109670. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Wang LY, Lu SN, Wu MH, Lou SL, Zhang YJ, Wang LW, Santella RM. Elevated aflatoxin exposure and increased risk of hepatocellular carcinoma. Hepatol. 1996;24:38–42. doi: 10.1002/hep.510240108. [DOI] [PubMed] [Google Scholar]
- Cotty PJ, Maime-Garcia R. Influences of climate on aflatoxin producing fungi and aflatoxin contamination. Int J Food Microbiol. 2007;119:109–115. doi: 10.1016/j.ijfoodmicro.2007.07.060. [DOI] [PubMed] [Google Scholar]
- Dominguez-Malagon H, Gaytan-Graham S. Hepatocellular carcinoma: an update. Ultrastruct Pathol. 2001;25:497–516. doi: 10.1080/019131201753343539. [DOI] [PubMed] [Google Scholar]
- Eaton DL, Gallagher EP. Mechanisms of aflatoxin carcinogenesis. Annu Rev Pharmacol Toxicol. 1994;34:135–172. doi: 10.1146/annurev.pa.34.040194.001031. [DOI] [PubMed] [Google Scholar]
- Gaylor DW. The ED01 study: summary and conclusions. J Environm Pathol Toxicol. 1979;3:179–186. [PubMed] [Google Scholar]
- Groopman JD, Scholl P, Wang JS. Epidemiology of human aflatoxin exposures and their relationship to liver cancer. Prog Clin Biol Res. 1996;395:211–222. [PubMed] [Google Scholar]
- Guengerich FP, Johnson WW, Ueng YF, Yamazaki H, Shimada T. Involvement of cytochrome P450, glutathione S-transferase, and epoxide hydrolase in the metabolism of aflatoxin B1 and relevance to risk of human liver cancer. Environm Hlth Perspect. 1996;104(Suppl 3):557–562. doi: 10.1289/ehp.96104s3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseman JK. An alternative perspective: a critical evaluation of the Waddell Threshold Extrapolation Model in chemical carcinogenesis. Toxicol Pathol. 2003;31:468–470. doi: 10.1080/01926230390228940. [DOI] [PubMed] [Google Scholar]
- Hendricks JD, Meyers TR, Shelton DW. Histological progression of hepatic neoplasia in rainbow trout (Salmo gairdneri) Natl Cancer Inst Monogr. 1984;65:321–336. [PubMed] [Google Scholar]
- Jiang H, Shen YM, Quinn AM, Penning TM. Competing roles of cytochrome P450 1A1/1B1 and aldo-keto reductase 1A1 in the metabolic activation of (±)-7,8-dihyroxy-7,8-dihydro-benzo[a]pyrene in human bronchoalveolar cell extracts. Chem Res Toxicol. 2005;18:365–374. doi: 10.1021/tx0497245. [DOI] [PubMed] [Google Scholar]
- Kew MC. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003;23:405–409. doi: 10.1111/j.1478-3231.2003.00869.x. [DOI] [PubMed] [Google Scholar]
- Kodell RL, Gaylor DW, Greenman DL, Littlefield NA, Farmer JH. Response to the Society of Toxicology Task Force re-examination of the ED01 study. Fundam Appl Toxicol. 1983;3:3a–12a. doi: 10.1016/s0272-0590(83)80150-x. [DOI] [PubMed] [Google Scholar]
- Kuang SY, Jackson PE, Wang JB, Lu PX, Munoz A, Qian GS, Kensler TW, Groopman JD. Specific mutations of hepatitis B virus in plasma predict liver cancer development. Proc Natl Acad Sci (USA) 2004;101:3575–3580. doi: 10.1073/pnas.0308232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Hendricks JD, Bailey GS. Toxicity of mycotoxins in the feed of fish. In: Smith JE, editor. Mycotoxins and Animal Feedstuff: Natural Occurrence, Toxicity and Control. CRC Press; 1991. pp. 607–626. [Google Scholar]
- Lensing SY, Kodell RL. Fitting the two-stage clonal expansion model based on exact hazard to the ED01 data using SAS NLIN. Risk Anal. 1995;15:233–245. doi: 10.1111/j.1539-6924.1995.tb00317.x. [DOI] [PubMed] [Google Scholar]
- Luch A. Polycyclic aromatic hydrocarbon-induced carcinogenesis – an introduction. In: Luch A, editor. The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons. Imperial College Press; London: 2005. pp. 1–18. [Google Scholar]
- Meijers JMM, Swaen GMH, Bloemen LJN. The predictive value of animal data in human risk assessment. Regul Toxicol Pharmacol. 1997;25:94–102. doi: 10.1006/rtph.1996.1080. [DOI] [PubMed] [Google Scholar]
- Mumford JL, Li X, Hu F, Lu XB, Chuang JC. Human exposure dosimetry of polycyclic aromatic hydrocarbons in urine from Xuan Wei, China with high lung cancer mortality associated with exposure to unvented coal smoke. Carcinogenesis. 1995;16:3031–3036. doi: 10.1093/carcin/16.12.3031. [DOI] [PubMed] [Google Scholar]
- Peto R, Gray R, Brantom P, Grasso P. Effects on 4080 rats of chronic ingestion of N-nitrosodiethylamine or N-nitrosodimethylamine: a detailed dose-response study. Cancer Res. 1991a;51:6415–6451. [PubMed] [Google Scholar]
- Peto R, Gray R, Brantom P, Grasso P. Dose and time relationships for tumor induction in the liver and esophagus of 4080 inbred rats by chronic ingestion of N-Nitrosodiethylamine or N-Nitrosodimethylamine. Cancer Res. 1991b;51:6452–6469. [PubMed] [Google Scholar]
- Primbs T, Simonich SL, Schmedding D, Wilson G, Jaffe D, Takami A, Kato S, Hatakeyama S, Kajii Y. Atmospheric outflow of anthropogenic semi-volatile organic compounds from East Asia in Spring 2004. Environm Sci Technol. 2007;41:3551–3558. doi: 10.1021/es062256w. [DOI] [PubMed] [Google Scholar]
- Ralston SL, Lau HHS, Seidel A, Luch A, Platt KL, Baird WM. The potent carcinogen dibenzo[a,l]pyrene is metabolically activated to fjord-region 11,12-diol 13,14-epoxides in human mammary carcinoma MCF-7 cell cultures. Cancer Res. 1994;54:887–890. [PubMed] [Google Scholar]
- Reisen F, Arey J. Atmospheric reactions influence seasonal PAH and Nitro-PAH concentrations in the Los Angeles Basin. Environm Sci Technol. 2005;39:64–73. [PubMed] [Google Scholar]
- Ruan Q, Kolbanovskiy A, Zhuang P, Chen J, Krzeminski J, Amin S, Geacintov NE. Synthesis and characterization of site-specific and stereoisomeric fjord dibenzo[a,l]pyrene diol epoxide-N(6)-adenine adducts: unusual thermal stabilization of modified DNA duplexes. Chem Res Toxicol. 2002;15:249–261. doi: 10.1021/tx010157k. [DOI] [PubMed] [Google Scholar]
- Schalburg KRV, Cooper GA, Leong J, Robb A, Lieph R, Rise ML, Davidson WS, Koop BF. Expansion of the genomics research on Atlantic salmon Salmo salar L. project (GRASP) microarray tools. J Fish Biol. 2008;72:2051–2070. [Google Scholar]
- Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, Essigmann JM. The aflatoxin B1 formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc Natl Acad Sci (USA) 2002;99:6655–6660. doi: 10.1073/pnas.102167699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S, Li X, Yang Y, Coveney RM, Lu X, Chen H, Shen W. Dispersion modeling of polycyclic aromatic hydrocarbons from combustion of biomass and fossil fuels and production of coke in Tianjin, China. Environm Sci Technol. 2006;40:4586–4591. doi: 10.1021/es060220y. [DOI] [PubMed] [Google Scholar]
- Tiemersma E, Omer RE, Bunschoten A, van’t Veer P, Kok FJ, Idris MO, Kadaru AMY, Fedail SS, Kampman E. Role of genetic polymorphism of glutathione-S-transferase T1 and microsomal epoxide hydrolase in aflatoxin-associated hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev. 2001;10:785–791. [PubMed] [Google Scholar]
- Tilton SC, Gerwick LG, Hendricks JD, Rosato CS, Corley-Smith G, Givan SA, Bailey GS, Bayne CJ, Williams DE. Use of a rainbow trout oligonucleotide microarray to determine transcriptional patterns in aflatoxin B1-induced hepatocellular carcinoma compared to adjacent liver. Toxicol Sci. 2005;88:319–330. doi: 10.1093/toxsci/kfi309. [DOI] [PubMed] [Google Scholar]
- Tilton SC, Hendricks JD, Orner GA, Pereira CB, Bailey GS, Williams DE. Gene expression analysis during tumor enhancement by the dietary phytochemical, 3,3′-diindolylmethane, in rainbow trout. Carcinogenesis. 2007;28:1589–1598. doi: 10.1093/carcin/bgm017. [DOI] [PubMed] [Google Scholar]
- Tilton SC, Orner GA, Benninghoff AD, Carpenter HM, Hendricks JD, Pereira CB, Williams DE. Genomic profiling reveals an alternate mechanism for hepatic tumor promotion by perfluorooctanoic acid in rainbow trout. Environm Hlth Perspect. 2008;116:1047–1055. doi: 10.1289/ehp.11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis CC, Zeng C, Nicholas J. Biological model of ED01 hepatocarcinogenesis. Toxicol Appl Pharmacol. 1996;140:19–29. doi: 10.1006/taap.1996.0193. [DOI] [PubMed] [Google Scholar]
- U.S. Environmental Protection Agency. Proposed guidelines for carcinogen risk assessment. Fed Reg. 1996;61:17960–18011. [Google Scholar]
- Waddell WJ. Thresholds of carcinogenicity in the ED01 study. Toxicol Sci. 2003;72:158–163. doi: 10.1093/toxsci/kfg004. [DOI] [PubMed] [Google Scholar]
- Walter RB, Timmins GS, Tilton SC, Orner GO, Benninghoff AD, Bailey GS, Williams DE. In: Carcinogenesis models: focus on Xiphophorus and Rainbow Trout, In Oceans and Human Health: Risks and Remedies from the Seas. Walsh PJ, Smith SL, Fleming LE, Solo-Gabriele HM, Gerwick WH, editors. Elsevier, Academic Press; London: 2008. pp. 585–611. [Google Scholar]
- Wang LY, Hatch M, Chen CJ, Levin B, You SL, Lu SN, Wu MH, Wu WP, Wang LW, Wang Q, Huang GT, Yang PM, Lee HS, Santella RM. Aflatoxin exposure and risk of hepatocellular carcinoma in Taiwan. Int J Cancer. 1996;67:620–625. doi: 10.1002/(SICI)1097-0215(19960904)67:5<620::AID-IJC5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Watts J. Doctors blame air pollution for China’s asthma increases. The Lancet. 2006;368:719–720. doi: 10.1016/S0140-6736(06)69267-2. [DOI] [PubMed] [Google Scholar]
- Wild CP. Aflatoxin exposure in developing countries: the critical interface of agriculture and health. Food Nutr Bull. 2007;28 (Suppl 2):S372–S380. doi: 10.1177/15648265070282S217. [DOI] [PubMed] [Google Scholar]
- Williams DE, Buhler DR. Purified form of cytochrome P-450 from rainbow trout with high activity toward conversion of aflatoxin B1 to aflatoxin B1-2,3-epoxide. Cancer Res. 1983;43:4752–4756. [PubMed] [Google Scholar]
- Williams DE, Bailey GS, Reddy A, Hendricks JD, Oganesian A, Orner G, Pereira CB, Swenberg JA. The rainbow trout tumor model: recent applications in low dose exposures to tumor initiators and promoters. Toxicol Pathol. 2003;31 (Suppl):58–61. [PubMed] [Google Scholar]
- Williams JH, Phillips TD, Jolly PE, Stiles JK, Jolly CM, Aggarwal D. Human aflatoxicosis in developing countries: a review of toxicology, exposure, potential health consequences, and interventions. Am J Clin Nutr. 2004;80:1106–1122. doi: 10.1093/ajcn/80.5.1106. [DOI] [PubMed] [Google Scholar]
- Wu F, Munkvold GP. Mycotoxins in ethanol co-products: modeling economic impacts on the livestock industry and management strategies. J Ag Food Chem. 2008;56:3900–3911. doi: 10.1021/jf072697e. [DOI] [PubMed] [Google Scholar]
- Xu S, Liu W, Tao S. Emission of polycyclic aromatic hydrocarbons in China. Environ Sci Technol. 2006;40:702–708. doi: 10.1021/es0517062. [DOI] [PubMed] [Google Scholar]
- Yu ITS, Chium Y-I, Au JSK, Wong T-W, Tang JL. Dose-response relationship between cooking fumes exposures and lung cancer among Chinese nonsmoking women. Cancer Res. 2006;66:4961–4967. doi: 10.1158/0008-5472.CAN-05-2932. [DOI] [PubMed] [Google Scholar]
- Zhang J, Smith KR. Household air pollution from coal and biomass fuels in China: Measurements, health impacts, and interventions. Environm Hlth Perspect. 2007;115:848–855. doi: 10.1289/ehp.9479. [DOI] [PMC free article] [PubMed] [Google Scholar]