

Abstract
Rationale
Numerous studies have proposed that glycogen synthase kinase-3β (GSK-3β) is a central regulator of the hypertrophic response of cardiomyocytes. However, all of this work has relied on over-expression of GSK-3β, expression of constitutively active mutants, or small molecule inhibitors with documented off-target effects. Genetic loss of function approaches have not been employed in the adult mouse because germ-line deletion of GSK-3β is embryonic-lethal.
Objective
This study was designed to define the role played by GSK-3β in pressure overload (PO)-induced hypertrophy and remodeling following myocardial infarction (MI).
Methods and Results
We employed a mouse model that allows inducible, cardiomyocyte-specific deletion of GSK-3β in the adult (KO). Surprisingly, we find that KO mice exposed to PO induced by thoracic aortic constriction exhibit a normal hypertrophic response. Thus, in contrast to virtually all prior published studies, GSK-3β appears to play at most a minor role in the hypertrophic response to PO stress. However, GSK-3β does regulate post-MI remodeling since the GSK-3β KOs had less LV dilatation and better-preserved LV function at up to 8 weeks post-MI despite demonstrating significantly more hypertrophy in the remote myocardium. Deletion of GSK-3β also led to increased cardiomyocyte proliferation following PO and MI.
Conclusion
Deletion of GSK-3β protects against post-MI remodeling and promotes stress-induced cardiomyocyte proliferation in the adult heart. These studies suggest that inhibition of GSK-3β could be a strategy to both prevent remodeling and to promote cardiac regeneration in pathologic states.
Keywords: cardiac hypertrophy, myocardial infarction, heart failure, myocardial regeneration, GSK-3
Introduction
Cardiomyocytes are terminally differentiated cells that respond to different stresses such as pressure stress (e.g. hypertension or aortic valvular disease) and cardiac injury leading to myocyte loss (e.g. myocardial infarction) by undergoing hypertrophy. As cardiomyocytes hypertrophy, they go through specific characteristic changes regardless of pathological stimulus. These alterations typically include conversion to a fetal gene expression profile, increased protein synthesis, and changes in metabolism1, 2. Over time, hypertrophy can progress to heart failure. Heart failure is characterized by progressive cell loss, extracelluar matrix remodeling and contractile dysfunction. It is unclear how (or if) hypertrophy contributes to these phenotypic manifestations of heart failure. Although the hypertrophic response normalizes cardiac wall stress, it seems that this response is not necessary to maintain contractile function3. Thus, targeting hypertrophic signaling pathways could possibly prevent or delay pathologic remodeling and heart failure progression, irrespective of the inciting stimulus.
Glycogen synthase kinase-3β (GSK-3β) is thought to be a key integrator of many of the pathways activated by hypertrophic stimuli4, 5. Although GSK-3 has 2 isoforms, GSK-3α and GSK-3β, the vast majority of studies have focused on GSK-3β. The near exclusive focus on GSK-3β as the predominant isoform may have originated in two landmark studies in Drosophila that established GSK-3β as the dominant isoform for body patterning6, 7. Both isoforms are ubiquitously expressed and share 97% homology within their catalytic domains but differ at their N and C-terminal regions8. The two isoforms of GSK-3 have both distinct and redundant functions in the cell9–13, but in cardiac development, GSK-3β is dominant14. GSK-3α knockout mice develop normally, consistent with full compensation by GSK-3β for loss of GSK-3α in cardiac development. In contrast, germ-line deletion of GSK-3β is embryonic lethal, due to hyperproliferation of cardiomyoblasts that largely obliterate the RV and LV cavities14.
The bias towards GSK-3β as the dominant isoform also applies to studies in the adult heart which have focused on GSK-3β as the key negative regulator of cardiac hypertrophy. GSK-3β is active in unstimulated cells. When GSK-3β phosphorylates its substrates, it typically leads to their inactivation. Multiple substrates have been identified, and several of these are proteins that appear to play central roles in cardiomyocyte hypertrophy and/or proliferation, including, eukaryotic protein synthesis initiation factor 2B (eIF2B)15, β-catenin16, 17, c-myc18, nuclear factor of activated T-cells (NF-AT)19, mammalian target of rapamyacin complex 1 (mTORC1)20, 21, and cyclin D122. Phosphorylation of these and other substrates by active GSK-3β is believed to blunt hypertrophy. Upon activation of hypertrophic signaling pathways such as PI3K/Akt, PKC, and PKA, GSK-3 is phosphorylated and inactivated 1, 4 and this inactivation releases the substrates from inhibition.
In reality, however, it is unclear whether the two isoforms share redundant or distinct functions with regard to hypertrophy and remodeling in the adult heart. Studies to date in the heart have led to confusing and sometimes contradictory conclusions concerning the role of the GSK-3 family in hypertrophy5. This appears to be due, in large part, to the methods employed in these studies, including the use of small molecule ATP-competitive inhibitors such as the indirubins with questionable selectivity (leading to off-target effects) and/or limited solubility23, 24, or reliance on transgenic over-expression of constitutively active (CA) or dominant negative (DN) mouse models.25–31 Therefore, the true roles of GSK-3β in the heart of adult mice in vivo remain to be defined.
To address this, we employ a cardiomyocyte-specific, conditional KO model to better define the role of GSK-3β in pathological hypertrophy and remodeling in the adult. Based on the majority of the published data discussed above, we expected that there would be exaggerated hypertrophy in response to thoracic aortic constriction (TAC) and, possibly, in response to MI. Herein, we demonstrate that GSK-3β, per se, plays at most a minor role in regulating pathological cardiac hypertrophy induced by pressure overload. In contrast, GSK-3β is central to the hypertrophic response following MI. More importantly, despite the exaggerated hypertrophic response to MI in the remote myocardium, we find that deletion of GSK-3β protects against the LV dilatation and dysfunction that follows a large MI. Finally, we find that cardiomyocyte-specific deletion of GSK-3β identifies a key role for the kinase in regulating cardiomyocyte proliferation in the setting of stress.
Methods
Generation of cardiac-specific, inducible GSK-3β knockout model
C57Bl6 mice with lox-p sites flanking exon 2 of the gsk-3β gene12 were crossed with mice carrying the Mer-Cre-Mer transgene driven by the α-MHC promoter (gift from Dr. J. Molkentin). For TAC studies, at 5 weeks of age, the mice were treated with tamoxifen (20mg/kg IP [Sigma-Aldrich] dissolved at a final concentration of 4mg/ml in 30% ethanol in sterile phosphate-buffered saline (PBS)) or vehicle (30% ethanol in PBS) for 5 days to induce excision of exon 2. At 7 weeks of age, mice underwent baseline echo followed by TAC. In contrast to a prior report32, baseline LV function with this protocol was not compromised in the conditional KO (Online Table I and data not shown). For MI studies, we wanted to avoid impacting on infarct size so we administered tamoxifen for 5 days, beginning 3 days after occlusion of the proximal left anterior descending coronary artery. An expanded Methods section can be found in the Online Supplement.
Results
Deletion of GSK-3β in the conditional knockout mouse
In order to clarify the role of GSK-3β in pressure overload-induced hypertrophy and post-MI remodeling, we first examined the degree of deletion of GSK-3β in our model. The GSK-3βfl/fl/Cre mice treated with vehicle (flox/Cre/V) demonstrated levels of GSK-3β protein expression that were comparable to the GSK-3βfl/fl mice lacking mer-cre-mer treated with tamoxifen (referred to as flox/−/T). This confirms that there was no significant “leaky” excision of exon 2 in the absence of tamoxifen in our model.
The GSK-3βfl/fl/Cre + tamoxifen treated mice (referred to as flox/Cre/T or KO) had a range of reduction of GSK-3β protein expression in their left ventricles (LV) (Figure 1). Residual GSK-3β protein ranged from 8% to 40% of control GSK-3β expression in our model and the average residual protein was 23% (n = 114 mice). The residual GSK-3β protein expression is due to incomplete recombination and the presence of non-myocytes in the hearts. Mice that had less than 30% residual GSK-3β protein expression by western blot were included in the study.
Figure 1. GSK-3β protein expression in the conditional KO mice.

Representative immunoblot of GSK-3β expression in conditional KO mice and controls. Five week old male mice received intra-peritoneal (IP) injection of tamoxifen (20mg/kg) as described in Methods. Pathological hypertrophy was induced at 7 weeks of age by TAC. Three weeks post TAC, expression of GSK-3β in the LV was determined. The three groups were as follows:
1) GSK-3βfl/fl, lacking mer-cre-mer (Cre −), treated with tamoxifen (tamoxifen +); n=87; (Control 1, lanes 1–3), hereinafter referred to as flox/−/T.
2) GSK-3βfl/fl, Cre+, tamoxifen +; n=114 (conditional KO, lanes 4–9), referred to as flox/Cre/T.
3) GSK-3βfl/fl, Cre+, treated with vehicle (tamoxifen −); n=17; (Control 2, lanes 10–11), referred to as flox/Cre/V.
GAPDH was used as a loading control.
Effect of deletion of GSK-3β on pressure-overload hypertrophy
The mice then underwent thoracic aortic constriction (TAC). Three weeks after TAC, mice were analyzed for cardiac function and morphometrics. Mice were catheterized with a high fidelity pressure transducer to determine the left ventricular systolic pressure (LVSP) (Online Table I). LVSP was comparable among the three groups, suggesting that the degree of TAC was also comparable in the groups. Heart weight (HW), left ventricle mass (LVM), and body weight (BW) were determined, and calculated HW/BW and LVM/BW (the primary morphometric measures of hypertrophy) were calculated. There were no significant differences in any of these parameters among the three groups that underwent TAC (Table 1). We then analyzed HW/BW ratio in relation to LVSP (Figure 2A). The regression lines relating LVSP to HW/BW were virtually identical, consistent with deletion of GSK-3β having little or no effect on the hypertrophic response. Next, we compared hypertrophy in mice with lower vs higher LVSPs to see if deletion of GSK-3β might modulate hypertrophy differently in these groups (Figure 2B). There was no significant difference in hypertrophy in the KO mice in any of the ranges of LVSP. To examine hypertrophy at the cellular level, we measured cross-sectional area of hematoxylin and eosin (H&E)- stained left ventricular sections and found no significant increase in cardiomyocyte size in the conditional KO (Figure 2C). We also measured the surface area of isolated cardiomyocytes and found no difference between the conditional KO and the control groups (Online Figure I). Finally, we examined the fetal gene program and found that expression of atrial natriuretic peptide (ANP) and β-myosin heavy chain (β-MHC) were approximately 5-fold higher in the control compared to the KO (Online Figure IIA). These findings, which are contrary to numerous studies employing transgenesis, suggest GSK-3β is not a central regulator of the hypertrophic response to pressure overload, whether the pressure load is mild, moderate or severe.
Table 1.
Morphometric measurements in the various groups 3 weeks post TAC or sham TAC.
| n | HW (mg) | BW (g) | LVM (mg) | HW/BW(mg/g) | LVM/BW(mg/g) | |
|---|---|---|---|---|---|---|
| Flox/−/T +TAC | 26 | 114 ± 11 | 23.2 ± 1.3 | 68 ± 17 | 5.1 ± 0.7 | 3.0 ± 0.7 |
| Flox/Cre/T +TAC (KO) | 26 | 120 ± 23 | 22.0 ± 2.0 | 70 ± 19 | 5.4 ± 1.0 | 3.4 ± 0.9 |
| Flox/Cre/V + TAC | 11 | 115 ± 17 | 23.1 ± 2.8 | 62 ± 10 | 5.0 ± 0.5 | 3.0 ± 0.4 |
| Flox/−/T + Sham | 5 | 100 ± 16 | 21.9 ± 1.0 | 52 ± 6.1 | 4.6 ± 1.0 | 2.4 ± 0.3 |
| Flox/Cre/T + Sham (KO) | 6 | 99 ± 16 | 22.0 ± 3.0 | 54 ± 7.8 | 4.5 ± 0.5 | 2.7 ± 0.4 |
HW, heart weight; BW, body weight; LVM, LV mass. All morphometric measurements (HW, BW, LVM, HW/BW, LVM/BW) from TAC groups were significantly different from sham groups (p < 0.05).
Figure 2. Deletion of GSK-3β does not alter the hypertrophic response to TAC.



A) Relationship between left ventricular systolic pressure (LVSP) and HW/BW in flox/Cre/T mice and flox/−/T mice subjected to TAC. At 3 weeks post TAC, mice underwent invasive hemodynamic assessment to determine LVSP. The regression lines relating LVSP to HW/BW were not significantly different between the groups, suggesting deletion of GSK-3β did not affect the hypertrophic response. For flox/Cre/T (KO) mice r = 0.373; for flox/−/T mice r = 0.386. n = 26 mice in each group.
B) Hypertrophy in mice with mild, moderate, or severe TAC. Mice were subjected to TAC and HW/BW was determined at sacrifice. There is no difference in hypertrophy between KO and controls at any LVSP. Mean LVSPs for the groups are shown below the figure.
C) Representative H&E-stained LV sections from mice with systolic pressures between 150–180mmHg, 3 weeks post TAC. Quantification of myocyte cross-sectional area is shown in the graph below. Cardiomyocyte size was not significantly different between the two control groups (flox/−/T mice and flox/Cre/V mice); therefore, these data were grouped together. n = 200 cardiomyocytes/mouse with 8 mice as controls and 6 conditional KO mice.
GSK-3β deletion does not affect cardiac function or extracellular matrix remodeling after pressure overload stress
We also assessed cardiac function by echocardiography both prior to and 3 weeks post TAC (Online Table II). As noted, there was no significant difference in the ejection fraction (EF) between the groups before TAC. Furthermore, there was no difference in EF between groups of mice exposed to more severe pressure overload (LVSP 150–190mmHg) (Online Figure IIIA) or in mice with less severe pressure overload (LVSP = 120–150mmHg) (Online Figure IIIB). Analysis of contractility also failed to detect any differences between the groups in + dP/dt (a more sensitive measure of contractility) and −dP/dt (a sensitive measure of relaxation)(Online Table I).
Studies have suggested that GSK-3β is anti-fibrotic30, 33. However, we found no difference in the percent area of fibrosis between the treatment groups in LVSP-matched hearts (150–180mmHg) at 3 weeks post TAC (Online Figure IV). Therefore, deletion of GSK-3β in cardiomyocytes does not appear to affect fibrosis, at least in the early phases of the response to pressure overload in the heart.
GSK-3β deletion affects several signaling pathways
Taken together, the data presented above suggest two possibilities: 1) the small amount of residual expression of GSK-3β in the conditional KO is sufficient to maintain normal functions of GSK-3β, or 2) GSK-3β plays at most a minor role in the response to pressure overload. Therefore we examined signaling pathways regulated by GSK-3β reasoning that if possibility 1 were the case, signaling pathways would not be significantly dysregulated in the KO. We first analyzed phosphorylation of glycogen synthase at serine 641 and found a striking reduction in the KO (Figure 3). This finding identifies GSK-3β as the dominant isoform regulating glycogen synthesis in the heart, but more importantly demonstrates that the magnitude of reduction in GSK-3β expression levels is sufficient to lead to dysregulation of signaling pathways.
Figure 3. GSK3β regulates glycogen synthase.
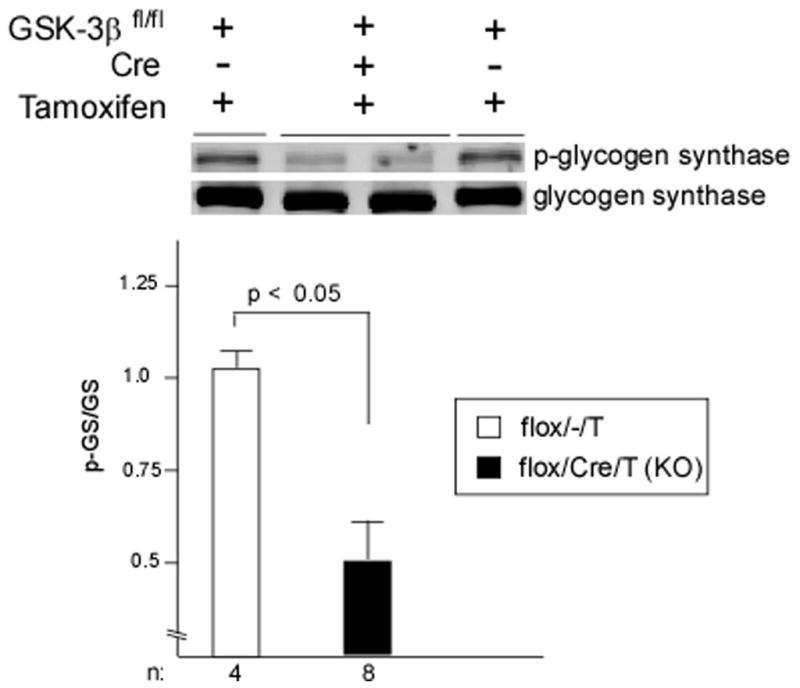
Immunoblot of phospho-glycogen synthase (p-GS) and quantification in conditional KO and control. LV lysates from flox/Cre/T and flox/−/T mice fasted for 24 hours prior to sacrifice (to maximize phosphorylation of GS) show significantly decreased p-GS in mice with GSK-3β deleted. In the graph, p-GS level was normalized to total GS.
To explore this further, we examined activity status of GSK-3α and downstream targets of GSK-3β known to play a role in the hypertrophic response. We found that GSK-3α protein levels and activity level (the latter as determined by phosphorylation at serine 21) were not different between GSK-3β KO and WT hearts, confirming no compensation (data not shown). Further, we examined the status of several GSK-3β targets that are known regulators of hypertrophy. We found a statistically significant decrease in phosphorylation of eiF2B (a direct target of GSK-3β), and an increase in c-myc expression (which is normally down-regulated by GSK-3β-triggered proteasomal degradation) in LV lysates obtained 24 hours post-TAC (Online Figures VA, B.). Thus, the decreased levels of GSK-3β were impacting on target activity, but this was not sufficient to lead to enhanced hypertrophy. One reason for this may be that deletion of GSK-3β did not lead to activation of mTORC1, a central regulator of the hypertrophic response (data not shown). In summary, we conclude that GSK-3β is not a central regulator of PO-induced hypertrophy.
GSK-3β regulates post-MI remodeling
We next asked whether GSK-3β might regulate the other major form of remodeling in the heart: post-MI remodeling. We subjected KO mice and controls to permanent ligation of the proximal LAD. Since GSK-3β has been implicated in regulating acute ischemic injury (reviewed in 34, 35), and we specifically wanted to examine post-MI remodeling, tamoxifen (or vehicle) injections were begun 3d post MI, at a time when the infarct was completed. We followed the mice for 8wks with periodic echocardiography. The infarcts produced were quite extensive in size (Figure 4A). Of note, the absolute length of the scar was not different between the KO and various controls, suggesting infarct size was comparable among the groups (KO: 1226 ± 64 pixels, n = 7 mice; Control: 1257 ± 88 pixels, n = 12 mice; p = NS). Also supporting this conclusion, ejection fraction was virtually identical among the groups at one week post-MI (Figure 4B). In fact, at one week post-MI, LVEDD was greater in the KO, suggesting infarct size might have been slightly larger in the KO. However, beginning at 2 wks post-MI, LVEF tended to be greater, and LVEDD tended to be smaller in the KO (Figure 4B). At 4 wks post-MI and after, LVEF was better maintained in the KO, and at ≥ 6 weeks, LV dilatation (LVEDD) was significantly less. Thus deletion of GSK-3β protects against post-MI remodeling.
Figure 4. Post-MI remodeling in the GSK-3β conditional KO.


A. Representative images of sagital sections of Control (flox/−/T) and KO hearts at 8 weeks post-MI.
B. Echocardiographic findings in the post-MI heart. KO and the various controls underwent baseline echo examination and then were subjected to occlusion of the proximal LAD. Follow-up echo examinations were done at the times shown and ejection fraction and LVEDD were determined at each time-point. There were no significant differences in either parameter for the three control groups. p values reflect the following comparisons:
P1: KO vs. flox/Cre/V
P2: KO vs flox/−/V
P3: KO vs flox/−/T
There were n = 32 KOs; 32 flox/Cre/V-; 31 flox/−/V-; 31 flox/−/T
We wanted to examine contractile function more closely, so we performed invasive hemodynamic testing at 8 wks post-MI. For these studies we present the various controls as one group since there were no significant differences between them in any parameter. We found no significant differences between KO and controls in LVSP or LVEDP, but +dP/dt and −dP/dt were significantly increased in the KO (Figure 5A–D). Thus LV function, both systolic and diastolic, are better maintained in the KO.
Figure 5. Hemodynamic parameters at 8 weeks post-MI.
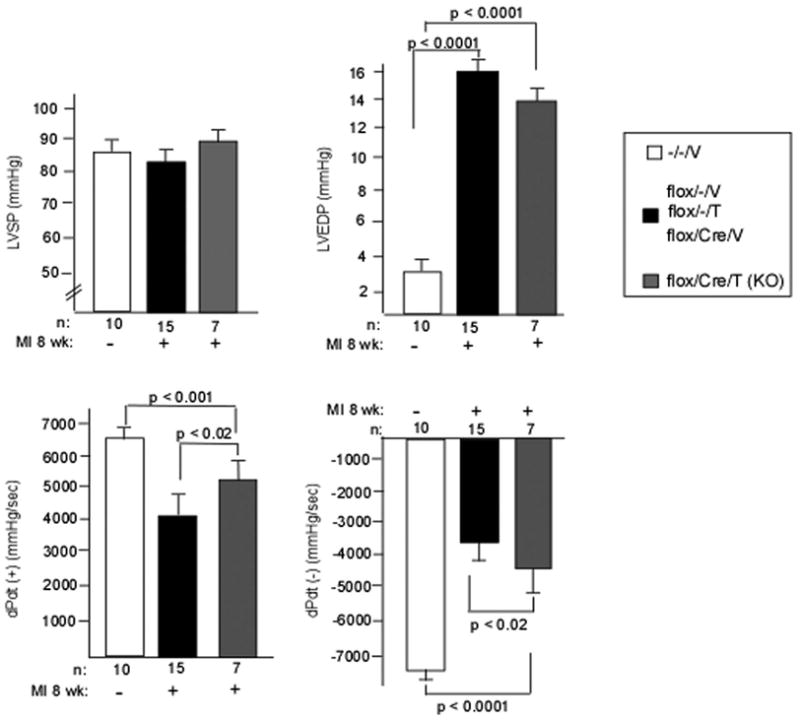
LVSP (panel A), LVEDP (panel B), +dP/dt (panel C), and −dP/dt (panel D) were determined in the KO and various controls by catheterization at 8 weeks post-MI as described in Methods. Sham (MI-) is also shown. Again, there were no differences between the various controls so they were grouped together.
We next wanted to understand potential mechanisms of this protective effect of deletion of GSK-3β. Since smaller infarcts did not seem to account for the differences, we focused on the non-infarcted remote myocardium. Surprisingly, we found that overall HW/BW was significantly increased in the KO, suggesting that in the setting of post-MI remodeling, as opposed to TAC, GSK-3β is indeed anti-hypertrophic (Figure 6A). This increased hypertrophy was due, in large part, to cardiomyocyte hypertrophy since cross-sectional area of the KO myocytes was significantly increased (Figure 6B). Despite the increased hypertrophy in the KO, fibrosis was not increased (data not shown) and apoptosis was significantly reduced (Figure 6C). As for the fetal gene program, β-MHC was increased in the KO, consistent with the increased hypertrophy, but ANP and BNP, which are also markers of hemodynamic stress, were not increased in the KO (Online Figure IIB). The above data, taken together, suggest that the hypertrophy seen in the remote myocardium of the KO is more consistent with physiologic, as opposed to pathologic, hypertrophy.
Figure 6. Characterization of the remote myocardium of the KO.



A/B. Hypertrophy in the remote myocardium. KO and various controls were subjected to MI vs. sham surgery (MI -). At 8 weeks post MI, HW/BW (panel A) and cardiomyocyte cross-sectional area (CSA; panel B) were determined. n’s for each group are shown below the graph. HW/BW and CSA were significantly greater in the KO compared to all other conditions. For determination of CSA, a minimum of 100 cardiomyocytes per heart were measured.
*p < 0.01 vs all other conditions; # p < 0.01 vs all other conditions.
C. Apoptosis in the remote myocardium. Heart sections from the remote myocardium of KO and control mice were stained with troponin I (TnI) and TUNEL, and then counter-stained with DAPI (nuclear stain). Rates of apoptosis are expressed as a percent of total cardiomyocytes (TnI positive cells). n for each group is shown below the graph. A minimum of 700 myocytes per heart were examined.
GSK-3β deletion increases cardiomyocyte proliferation
Deletion of GSK-3β blocks differentiation and promotes proliferation in mouse ES cells in culture.9, 14, 36 Although a small molecule inhibitor of GSK-3 has been reported to lead to increased proliferation of neonatal rat ventricular myocytes (NRVMs) and adult rat ventricular myocytes (ARVMs) in culture37, the only in vivo study to date, which utilized expression of a constitutively active mutant, reported that GSK-3β did not regulate cardiomyoycte proliferation in the adult heart.30 To examine this further, we analyzed cardiomyocyte proliferation in LV heart sections both post TAC and post-MI by injecting mice with bromodeoxyuridine (BrdU) and then immuno-staining sections for BrdU. In the KO mice subjected to TAC, we found a significant increase in cells that were positive for BrdU (Figure 7A). All of the BrdU positive nuclei overlapped with DRAQ5, a nuclear stain. Confocal analysis confirmed that the majority of the proliferating cells were from a cardiomyocyte lineage, based on positive staining for TnI (Figure 7B). The increased cardiomyocyte proliferation in the KO was confirmed with staining for the M-phase marker, phosphorylated histone H3. Rates of positivity with this technique were 0.63 ± 0.11% for KO (n = 7 mice); 0.07 ± 0.03% for flox/−/T (n = 5 mice); and 0.05 ± 0.03 for flox/Cre/V (n = 3 mice) p < 0.001 for KO vs. both controls. We also examined cardiomyocyte proliferation in the KO hearts post-MI (at 4 weeks) and found a similar increase in BrdU positive/TnI positive cells compared to controls (Figure 7C). Thus, GSK-3β inhibits cardiomyocyte proliferation in the stressed adult heart.
Figure 7. GSK-3β deletion increases cardiomyocyte proliferation.



A) Cardiomyocyte proliferation in the post-TAC heart. There is a significant increase in BrdU positive nuclei in conditional KO hearts subjected to TAC, compared to the various controls. There was also a small, but statistically significant, increase in BrdU positive nuclei in the conditional KO mice subjected to sham surgery vs. control mice undergoing sham surgery. BrdU positive cardiomyocytes are expressed as a percent of total cardiomyocytes.
B) Representative image of a KO heart section immunostained with BrdU and TnI, and with DRAQ5. Images are from a confocal Z-stack of a KO mouse 3 weeks post TAC. Top. Sections were stained with BrdU and counter-stained with DRAQ5 and the merged image is also shown. Bottom. Sections were stained with BrdU and TnI. Arrows highlight a BrdU positive nucleus that is within the borders of a TnI-positive cardiomyocyte.
C) Cardiomyocyte proliferation in the post-MI heart. KO and the various controls underwent LAD occlusion. Four weeks later mice were injected twice daily with BrdU for 5 days prior to sacrifice and immunostaining for BrdU, TnI, and counterstain with DAPI as described in Methods.
n’s are shown below the graph. Twelve separate areas of each heart section were examined for each mouse.
Discussion
Our results indicate that GSK-3β does not play a central role in regulating the hypertrophic response to pressure overload but does inhibit hypertrophy following MI. Following deletion of GSK-3β, despite the fact that hypertrophy is increased in the remote myocardium, apoptosis is reduced and cardiomyocyte proliferation is enhanced. At the organ level, LV dilatation is reduced and LV function is better-preserved.
Previous studies point to GSK-3β as a critical regulator of cardiac hypertrophy.4, 15, 19, 25–31 (and reviewed in1, 5), but all of the in vivo models used to reach this conclusion involved over-expression of WT, CA, or DN mutants or knock-in of constitutively active mutants. Transgenic models can lead to non-physiologic protein-protein interactions, aberrant sub-cellular localization, and phosphorylation of protein substrates that are not physiologic substrates. In addition, over-expressed GSK-3β can subsume the physiological roles of GSK-3α, making it impossible to decipher isoform-specific functions. Finally, the constitutively active GSK-3β mutant (with a Ser 9 to Ala mutation) can still be inhibited by the canonical Wnt pathway and by a novel p38-MAPK mediated mechanism, clouding interpretation of findings.38–40
To attempt to clarify distinct GSK-3β isoform functions in pathological hypertrophy, we employed a cardiac-specific, inducible GSK-3β knockout. Our model allows temporal control of GSK-3β deletion so we can specifically focus on the role of GSK-3β in the adult. We restricted our studies to mice with greater than 70% knockdown of GSK-3β protein expression in the heart. Average residual GSK-3β expression in these mice was ~20%. Considering that non-myocytes account for roughly 10% of heart mass, and GSK-3β is not deleted in these cells, residual GSK-3β expression in cardiomyocytes is more likely about 10–15% of WT.
In the conditional KO, we did not see the expected exaggerated PO-induced hypertrophic phenotype. This is likely not due to inadequate deletion of GSK-3β since this level of deletion of GSK-3β did lead to increased hypertrophy in the post-MI model. This dichotomy between hypertrophic responses following TAC vs. MI clearly suggests a complex pattern of isoform specificity vs redundancy for GSK-3α and GSK-3β in regulating these processes.10 In the case of PO-induced hypertrophy, GSK-3β is not a central regulator.
In distinct contrast, GSK-3β appears to be the dominant isoform regulating post-MI hypertrophy and remodeling. Although infarct sizes were virtually identical in WT vs. KO, hypertrophy was increased in the remote myocardium of the KO. The underlying mechanisms by which GSK-3β mediates post-MI hypertrophy but not PO hypertrophy are not clear but the data point to a very different milieu of cytokines, signaling factors, etc. recruited by the different hypertrophic stimuli. Of note, given the reduction in LV dilatation, better-preserved EF, no increase in fibrosis, and no increase in ANP or BNP in the KO, this hypertrophy may be viewed as more “physiological” than “pathological.”
The third key finding of this paper is that GSK-3β regulates cardiomyocyte proliferation. One prior study concluded that GSK-3β regulates cardiomyocyte proliferation; however, this study was done in vitro and employed a commercially available small molecule inhibitor37 (BIO). This agent has a poorly-defined selectivity profile and also inhibits both isoforms of GSK-3, making it impossible to decipher isoform-specific effects.
Herein, we found that deletion of GSK-3β in cardiomyocytes in vivo in the setting of pressure overload led to a 3- to 4-fold increase in BrdU positive cardiomyocytes compared to control mice subjected to TAC. In the post-MI KO hearts, absolute rates of BrdU positivity were higher compared to TAC, but the relative fold increase in KO compared to controls was similar. It is impossible at this time to determine what, if any, role this enhanced proliferation had on the phenotypes of the KOs, but it is clear that GSK-3β deletion increases cardiomyocyte proliferation in the setting of pressure overload and MI just as it does in ES cells and in cardiomyocytes in the developing heart.14 We did not examine proliferation in progenitor cell populations committed to the cardiomyocyte lineage, but it seems likely that cell cycling in these populations would be increased as well and would contribute to the BrdU+/TnI+ population we measured.
Our findings may have potential implications for treatment of patients post-MI and for regenerative therapies. Traditional therapeutics to prevent post-MI remodeling following a large MI (e.g. angiotensin converting enzyme inhibitors (ACEI) and angiotensin receptor antagonists) are effective to some degree but progression to congestive heart failure or death, despite standard approaches, is common. Deletion of GSK-3β beginning ~ 1 week post MI partially prevented the decline in EF, +dP/dt, and −dP/dt. Thus while the difference in LVEF in our study at 8wks post-MI was modest (6–7 EF points), it is important to note that in the large ACEI trials such as SOLVD, SAVE, and GISSI-3, even a 5 point difference in EF was associated with a significant reduction in mortality.41–43 Mortality at 8 weeks post-MI in our series was reduced by 25% in the knockout, although this did not achieve statistical significance by Kaplan-Meier analysis. Maybe more importantly, LV dilatation is a major predictor of progression to CHF and death. Whereas the LVEDD in the WT mice increased by 78%, the increase in the KO was only 48%, consistent with much less remodeling. Again, in the large clinical trials, increases of ~10% in LV volume were associated with significantly worse outcomes.41–43 While direct comparisons between our studies and clinical trials are obviously impossible, it is important to understand how critical even modest differences in LVEF and LV volume can be. Given the near-total lack of important drug development in heart failure in the past 10+ years, novel strategies need to be identified. It is conceivable that GSK-3β inhibition could be one such strategy.
Finally, the findings related to GSK-3 isoform specificity vs redundancy in function could translate to superior strategies for regenerative therapies. Cardiomyocyte proliferation occurs at a very low level throughout life44, but increases in the heart in response to stress (reviewed in 45–46). Cardiac progenitor cells and/or small immature cardiomyocytes proliferate following pressure overload and myocardial infarction. The current beliefs held in the field are that while these cells proliferate and progenitor cells do differentiate into cardiomyocytes, this occurs at too slow of a rate to keep up with the rate of cell death so that the net decrease of cells plays a role in the progression of heart failure. Strategies to manipulate progenitor cell behavior in situ by modulating activity of various kinases are being explored.47, 48 GSK-3β inhibition could increase cardiomyocyte proliferation and perhaps keep pace with cell death rates, thereby preventing progression to heart failure. In addition, based on our earlier findings which show that inhibition of GSK-β promotes ES cell proliferation whereas active GSK-β promotes differentiation into the cardiomyocyte lineage,9, 10, 14, 49 periodic withdrawal of a GSK-β inhibitor could help drive progenitor differentiation into cardiomyocytes. That said, targeting both GSK-3α and GSK-β long-term with small molecule inhibitors raises concerns over promoting cancer by enhancing β-catenin signaling. Selective targeting of individual isoforms (i.e. GSK-3β) could, however, maintain normal β-catenin signaling9 while allowing increased cardiomyocyte proliferation.
Novelty and Significance.
What is known?
Large myocardial infarctions typically progress to remodeling of the ventricle and, in many cases, to heart failure.
Molecular mechanisms underlying these processes are only beginning to be identified.
Current therapeutic options for the sub-acute to chronic phases of MI are limited.
What new information does this article contribute?
We identify GSK-3β as one key factor that drives the post-MI remodeling process; we find that deleting GSK-3β preserves LV structure and function.
We also find that deletion of GSK-3β leads to proliferation of cardiomyocytes in situ.
Contrary to numerous prior publications, we find that GSK-3β does not appear to importantly modulate pressure overload hypertrophy.
The molecular mechanisms that regulate the adverse remodeling that occurs following an MI are not known. Identification of these mechanisms could lead to novel therapeutic strategies to limit remodeling and progression to heart failure. In the first studies to examine the role of GSK-3β in the heart using gene targeting, we report that deletion of GSK-3β specifically in cardiomyocytes limits adverse remodeling and leads to cardiomyocyte proliferation post-MI. In contrast, despite numerous publications that have employed transgenesis to suggest that GSK-3β regulates the hypertrophic response to pressure overload, we find that GSK-3β plays at most a minor role in this process. Thus we have identified a unique role of the β isoform of GSK-3 in the heart, which could have direct implications for patient care. Specifically, small molecule inhibitors of GSK-3 could be used in the post-infarct period to reduce remodeling and possibly to induce reparative cardiomyocyte proliferation. Given the limited treatment options currently available for patients who have suffered a large MI, and the dearth of new classes of drugs to treat these patients beyond the acute phase, GSK-3β inhibition could offer a novel strategy to prevent remodeling and heart failure.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by grants HL061688 from the U.S. National Heart Lung and Blood Institute and by donations from The Kahn Foundation and the Scarperi family (to T.F.), the Canadian Institutes of Health Research Grants MOP12858 and 74711 (to J.R.W.), and a grant 0715281U from the American Heart Association (to K.C.W.).
Non-Standard Abbreviations and Acronyms
- GSK-3β
Glycogen synthase kinase-3β
- MI
Mycardial infarction
- KO
Knockout
- TAC
Thoracic aortic constriction
- LV
Left ventricle
- RV
Right ventricle
- CA
Constitutively active
- DN
Dominant negative
- α-MHC
α-myosin heavy chain
- LVEDD
Left ventricular end diastolic diameter
- LVESD
Left ventricular end systolic diameter
- FS
Fractional shortening
- EF
Ejection fraction
- LVM
Left ventricular mass
- LVEDV
Left ventricular end diastolic volume
- LVESV
Left ventricular end systolic volume
- PWT
Posterior wall thickness
- TnI
Troponin I
- Fl/Cre/V
GSK-3βfl/fl mice, expressing Cre, treated with vehicle
- Fl/−/T
GSK-3βfl/fl mice treated with tamoxifen
- Fl/Cre/T
GSK-3βfl/fl mice, expressing Cre, treated with tamoxifen
- Fl/−/V
GSK-3βfl/fl mice treated with vehicle
Footnotes
Disclosures
Conflicts of interest: None
References
- 1.Dorn GW, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 3.Hill JA, Karimi M, Kutschke W, Davisson RI, Zimmerman K, Wang Z, Kerber RE, Wieiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 4.Hardt SE, Sadoshima J. Glycogen synthase kinase-3s: a novel regulator of cardiac hypertrophy and development. Circ Res. 2002;90:1055–1063. doi: 10.1161/01.res.0000018952.70505.f1. [DOI] [PubMed] [Google Scholar]
- 5.Sugden PH, Fuller SJ, Weiss SC, Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br J Pharmacol. 2008;153 (Suppl 1):S137–153. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruel L, Bourouis M, Heitzler P, Pantesco V, Simpson P. Drosophila shaggy kinase and rat glycogen synthase kinase-3 have conserved activities and act downstream of Notch. Nature. 1993;362:557–560. doi: 10.1038/362557a0. [DOI] [PubMed] [Google Scholar]
- 7.Siegfried E, Chou TB, Perrimon N. Wingless signaling acts through zeste-white 3, the Drosophila homolog of glycogen synthase kinase-3, to regulate engrailed and establish cell fate. Cell. 1992;71:1167–1179. doi: 10.1016/s0092-8674(05)80065-0. [DOI] [PubMed] [Google Scholar]
- 8.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Force T, Woodgett JR. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J Biol Chem. 2009;284:9643–9647. doi: 10.1074/jbc.R800077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314–6328. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanabe K, Liu Z, Patel S, Doble BW, Li L, Cras-Meneur C, Martinez SC, Welling CM, White MF, Bernal-Mizrachi E, Woodgett JR, Permutt MA. Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol. 2008;6:e37. doi: 10.1371/journal.pbio.0060037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T, Huggins GS. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest. 2008;118:3609–3618. doi: 10.1172/JCI36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardt SE, Tomita H, Katus HA, Sadoshima J. Phosphorylation of eukaryotic translation initiation factor 2Bε by glycogen synthase kinase-3β regulates b-adrenergic cardiac myocytes hypertrophy. Circ Res. 2004;94:926–935. doi: 10.1161/01.RES.0000124977.59827.80. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Shevtsov S, Hsich E, Cui L, Haq S, Aronovitz MJ, Kerkela R, Molkentin JD, Liao R, Salomon R, Patten RD, Force T. The β-catenin/T-cell factor/Lymphocyte enhancer factor signaling pathway is required for normal and stress-induced cardiac hypertrophy. Mol Cell Biol. 2006;26:4462–4473. doi: 10.1128/MCB.02157-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haq S, Michael A, Andreucci M, Bhattacharya K, Dotto P, Walters B, Woodgett J, Kilter H, Force T. Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci U S A. 2003;100:4610–4615. doi: 10.1073/pnas.0835895100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2502–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haq S, Choukroun G, Kang ZB, Lee K-H, Ranu H, Matsui T, Rosenzweig A, Alessandrini A, Molkentin JD, Woodgett J, Hajjar R, Force T. Glycogen synthase kinase-3s is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151:117–129. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 21.Proud CG. Ras, PI3-kinases and mTOR signaling in cardiac hypertrophy. Cardiovasc Res. 2004;63:403–413. doi: 10.1016/j.cardiores.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3s regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase kinase-3s suppresses cardiac hypertrophy in vivo. Proc Nat Acad Sci. 2002;99:907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michael A, Haq S, Chen X, Hsich E, Cui L, Walters B, Shao Z, Bhattacharya K, Kilter H, Huggins G, Andreucci M, Periasamy M, Solomon RN, Liao R, Patten R, Molkentin JD, Force T. Glycogen synthase kinase-3beta regulates growth, calcium homeostasis, and diastolic function in the heart. J Biol Chem. 2004;279:21383–21393. doi: 10.1074/jbc.M401413200. [DOI] [PubMed] [Google Scholar]
- 27.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- 28.Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 29.Hirotani S, Zhai P, Tomita H, Galeotti J, Marquez JP, Gao S, Hong C, Yatani A, Avila J, Sadoshima J. Inhibition of glycogen synthase kinase 3beta during heart failure is protective. Circ Res. 2007;101:1164–1174. doi: 10.1161/CIRCRESAHA.107.160614. [DOI] [PubMed] [Google Scholar]
- 30.Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto K, Takagi H, Tamamori-Adachi M, Kitajima S, Sadoshima J. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci U S A. 2008;105:20900–20905. doi: 10.1073/pnas.0808315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhai P, Gao S, Holle E, Yu X, Yatani A, Wagner T, Sadoshima J. Glycogen synthase kinase-3alpha reduces cardiac growth and pressure overload-induced cardiac hypertrophy by inhibition of extracellular signal-regulated kinases. J Biol Chem. 2007;282:33181–33191. doi: 10.1074/jbc.M705133200. [DOI] [PubMed] [Google Scholar]
- 32.Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ Res. 2009;105:12–15. doi: 10.1161/CIRCRESAHA.109.198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maes C, Goossens S, Bartunkova S, Drogat B, Coenegrachts L, Stockmans I, Moermans K, Nyabi O, Haigh K, Naessens M, Haenebalcke L, Tuckerman JP, Tjwa M, Carmeliet P, Mandic V, David JP, Behrens A, Nagy A, Carmeliet G, Haigh JJ. Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J. 2010;29:424–441. doi: 10.1038/emboj.2009.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy E, Steenbergen C. Does inhibition of glycogen synthase kinase protect in mice? Circ Res. 2008;103:226–228. doi: 10.1161/CIRCRESAHA.108.181602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doble BW, Woodgett JR. Exploring pluripotency with chemical genetics. Cell Stem Cell. 2009;4:98–100. doi: 10.1016/j.stem.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng AS, Engel FB, Keating MT. The GSK-3 inhibitor BIO promotes proliferation in mammalian cardiomyocytes. Chem Biol. 2006;13:957–963. doi: 10.1016/j.chembiol.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 38.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shapiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signaling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bikkavilli RK, Feigin ME, Malbon CC. p38 mitogen-activated protein kinase regulates canonical Wnt-beta-catenin signaling by inactivation of GSK3beta. J Cell Sci. 2008;121:3598–3607. doi: 10.1242/jcs.032854. [DOI] [PubMed] [Google Scholar]
- 40.Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, Sabio G, Davis RJ, Matthews DE, Doble B, Rincon M. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science. 2008;320:667–670. doi: 10.1126/science.1156037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicolosi GL, Latini R, Marino P, Maggioni AP, Barlera S, Franzosi MG, Geraci E, Santoro L, Tavazzi L, Tognoni G, Vecchio C, Volpi A. The prognostic value of predischarge quantitative two-dimensional echocardiographic measurements and the effects of early lisinopril treatment on left ventricular structure and function after acute myocardial infarction in the GISSI-3 Trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico. Eur Heart J. 1996;17:1646–1656. doi: 10.1093/oxfordjournals.eurheartj.a014747. [DOI] [PubMed] [Google Scholar]
- 42.St John Sutton M, Pfeffer MA, Plappert T, Rouleau JL, Moye LA, Dagenais GR, Lamas GA, Klein M, Sussex B, Goldman S, et al. Quantitative two-dimensional echocardiographic measurements are major predictors of adverse cardiovascular events after acute myocardial infarction. The protective effects of captopril. Circulation. 1994;89:68–75. doi: 10.1161/01.cir.89.1.68. [DOI] [PubMed] [Google Scholar]
- 43.Greenberg B, Quinones MA, Koilpillai C, Limacher M, Shindler D, Benedict C, Shelton B. Effects of long-term enalapril therapy on cardiac structure and function in patients with left ventricular dysfunction. Results of the SOLVD echocardiography substudy. Circulation. 1995;91:2573–2581. doi: 10.1161/01.cir.91.10.2573. [DOI] [PubMed] [Google Scholar]
- 44.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kajstura J, Urbanek K, Rota M, Bearzi C, Hosoda T, Bolli R, Anversa P, Leri A. Cardiac stem cells and myocardial disease. J Mol Cell Cardiol. 2008;45:505–513. doi: 10.1016/j.yjmcc.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 46.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–856. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 47.Cottage CT, Bailey B, Fischer KM, Avitable D, Collins B, Tuck S, Quijada P, Gude N, Alvarez R, Muraski J, Sussman MA. Cardiac Progenitor Cell Cycling Stimulated by Pim-1 Kinase. Circ Res. 2010 Jan 14; doi: 10.1161/CIRCRESAHA.109.208629. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sussman M. “AKT”ing lessons for stem cells: regulation of cardiac myocyte and progenitor cell proliferation. Trends Cardiovasc Med. 2007;17:235–240. doi: 10.1016/j.tcm.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.