

Abstract
The study of membrane protein structure and enzymology has traditionally been hampered by the inherent insolubility of membrane proteins in aqueous environments and experimental challenges in emulating an in vivo lipid environment. Phospholipid bilayer nanodiscs have recently been shown to be of great use for the study of membrane proteins since they offer a controllable, stable, and monodisperse model membrane with a native-like lipid bilayer. Here we report the integration of nanodiscs with hydrogen exchange (HX) mass spectrometry (MS) experiments, thereby allowing for analysis of the native conformation of membrane proteins. Gamma-glutamyl carboxylase (GGCX), an ~94 kDa transmembrane protein, was inserted into nanodiscs and labeled with deuterium oxide under native conditions. Analytical parameters including sample-handling and chromatographic separation were optimized to measure the incorporation of deuterium into GGCX. Coupling nanodisc technology with HX MS offers an effective approach for investigating the conformation and dynamics of membrane proteins in their native environment and is therefore capable of providing much needed insight into the function of membrane proteins.
Keywords: deuterium, gamma-glutamyl carboxylase, protein conformation
One of the great challenges in structural biology has been (and remains) the study of membrane proteins. Membrane proteins play a role in numerous biological processes including signal transduction, transport, and a variety of enzymatic and metabolic pathways. Integral membrane proteins alone are estimated to account for up to 30% of the human proteome.1, 2 Further, membrane proteins are the most widely targeted class of proteins for current and emerging drug therapies; 60% of drug targets are located on the surface of the plasma membrane3. Despite their abundance, there are only ~180 unique structures of membrane proteins in the PDB4 and their biophysical characterization in general is significantly underrepresented in the literature. Difficulties accompanying in vitro reproduction of the naturally occurring lipid environment of membrane proteins under conditions that are also compatible with common biophysical methods for protein characterization has significantly hindered interrogation of membrane proteins.
In in vitro systems, membrane proteins are most often studied in detergent micelles or liposomes but these in vitro systems have a number of limitations. As model membranes, micelles have the inherent disadvantage of being unilamellar which can produce a poorly defined membrane environment. Micelles are also prone to self-assembly5 and oftentimes the structure and activity of a protein is perturbed in the presence of detergent resulting in inactive or denatured entities.6–8 Liposomes, on the other hand, are vesicles of amphiphilic lipid bilayers and may form small (SUV) or large (LUV) unilamellar vesicles in addition to multilamellar vesicles (MLVs) with several concentric bilayers. Numerous methods of liposome preparation have been described,9 however, most of these procedures generate a heterogeneous population of vesicles resulting in unavoidable variability in subsequent analyses. Although liposomes have the advantage of providing a compartmentalized membrane environment, the challenges involved with aggregate formation, controlled stoichiometry, polydispersity, and maintaining the correct membrane protein directionality add another layer of variability when investigating the conformational properties of membrane proteins. For these reasons, alternative techniques for in vitro preparation of membrane proteins have been pursued.
Nanodiscs, first pioneered by the Sligar laboratory,10, 11 offer a unique method for addressing the challenges of studying membrane proteins in a more physiologically-relevant context. In this system, a target membrane protein is transferred to a monodisperse and reproducible phospholipid environment maintained by a membrane scaffold protein (MSP). MSP’s are modeled after apolipoprotein A-1 which transports lipids by the formation of high density lipoprotein (HDL) particles.12 Upon removal of detergent from a solubilized lipid mixture, the target membrane protein self-assembles with the phospholipid into a discoidal bilayer encircled by the MSP. Two-copies of the MSP wrap around the periphery of the phospholipid bilayer domain to stabilize the nanodisc structure in a belt-like configuration (Figure 1), where the size of the nanodisc is strictly controlled by the length of the MSP protein.11, 13, 14 The resulting structure creates a uniformly sized nanoscale bilayer which mimics a stabilized native environment for a target membrane protein of choice to be incorporated.15 Multiple target proteins in nanodiscs have been studied including bacteriorhodopsin,16 tissue factor,17 and cytochrome P450.18
Figure 1.

The integrated nanodisc-HX MS workflow. Loaded nanodiscs were assembled from a mixture of membrane scaffold protein (MSP), lipids, and the target membrane protein solubilized with detergent. Nanodiscs self-assembles as detergent was removed. Loaded nanodiscs were purified with SEC (see Supporting Information, Figure S1a) and exposed to deuterated buffer for various times before quenching the exchange reaction. Cholate was immediately added to the quenched reaction to begin disassembly of the nanodiscs. Protein was digested with pepsin for 5 minutes on ice. In the last minute of digestion, ZrO2 resin was added to the digestion mixture to selectively remove phospholipid. Filtration removed immobilized-pepsin beads and the ZrO2 resin. UPLC/ESIMS were used to measure the incorporation of deuterium.
Although it is highly desirable to use X-ray crystallography and NMR for structural determination and analysis of proteins, these methods are not yet routinely used for membrane proteins, even those embedded in nanodiscs. The nanodisc structure does not readily crystallize and the size of the molecular assembly (nanodisc + membrane protein) has so far limited NMR analyses (several recent examples have applied solid-state NMR and magic-angle spinning 19, 20). Thus, despite the ability of nanodisc technology to preserve membrane proteins in a physiologically-relevant environment, few nanodisc-embedded membrane proteins have been subjected to detailed structural investigation.
Other biophysical methods offer the potential to provide a great deal of information about membrane proteins in nanodiscs, although not at the level of detail of x-ray crystallography or NMR. To extend the analysis of membrane protein conformation and dynamics, we have combined nanodisc technology with hydrogen exchange (HX) mass spectrometry (MS).21, 22 Relative to other methods for membrane protein characterization, HX MS provides some advantages, including low sample and concentration requirements as well as peptide level or even single-residue resolution.23 Previous studies investigating membrane proteins by HX MS have involved lipid vesicles or detergent micelles (for example, Refs 24–30), and confront the challenges described above. We sought to design a general method that integrates the advantages of nanodiscs with the benefits of HX MS. To ensure method compatibility with large and complex membrane proteins, we employed a model system of nanodiscs loaded with human gamma-glutamyl carboxylase (GGCX). This 94 kDa (including all modifications, such as the five N-linked glycosylations), is a 5-pass transmembrane protein and a key regulator of blood coagulation31. However, little detailed structural information is so far available for GGCX.
Figure 1 outlines our final, optimized method flow for HX MS analysis of a membrane protein embedded in a nanodisc. Nanodiscs loaded with GGCX were prepared by a similar procedure as has been described for other protein embedded nanodiscs.16, 32 Briefly (see Supporting Information for detailed Materials and Methods), a phospholipid/detergent solubilized mixture was added to purified GGCX and allowed to incubate for one hour at 4°C. Purified MSP protein was added to the reconstitution mixture in a 20:1 MSP:GGCX active target protein ratio encouraging formation of one target molecule per nanodisc. To initiate nanodisc self-assembly, detergent was removed with a 2 hour gentle rotation over damp Biobeads SM-2 (BioRad) at 4°C (see Supporting Information for experimental details). The disk preparations were purified by size exclusion chromatography to separate loaded discs from unloaded discs (see Supporting Information, Figure S1A). SDS-PAGE was used to confirm the presence and purity of GGCX containing nanodiscs and fractions containing purified nanodiscs were isolated and concentrated (see Supporting Information, Figure S1B). Deuterium labeling of the nanodisc-embedded GGCX membrane protein was performed according to standard HX practices 21, 22, 33: purified nanodisc samples were diluted into deuterated buffer to initiate the exchange reaction and the reaction was quenched at defined time-intervals by acidification. The resulting mixture contained lipids, digested MSP and digested membrane protein; therefore, high-quality mass measurement of the peptides from the membrane protein required significant sample cleanup, digestion enhancement and chromatographic optimization. Because deuterium loss from the membrane protein to solvent (back-exchange) occurred continually in the quenched sample, despite low pH and temperature22, the entire process needed to be performed as quickly as possible after quenching to retain as much deuterium as possible. We devised a workflow that makes complex nanodisc samples compatible with mass analysis while meeting the time-limitations of the HX MS experiment. The method used three key steps, all of which were required for ideal performance: (1) disassembly of the nanodisc by the addition of cholate (2) removal of phospholipid from the sample mixture by selective binding to zirconium oxide (ZrO2) beads, and (3) optimized chromatography (UPLC) to separate membrane protein peptides from the interfering scaffold protein peptides.
The final workflow was established after extensive preliminary experiments on empty nanodiscs. HX MS experiments using conventional HPLC separation (i.e., 3.5 micron C-18 particles with 10–50% acetonitrile gradient elution in 15 min) to analyze empty nanodiscs (MSP protein and phospholipid only) showed that MSP peptides eluted by 40% acetonitrile followed by undigested protein at 50% acetonitrile (Fig. 2a, blue trace). The presence of a large signal of undigested protein indicated that the protein component of the nanodisc did not digest well, likely due to inefficient nanodisc disassembly under quench conditions. Previous studies have shown that cholate molecules can destabilize the MSP scaffold protein encapsulating the circular edge of the lipid bilayer.34 We found that addition of cholate to nanodisc samples in a 25:1 cholate to DOPC ratio greatly facilitated the disassembly of nanodiscs, as observed by SEC (data not shown) and in the context of an HX MS experiment. Cholate addition resulted in an overall increase in MSP peptide intensity upon digestion of empty nanodiscs, relative to an identical experiment performed in the absence of cholate (Figure 2a, red and blue traces respectively). The increase in MSP peptide intensity was coupled to a decrease in the amount of undigested protein, further indicative of nanodisc disassembly and enhanced digestion.
Figure 2.

Chromatographic traces of nanodisc peptic digests. (A) HPLC separation of 44 pmol of empty nanodisc (88 pmol MSP) with (red) and without (blue) addition of 400 μM sodium cholate. (B) UPLC separation of 50 pmol loaded nanodiscs (50 pmol GGCX, 100 pmol MSP) with (black) and without (green) addition of 480 μM sodium cholate. The more simple mixture of just empty nanodiscs was not analyzed with UPLC. Rather we went immediately to the much more complex loaded nanodiscs sample (MSP + GGCX mixture), which was the actual mixture that must be analysed for the final hydrogen exchange experiments. UPLC separation of loaded nanodiscs under HX MS analysis conditions included separation of peptic peptides from both MSP (24 kDa) and GGCX (94 kDa); a combined total mass of 118 kDa of unique sequence was digested. The average number of peptides identified in UPLC experiments with and without cholate is shown in the inset. HPLC and UPLC are shown on the same time scale to illustrate that reasonable HPLC performance required a longer gradient; even so, UPLC performance was superior for a more complex sample (loaded nanodisc) in a shorter time. In both chromatography systems, cholate was retained longer than the peptides and did not interfere with peptide ionization and identification (most runs, the UPLC was disconnected from the MS before the bulk of cholate eluted; cholate is shown in this trace for example purposes only.
The presence of large amounts of phospholipid (DOPC) did not result in ion suppression during mass analysis and thus did not directly interfere with analysis of peptides from a membrane protein (also shown previously Ref 27). Consistent with prior observations,35 we found that column lifetime and retention time reproducibility, particularly in UPLC, were drastically reduced by loading the large amounts of DOPC present in digest mixtures from nanodisc samples. This prompted efforts to remove the majority of lipid prior to chromatographic separation and peptide elution into the mass spectrometer. Preliminary experiments using a VanGuard C18 pre-column as a lipid reservoir showed that the guard column offered considerable entrapment of lipid but the lifetime of the guard column was limited to only a few injections (data not shown). Attempts to extract the lipid with organic solvents (i.e., ether, chloroform, etc) showed little efficacy in selectively removing phospholipid from our experimental setup (data not shown). To improve chromatographic performance, lipid removal was facilitated with zirconium oxide. In the presence of lipid, zirconium oxide acts as a Lewis acid that will interact strongly with the phosphate group inherent in all phospholipids.36–38 We observed that adding ZrO2-coated silica beads to quenched samples of digested nanodiscs provided a very rapid means of selectively binding phospholipid released from the discs. Further, the formic acid present in the quench solution acted to neutralize silanol groups and thus improve analyte recovery by preventing non-specific interactions. Incubation with ZrO2 was time-sensitive: at longer incubation periods, peptide adsorption to the silica becomes problematic resulting in peptide loss, whereas at very short intervals, phospholipid trapping was insufficient (data not shown). To balance lipid removal versus unwanted loss of deuterium label on peptides in the protiated solvent of the quench buffer, the optimum ZrO2 incubation time was found to be 1 minute.
HX MS experiments using HPLC chromatography (Figure 2a) revealed that the large abundance of peptides from the scaffold protein hindered the mass analysis of peptides from the membrane protein (two copies of the MSP protein are required to make a nanodisc, thus peptides from MSP will always be present in a 2:1 molar ratio to those of the inserted membrane protein). We therefore evaluated the use of higher performance separations, such as those afforded by UPLC. Using a UPLC system optimized for HX MS39, we observed dramatically improved separation in a shortened time-frame relative to conventional HPLC. This setup enhanced membrane peptide mass analysis by improving peak discrimination from interfering peptides. Figure 2b shows the application of UPLC chromatography to a loaded nanodisc digest with offline cholate disassembly and phospholipid removal by zirconium oxide beads. The superior chromatography attainable by UPLC (compare Figure 2a and b) coupled with efficient nanodisc cholate induced disassembly made it possible to sort out the complex MSP peptide mixture in a time frame (5–8 min) and under temperature conditions (0°C) that minimized undesirable deuterium loss (back-exchange) during the HX-MS experiment. To achieve even reasonable HPLC performance with this system required a much longer gradient and therefore resulted in more back-exchange. UPLC separation (Figure 2b) could be performed on a more complex mixture (MSP+GGCX) in a shorter time and with better resolution than HPLC (Figure 2a) of just one protein (MSP).
Based on the collective optimizations from experiments on empty nanodiscs, we applied our nanodisc-HX MS procedure (see Figure 1) to nanodiscs loaded with the large, transmembrane protein, GGCX. Using the optimized procedure, GGCX embedded in nanodiscs, was labeled with deuterium and efficiently and rapidly digested for mass analysis. Figure 3 illustrates the chromatographic separation of peptides from GGCX, along with representative raw mass spectra and corresponding deuterium uptake curves. The time-resolved deuterium incorporation into each peptide was determined, in duplicate experiments, and showed very good reproducibility. Deuterium incorporation was followed in 71 peptic peptides (mostly non-overlapping), corresponding to 45% of the sequence of the 770 residue GGCX membrane protein (see Figure 4). The significance and biological interpretation of the deuteration of GGCX will be discussed elsewhere as will the conformational information on the scaffold MSP protein (manuscripts in preparation). Coupling nanodisc technology with HX MS offers a powerful approach for investigating the conformation of GGCX and encourages future studies aimed at examining dynamic structural changes upon substrate binding.
Figure 3.

Representative data for a nanodisc-HX MS experiment. (A) UPLC traces for empty (E) and loaded (L) nanodisc peptic digests at the deuterium exchange times indicated (UN – undeuterated). There were few differences in the empty and loaded traces as the most intense ions were always derived from the MSP (2:1 MSP:GGCX molar ratio). The bar at ~5.25 min corresponds to the scans combined to generate panel B. (B) Spectra for the m/z range 500–550. GGCX peptides (red) were not present in the empty nanodisc spectrum (top) and MSP peptides (black) are seen in both empty and loaded samples. Due to the high quality UPLC separations, very few peptides overlapped with one another, as shown here and found in most other spectra (data not shown). The bar corresponds to the zoomed region shown in panel C. (C) Magnification of the m/z range 532–545 showing typical mass spectral quality. (D) Deuterium uptake plots for the peptides shown in panel C. In these plots, the error of determining the deuterium level did not exceed ±0.3 daltons (see Supporting Information).
Figure 4.
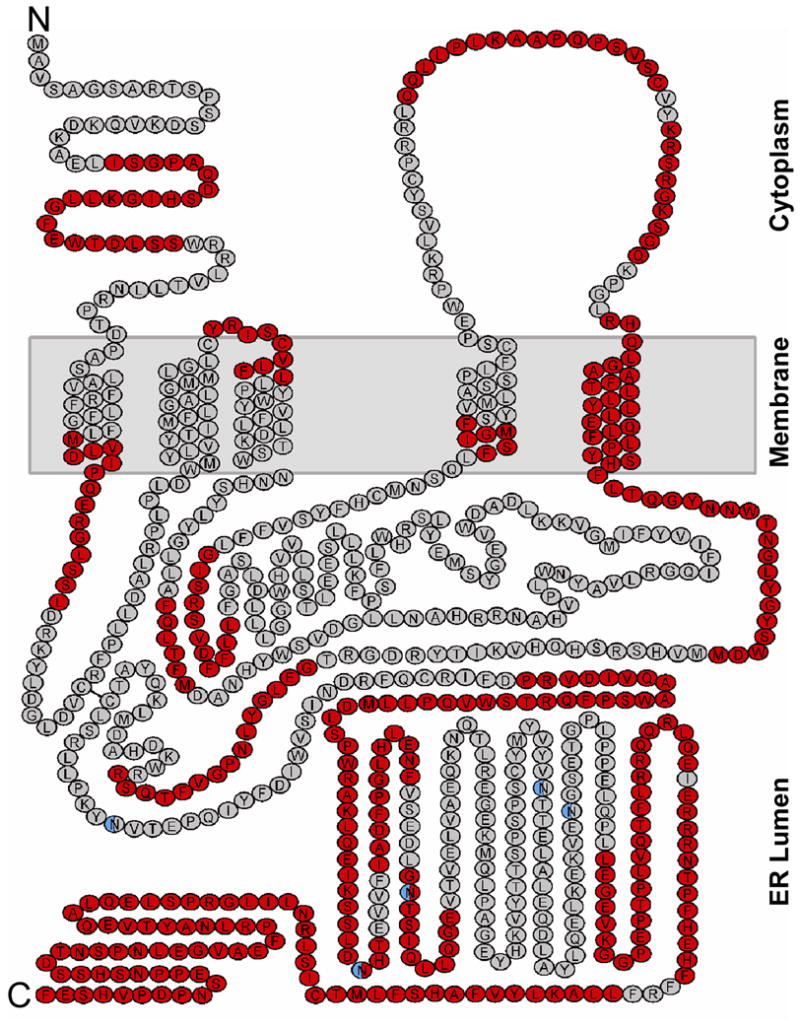
Sequence coverage plotted on the predicted topology map of gamma-glutamyl carboxylase40. The protocol described in Figure 1 resulted in 71 unique, mostly non-overlapping GGCX peptic peptides that were identified and followed in duplicate nanodisc HX MS experiments. Residues within these peptides are shown in red and those not covered by these peptides are shown in gray. The five N-linked glycosylation sites are colored in blue. Recovery of peptic peptides containing post-translationally modified sites was not performed in the current HX-MS experiments..
To our knowledge, this study represents the first successful application of HX MS analyses on a native membrane protein incorporated into phospholipid bilayer nanodiscs. Nanodisc technology provides a universal technique in which membrane proteins are assembled into a controlled and reproducible environment that can be tailored for a transmembrane protein of interest. Nanodiscs are preferred over detergent or lipid vesicle systems because nanodiscs are simultaneously very similar to native cellular membranes and very reliably and reproducibly prepared. In addition, nanodiscs are much more compatible with analytical techniques based on mostly water solvent systems, allowing for purification, and preparation with methods such as gel filtration. As we have illustrated, the complex composition of membrane protein-containing nanodiscs becomes compatible with the strict sample handling- and chromatographic requirements of HX MS experiments only upon addition of several key steps that facilitate efficient nanodisc disassembly, selective phospholipid removal and optimized chromatographic separations. Nanodisc technology, combined with HX MS, opens the door for characterizing membrane protein conformation and dynamics in the context of a controlled and native lipid-environment, thus addressing critical questions regarding membrane protein function. Such experiments are poised to reveal a wealth of information for the mostly uncharted territory of membrane proteins, and indeed such experiments are already underway.
Supplementary Material
Acknowledgments
We thank James H. Morrissey at the University of Illinois at Urbana-Champaign and David L. Straight at the University of North Carolina at Chapel Hill for useful discussions. We gratefully acknowledge financial support from the National Institutes of Health (GM086507 to J.R.E., HL48318 to D.W.S.), the Danish Natural Science Research Council (Grant 272-07-0276 to K.D.R.), and the Waters Corporation (J.W.J. and J.R.E.). C.M.H. was supported in part by a fellowship from Merck Research Laboratories. This is contribution 966 from the Barnett Institute.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wallin E, von Heijne G. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng YZ, Foster LJ. J Proteomics. 2009;72:12–22. doi: 10.1016/j.jprot.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Overington JP, Al-Lazikani B, Hopkins AL. Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 4.White SH. Nature. 2009;459:344–346. doi: 10.1038/nature08142. [DOI] [PubMed] [Google Scholar]
- 5.Seddon AM, Curnow P, Booth PJ. Biochim Biophys Acta. 2004;1666:105–117. doi: 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Garavito RM, Ferguson-Miller S. J Biol Chem. 2001;276:32403–32406. doi: 10.1074/jbc.R100031200. [DOI] [PubMed] [Google Scholar]
- 7.Gohon Y, Popot JL. Curr Opin Colloid Interface Sci. 2003;8:15–22. [Google Scholar]
- 8.le Maire M, Champeil P, Moller JV. Biochim Biophys Acta. 2000;1508:86–111. doi: 10.1016/s0304-4157(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 9.Rigaud JL, Pitard B, Levy D. Biochim Biophys Acta. 1995;1231:223–246. doi: 10.1016/0005-2728(95)00091-v. [DOI] [PubMed] [Google Scholar]
- 10.Bayburt TH, Grinkova YV, Sligar SG. Nano Letters. 2002;2:853–856. [Google Scholar]
- 11.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 12.Atkinson D, Small DM. Annu Rev Biophys Biophys Chem. 1986;15:403–456. doi: 10.1146/annurev.bb.15.060186.002155. [DOI] [PubMed] [Google Scholar]
- 13.Nath A, Atkins WM, Sligar SG. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- 14.Shih AY, Arkhipov A, Freddolino PL, Sligar SG, Schulten K. J Phys Chem B. 2007;111:11095–11104. doi: 10.1021/jp072320b. [DOI] [PubMed] [Google Scholar]
- 15.Shaw AW, McLean MA, Sligar SG. FEBS Lett. 2004;556:260–264. doi: 10.1016/s0014-5793(03)01400-5. [DOI] [PubMed] [Google Scholar]
- 16.Bayburt TH, Sligar SG. Protein Sci. 2003;12:2476–2481. doi: 10.1110/ps.03267503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw AW, Pureza VS, Sligar SG, Morrissey JH. J Biol Chem. 2007;282:6556–6563. doi: 10.1074/jbc.M607973200. [DOI] [PubMed] [Google Scholar]
- 18.Nath A, Grinkova YV, Sligar SG, Atkins WM. J Biol Chem. 2007;282:28309–28320. doi: 10.1074/jbc.M703568200. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Berthold DA, Gennis RB, Rienstra CM. Protein Sci. 2008;17:199–204. doi: 10.1110/ps.073225008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Kijac AZ, Sligar SG, Rienstra CM. Biophys J. 2006;91:3819–3828. doi: 10.1529/biophysj.106.087072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoofnagle AN, Resing KA, Ahn NG. Annu Rev Biophys Biomol Struct. 2003;32:1–25. doi: 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- 22.Wales TE, Engen JR. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 23.Rand KD, Zehl M, Jensen ON, Jorgensen TJ. Anal Chem. 2009;81:5577–5584. doi: 10.1021/ac9008447. [DOI] [PubMed] [Google Scholar]
- 24.Akashi S, Takio K. J Am Soc Mass Spectrom. 2001;12:1247–1253. doi: 10.1016/S1044-0305(01)00314-2. [DOI] [PubMed] [Google Scholar]
- 25.Busenlehner LS, Salomonsson L, Brzezinski P, Armstrong RN. Proc Natl Acad Sci U S A. 2006;103:15398–15403. doi: 10.1073/pnas.0601451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouchard M, Benjamin DR, Tito P, Robinson CV, Dobson CM. Biophys J. 2000;78:1010–1017. doi: 10.1016/S0006-3495(00)76659-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demmers JA, van Duijn E, Haverkamp J, Greathouse DV, Koeppe RE, 2nd, Heck AJ, Killian JA. J Biol Chem. 2001;276:34501–34508. doi: 10.1074/jbc.M101401200. [DOI] [PubMed] [Google Scholar]
- 28.Hansen RK, Broadhurst RW, Skelton PC, Arkin IT. J Am Soc Mass Spectrom. 2002;13:1376–1387. doi: 10.1016/S1044-0305(02)00702-X. [DOI] [PubMed] [Google Scholar]
- 29.Pinheiro TJ, Cheng H, Seeholzer SH, Roder H. J Mol Biol. 2000;303:617–626. doi: 10.1006/jmbi.2000.4159. [DOI] [PubMed] [Google Scholar]
- 30.Man P, Montagner C, Vernier G, Dublet B, Chenal A, Forest E, Forge V. J Mol Biol. 2007;368:464–472. doi: 10.1016/j.jmb.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 31.Stafford DW. J Thromb Haemost. 2005;3:1873–1878. doi: 10.1111/j.1538-7836.2005.01419.x. [DOI] [PubMed] [Google Scholar]
- 32.Bayburt TH, Sligar SG. Proc Natl Acad Sci U S A. 2002;99:6725–6730. doi: 10.1073/pnas.062565599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brier S, Engen JR. In: Mass spectrometry analysis for protein-protein interactions and dynamics. Chance M, editor. Wiley-Blackwell; New York: 2008. pp. 11–43. [Google Scholar]
- 34.Shih AY, Freddolino PL, Sligar SG, Schulten K. Nano Lett. 2007;7:1692–1696. doi: 10.1021/nl0706906. [DOI] [PubMed] [Google Scholar]
- 35.Burke JE, Karbarz MJ, Deems RA, Li S, Woods VL, Jr, Dennis EA. Biochemistry. 2008;47:6451–6459. doi: 10.1021/bi8000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kweon HK, Hakansson K. Anal Chem. 2006;78:1743–1749. doi: 10.1021/ac0522355. [DOI] [PubMed] [Google Scholar]
- 37.Petersen G, Chapman KD, Hansen HS. J Lipid Res. 2000;41:1532–1538. [PubMed] [Google Scholar]
- 38.Pucci V, Di Palma S, Alfieri A, Bonelli F, Monteagudo E. J Pharm Biomed Anal. 2009;50:867–871. doi: 10.1016/j.jpba.2009.05.037. [DOI] [PubMed] [Google Scholar]
- 39.Wales TE, Fadgen KE, Gerhardt GC, Engen JR. Anal Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tie J, Wu SM, Jin D, Nicchitta CV, Stafford DW. Blood. 2000;96:973–978. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.