

Abstract
Primary cilia are present on most mammalian cells and are implicated in transducing Hedgehog (Hh) signals during development; however, the prevalence of cilia on human tumors remains unclear, and the role of cilia in cancer has not been examined. Here we show that human basal cell carcinomas (BCCs) are frequently ciliated, and we test the role of cilia in BCC by conditionally deleting Kif3a (encoding kinesin family member 3A) or Ift88 (encoding intraflagellar transport protein 88), genes required for ciliogenesis, in two Hh pathway–dependent mouse tumor models. Ciliary ablation strongly inhibited BCC-like tumors induced by an activated form of Smoothened. In contrast, removal of cilia accelerated tumors induced by activated Gli2, a transcriptional effector of Hh signaling. These seemingly paradoxical effects are consistent with a dual role for cilia in mediating both the activation and the repression of the Hh signaling pathway. Our findings demonstrate that cilia function as unique signaling organelles that can either mediate or suppress tumorigenesis depending on the nature of the oncogenic initiating event.
Elevated Hh pathway activity is associated with diverse tumors, including basal cell carcinoma, the most commonly diagnosed cancer in North America1,2. Hh signaling is normally restrained by the tumor suppressor Patched (Ptch1), which inhibits the function of the protooncogene Smoothened (Smo), a central activator of the pathway3. Binding of Hh ligand to Ptch1 relieves its inhibition of Smo, allowing Smo to induce downstream Gli activators and inhibit the formation of Gli repressors. In BCCs, loss of function mutations in PTCH1, gain of function mutations in SMO, and upregulation of GLI1 and GLI2 are frequently observed, suggesting that dysregulated Hh signaling is the underlying cause of this disease4–8.
Recent studies have shown that the primary cilium has a prominent role in modulating mammalian Hh signaling9. Ptch1 suppresses the Hh pathway, at least in part, by preventing the trafficking of Smo into the primary cilium10. Upon binding of Hh ligand to Ptch1, Smo moves to the cilium11. This event seems to be crucial for activating the Hh pathway, as a mutant form of Smo that fails to localize to the cilium cannot fully transduce Hh signals, whereas an activated mutant form of Smo isolated from individuals with BCC localizes constitutively to the cilium11.
In addition to its essential role in transducing Hh signals, the cilium also negatively regulates the pathway. Genetic analyses have shown that the cilium is required for the proteolytic processing of Gli3 into a form that represses the Hh transcriptional program (Gli3-R)12–15. Cilium-dependent formation of Gli3-R occurs in the absence of Hh and is suppressed upon Smo movement to the cilium. Thus, the cilium exerts both positive and negative control over the Hh pathway.
Although cilia are present on many vertebrate cells, their involvement in cancer has not been explored. Early ultrastructural studies have indicated that individual cells from human BCCs can be ciliated16; however, the prevalence of these ciliated cells has remained unclear. To determine whether cancer cells frequently possess cilia, we examined clinical biopsies from eight human BCCs. We observed that five BCCs contained numerous ciliated cells (Fig. 1a and Supplementary Fig. 1). In addition, ciliated cells were present in BCC-like lesions that arose in mouse skin expressing the constitutively active M2 allele of Smo (SmoM2cond)4,17 (Fig. 1b). We also observed ciliated cells in normal mouse skin, including the follicular outer root sheath, the mesenchymal dermal papilla and the interfollicular epidermis, as well as in cultured keratinocytes (Fig. 1c,d, data not shown and as recently reported18).
Figure 1.

BCC and normal skin cells possess primary cilia. (a) Immunofluorescence imaging of the ciliary marker acetylated tubulin (green) and rootletin (red), a ciliary rootlet component. Five of eight independent human clinical BCCs, including nodular and superficial subtypes, contain numerous ciliated cells (arrows). Insets, H&E staining of adjacent biopsies. Right images are views of the left and middle panels. (b) Immunofluorescence imaging of the ciliary marker detyrosinated tubulin (green) and the centrosomal marker γ -tubulin (red) (dotted line, epidermal basal layer;inset, enlarged view). Arrows indicate ciliated cells present in SmoM2-induced regions of mouse epidermal hyperplasia. (c) Cilia extending from normal skin cells (arrows). Left images, low magnification;right images, high magnification of boxed areas. (d) Cilia extending from cultured keratinocytes (arrows). All scale bars are 50 μm except that in d (10 μm).
To test the role of cilia in skin tumorigenesis, we generated mice that harbor both a conditionally-activatable allele of SmoM2 and a conditional loss of function allele of Kif3a (Kif3aflox), which encodes a subunit of kinesin-II, a heterotrimeric motor required for assembly of the primary cilium19. Deletion of Kif3a and induction of SmoM2 was mediated by a Ker14 gene (encoding keratin-14) promoter–driven Cre recombinase (Ker14-CreERT), which is active only in the presence of tamoxifen and induces recombination throughout the skin20 (Supplementary Fig. 2). Thus, adult skin cells harboring these conditional alleles both activate SmoM2 expression and delete the floxed allele of Kif3a in response to tamoxifen, allowing spatial and temporal control of tumor initiation and ciliation (Fig. 2a).
Figure 2.

Cilia are essential for SmoM2-induced neoplasia. (a) Schematic showing that skin epithelial cells exposed to tamoxifen (TAM) both delete Kif3aflox and activate SmoM2cond (red cells), whereas unexposed cells retain Kif3aflox and do not activate SmoM2cond (gray cells). Cilia are lost in cells from Kif3aflox/− but not Kif3aflox/+ mice exposed to tamoxifen. (b) H&E staining of dorsal skin biopsies, showing that removal of Kif3a (Kif3aflox/−) blocks SmoM2-initiated tumorigenesis (arrows). (c) Immunofluorescence imaging showing that SmoM2 localizes to cilia (arrows) in neoplastic epidermal downgrowths. (d) Quantification of interfollicular epidermis thickness. Seven or eight independent Ker 14-CreERT;SmoM2cond;Kif3aflox/+ and Ker14-CreERT;SmoM2cond;Kif3aflox/− mice per genotype were analyzed. Four or five mice were analyzed for all other genotypes. (e) Protection against SmoM2-induced ear skin hyperplasia upon loss of Kif3a (arrow). (f) H&E staining of representative ear sections. (g) In situ staining showing that SmoM2-induced skin lesions upregulate Hh target genes Gli1 and Ptch1 (arrows). (h) Immunofluorescence imaging showing that Gli2 is upregulated in SmoM2-induced tumors that express Kif3a (stainings are representative of the mice analyzed in d and j). (i) Immunofluorescence imaging showing that Gli2 is upregulated upon expression of Myc-tagged SmoA1 in ciliated (wild type), but not unciliated (Kif3a−/− and Ift172−/−), transformed MEFs. (j) Cellular proliferation in skin, as assessed by Ki67 staining. Eight Ker14-CreERT;SmoM2cond;Kif3aflox/+ and Ker14-CreERT;SmoM2cond;Kif3aflox/− mice per genotype were analyzed, as well as four or five mice for the other genotypes. All scale bars are 50 μm except that in e (5 mm). All error bars shown, ± s.e.m.
We treated Ker14-CreERT;SmoM2cond mice that were either Kif3aflox/+ or Kif3aflox/− with tamoxifen at approximately 30 d of age and collected skin biopsies 5, 10 and 20 weeks after induction. We observed that Ker14-CreERT;SmoM2cond;Kif3aflox/+ mice developed epidermal hyperplasia beginning 5 weeks after induction (data not shown). By 10–20 weeks, the neoplastic epithelium extended numerous downgrowths into the underlying dermis (Fig. 2b). Neoplastic cells showed high nuclear to cytoplasmic ratios and manifested histopathological features of BCC, such as palisading (Supplementary Fig. 3). Consistent with our previously reported results showing that an oncogenic form of Smo constitutively localizes to cilia in vitro11, SmoM2 localized to tumor cilia in vivo (Fig. 2c). As expected, ablation of Kif3a in Ker14-CreERT;Kif3aflox/− mice reduced the number of ciliated cells in the follicular epithelium (Supplementary Fig. 4).
Strikingly, loss of cilia protected against SmoM2-induced tumorigenesis (Fig. 2b,d). The average thickness of the interfollicular epidermis from Ker14-CreERT;SmoM2cond;Kif3aflox/− mice was nearly indistinguishable from that of control nontumorigenic skin of Ker14-CreERT;Kif3aflox/− or Ker14-CreERT;Kif3aflox/+ mice 10 weeks after induction (Fig. 2d). Within 10 weeks after SmoM2 induction, Kif3aflox/+ mice also developed hyperplastic ear and tail skin, which was markedly alleviated in mice that had deleted Kif3a (Fig. 2e–f and Supplementary Figs. 5 and 6).
Genetic analyses in the embryonic neural tube have previously shown that cilia are required for Smo-mediated induction of Hh target genes14,15,21. Consistent with this, in situ analyses in skin revealed that SmoM2-induced upregulation of Gli1 and Ptch1, direct targets of Hh signaling, was blocked by ciliary ablation (Fig. 2g). Furthermore, loss of cilia prevented SmoM2-induced upregulation of Gli2 (Fig. 2h). We confirmed this observation in vitro by expressing the mouse homolog of SmoM2 (SmoA1) in transformed mouse embryonic fibroblasts (MEFs). Whereas SmoA1 increased Gli2 protein levels in ciliated transformed MEFs, we did not observe such elevation in unciliated MEFs deficient in either Kif3a or Ift172, another gene required for ciliogenesis21 (Fig. 2i).
As previous studies have shown that Hh signaling promotes proliferation of the follicular epithelium22–24, we examined whether cilia are required for SmoM2-induced proliferation in the skin. Indeed, ciliated Kif3aflox/+ tumors showed significantly higher proliferation relative to unciliated Kif3aflox/− lesions (Fig. 2j). Thus, these results indicate that cilia are essential for Smo-mediated skin tumorigenesis.
In a variety of organs including skin, Hh pathway activity downstream of Smo is mediated primarily by Gli2 (ref. 24). To further define how cilia function in the Hh pathway, we tested the requirement for Kif3a in a second skin tumorigenesis model that uses conditional expression of a Myc-tagged, constitutively active human GLI2 lacking its amino-terminal repressor domain Tg(CAG-loxP-eGFP-loxP-GLI2ΔN-Myc), referred to here as CLEG2cond (ref. 25). We predicted that if cilia function specifically at the level of Smo for Hh pathway activation, loss of Kif3a or cilia should not affect GLI2ΔN-induced skin tumorigenesis.
As in the SmoM2 experiments, we injected Ker14-CreERT;CLEG2cond mice that were either Kif3aflox/+ or Kif3aflox/− with tamoxifen. By 4–5 weeks after induction, GLI2ΔN-driven Kif3aflox/+ epidermal lesions were mildly hyperplastic and rarely extended into the dermis (Fig. 3a,b). However, in contrast to both our predictions and the effects that we observed in SmoM2-dependent tumorigenesis, loss of Kif3a markedly accelerated GLI2ΔN-induced skin neoplasia (Fig. 3a–c). Ker14-CreERT;CLEG2cond;Kif3aflox/− mice showed gross changes in the skin, including hair loss, disorderly pelage hair and scaling, after induction (Fig. 3a). Histological analysis revealed that the majority of these Ker14-CreERT;CLEG2cond;Kif3aflox/− mice developed BCC-like lesions that extended strands and nests of cells into the dermis (Fig. 3b). As expected, lesions that arose in Kif3a flox/− mice did not contain ciliated cells (Supplementary Fig. 7).
Figure 3.
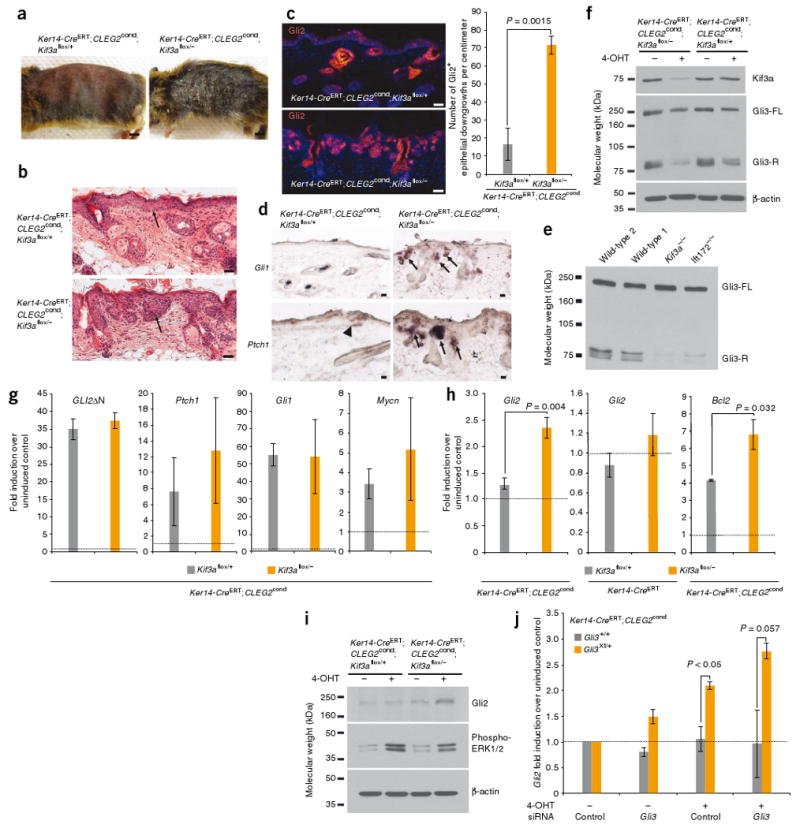
Loss of cilia accelerates GLI2ΔN-induced neoplasia. (a) Gross morphology of mice upon induction of CLEG2cond. (b) H&E staining showing that loss of Kif3a increases the severity of GLI2ΔN-induced skin lesions (arrows). Images are representative of five or six mice analyzed per genotype. (c) Immunofluorescence imaging showing that loss of Kif3a increases the number of Gli2+ downgrowths in the skin. (d) In situ staining revealing that Ptch1 is upregulated in small regions of CLEG2-expressing Kif3aflox/+ skin (arrowhead), whereas Gli1 and Ptch1 are more frequently upregulated in CLEG2-induced lesions lacking Kif3a (arrows). (e) Formation of Gli3-R in ciliated (wild type) and non-ciliated (Kif3a−/−, Ift172−/−) transformed MEFs (Gli3-FL, full length Gli3). (f) Western blot indicating that Kif3a protein expression is lower and Gli3-R formation is impaired in 4-OHT–treated Ker14-CreERT;CLEG2cond;Kif3aflox/−keratinocytes, relative to control cells. (g) Expression of constitutively active human GLI2 (GLI2ΔN), Ptch1, Gli1 and Mycn in Kif3aflox/+ and Kif3aflox/−keratinocytes treated with 4-OHT. (All expression values expressed relative to those of vehicle-treated control cells, set to a baseline of ‘1’, dotted line.) (h) Quantiative RT-PCR assessment of GLI2ΔN-mediated induction of endogenous Gli2 and Bcl2 in ciliated and unciliated Kif3aflox/− keratinocytes. (i) Endogenous Gli2 and phosphorylated ERK1/2 protein levels in 4-OHT–treated Ker14-CreERT;CLEG2cond keratinocytes that are either Kif3aflox/+ or Kif3aflox/−. (j) Expression of endogenous Gli2 in GLI2ΔN-induced Gli3Xt/+ or Gli3+/+ keratinocytes, treated with siRNA against Gli3 or control. All expression values are normalized to those of vehicle-treated cells transfected with control siRNAs (baseline set to ‘1’, dotted line). All scale bars are 50 μm except that in a (5 mm). All error bars show means ± s.e.m.
Further molecular characterization revealed that the number of lesions with high endogenous Gli2 protein expression was significantly expanded in Kif3aflox/− mice compared to Kif3aflox/+ mice (Fig. 3c). In addition, we observed that Gli1 and Ptch1 were both upregulated in Kif3a flox/− lesions compared to normal skin, although some Kif3aflox/+ lesions also showed increased Hh signaling (Fig. 3d). Together, these results indicate that cilia suppress GLI2ΔN-induced tumorigenesis in the skin.
As noted above, in addition to being required for full Hh pathway activation, the cilium also negatively regulates the Hh transcriptional program by mediating Gli3 processing into Gli3-R12–15. Transformed MEFs lacking cilia formed less Gli3-R than did ciliated transformed MEFs, confirming the role of cilia in Gli3-R formation (Fig. 3e). To determine whether cilia also mediate Gli3 processing in skin cells, we deleted Kif3a and induced expression of GLI2ΔN by treating keratinocytes isolated from Ker14-CreERT;CLEG2cond mice that were either Kif3aflox/+ or Kif3aflox/− with the tamoxifen derivative 4-hydroxytamoxifen (4-OHT). 4-OHT induced recombination in ∼60% of cells (Supplementary Fig. 8), and, consistent with results from MEFs, loss of Kif3a decreased Gli3-R formation (Fig. 3f). These findings suggest that loss of cilia may promote GLI2ΔN-mediated tumorigenesis by inhibiting Gli3-R formation.
To elucidate how the cilium restrains GLI2ΔN activity, we examined GLI2ΔN induction of Hh target genes in 4-OHT–treated keratinocytes. 4-OHT increased expression of GLI2ΔN to comparable levels regardless of Kif3a status, indicating that the cilium itself does not affect expression of the transgene (Fig. 3g). On average, GLI2ΔN expression induced ten-, 55- and threefold increases in expression of the Hh target genes Ptch1, Gli1 and Mycn, respectively, over that of cells treated with vehicle control (Fig. 3g). As with GLI2ΔN, similar levels of these genes were induced regardless of Kif3a status.
However, in agreement with our immunohistochemical results, GLI2ΔN significantly increased expression of endogenous Gli2 in Kif3aflox/− keratinocytes relative to similarly-treated Kif3aflox/+ cells (Fig. 3h). Loss of Kif3a by itself was not sufficient to upregulate Gli2 (Fig. 3h). Expression of Bcl2, another Hh target gene, was also increased to a greater extent in GLI2ΔN-induced keratinocytes that had lost Kif3a (Fig. 3h). Thus, loss of Kif3a increases GLI2ΔN-mediated induction of some Hh targets (Gli2 and Bcl2), but not others (Ptch1, Gli1 and Mycn).
In light of the increased Gli2 expression that we observed in GLI2ΔN-induced Kif3aflox/− cells, we examined whether endogenous Gli2 protein was also upregulated with an antibody that specifically recognizes mouse Gli2 (Supplementary Fig. 9). We observed that induction of GLI2ΔN, coupled with loss of Kif3a, increased endogenous Gli2 protein levels in keratinocytes (Fig. 3i). As cilia have been reported to be required for platelet-derived growth factor– induced mitogen-activated protein (MAP) kinase signaling26, we also probed keratinocyte lysates for phosphorylated extracellular signal–related kinase-1 and extracellular signal–related kinase-2 (here referred to as ERK1/2). We observed that GLI2ΔN induction consistently increased ERK1/2 phosphorylation to a slightly greater extent in cells that had retained Kif3a, relative to cells that had lost Kif3a (Fig. 3i). As Wnt signaling is involved during folliculogenesis27 and has recently been linked to BCC progression28, we also assessed the expression of four Wnt target genes but found that none differed significantly between GLI2ΔN-expressing Kif3aflox/+ and Kif3aflox/− cells (P > 0.05) (Supplementary Fig. 10). Together, these results suggest that augmented expression of some Hh target genes, including Gli2 and Bcl2, but not differences in MAP kinase or Wnt signaling, may contribute to the accelerated progression of GLI2ΔN-induced BCC-like lesions upon loss of cilia.
Because ciliary loss disrupted formation of Gli3-R and increased expression of Gli2, we tested whether inhibiting Gli3 expression in ciliated cells was sufficient to elevate endogenous Gli2 levels. We derived keratinocytes from Ker14-CreERT;CLEG2cond mice that were either wild type for Gli3 or heterozygous for a null allele of Gli3 (Gli3Xt) and observed that GLI2ΔN-expressing Gli3Xt/+ keratinocytes showed elevated Gli2 expression relative to that in GLI2ΔN-expressing Gli3+/+ cells upon induction with 4-OHT (Fig. 3j). Moreover, further down-regulation of Gli3 by RNA interference (RNAi) in Gli3Xt/+ keratinocytes augmented Gli2 expression (Fig. 3j).
Because Kif3a possesses nonciliary functions29,30, we repeated our studies using conditional deletion of Ift88, which encodes an intra-flagellar transport protein required for ciliogenesis31. Similarly to the strategies described above, we induced expression of SmoM2 or GLI2ΔN in Ker14-CreERT mice that were either Ift88flox/+ or Ift88flox/−. As observed previously with Kif3a, loss of Ift88 suppressed SmoM2-induced tumorigenesis (Fig. 4a,b). Also concordant with previous results, loss of Ift88 accelerated the formation of GLI2ΔN-induced BCCs-like lesions (Fig. 4c,d). These findings confirm that the cilium specifically modulates the Hh-related tumor phenotypes observed in this study.
Figure 4.

Loss of Ift88 restrains SmoM2-mediated tumorigenesis and promotes GLI2ΔN-induced BCC-like lesions in tamoxifen-treated mice. (a) H&E staining of dorsal skin biopsies, revealing that loss of Ift88 (Ift88flox/−) blocks SmoM2-induced epidermal hyperplasia and tumorigenesis. (b) Quantification of epidermal thickness for a. Three to five mice per genotype were analyzed 10 weeks after induction by tamoxifen, and five mice per genotype were assessed 20 weeks after tamoxifen treatment. (c) H&E staining revealing that loss of Ift88 promotes skin lesions induced by activated Gli2. All scale bars, 50 μm. (d) Quantification of the number of Gli2+ downgrowths in the skin. Four mice were analyzed per genotype. (e) Schematic showing that in nonpathological conditions and in the absence of Hh, the cilium mediates the formation of Gli3-R, a repressor of downstream Hh target genes. In the presence of Hh ligand, Ptch1-mediated inhibition of Smo is suppressed, allowing Smo to act through the cilium to inhibit Gli3-R formation and induce Gli activators (Gli-Act), which turn on the Hh transcriptional program. In cilium-dependent oncogenesis (for example, SmoM2-induced tumors), the cilium is essential for Gli-Act formation and, thus, tumorigenesis. In cilium-independent oncogenesis (for example, tumors induced by constitutively active GLI2 (Gli-Act*)), the cilium restrains tumorigenesis through production of counter-balancing Gli3-R. Loss of the cilium inhibits Gli3-R formation, allowing unopposed Gli-Act* to aggressively drive tumorigenesis. All error bars show means ± s.e.m.
We have shown here that tumor cells from human BCCs are frequently ciliated and that these cilia possess dual roles in modulating Hh pathway–dependent tumor phenotypes in the skin. Whereas the cilium normally suppresses the Hh pathway by mediating Gli3-R formation in the absence of Hh, the cilium is also required for Smo-mediated activation of the pathway in the presence of Hh (Fig. 4e). Our data suggest that tumors initiated by Smo depend upon the cilium both for transducing positive signals through Gli activators and for suppressing negative ones such as Gli3-R. For tumors initiated by constitutively active GLI2ΔN, loss of the cilium prevents Gli3-R formation, permitting unopposed activation of downstream signaling (Fig. 4e).
We have also shown here that in Smo-mediated tumorigenesis, upregulation of Gli2 requires cilia, whereas GLI2ΔN-induced elevation of endogenous Gli2 is augmented by loss of cilia, consistent with our hypothesis that ciliary control of Gli3 processing likely modulates gene expression in tumors. Indeed, we also observed that disruption of Gli3 increases Gli2 expression, concordant with previous findings that Gli3 binds the Gli2 promoter32. In addition to Gli2, Gli3-R likely represses other genes that promote tumorigenesis, such as Bcl2.
In addition to disrupting Gli3-R formation, loss of cilia might potentially inhibit formation of a Gli2 repressor, although evidence for a Gli2 repressor function is currently lacking. In a variety of cells, loss of cilia has also been associated with increased response to Wnt, although basal levels of Wnt signaling are unaffected30,33. As Wnt signaling regulates folliculogenesis27 and has recently been implicated in the pathogenesis of BCC28, we examined the expression of Wnt target genes in keratinocytes. Although we found no evidence that Wnt signaling is affected either by GLI2ΔN induction or by ciliary status, it remains possible that increased Wnt responsiveness may contribute to tumorigenesis after loss of cilia in vivo.
Because the majority of human BCCs possess mutations in either SMO or PTCH1, and loss of a single copy of PTCH1 underlies basal cell nevus (Gorlin's) syndrome, it is noteworthy that cells from three of eight human BCCs examined appeared poorly ciliated. One explanation for this may be that Hh signaling in a small population of ciliated cells is sufficient for tumor growth. Alternatively, unciliated cells may be supported by signaling pathways functioning independently of cilia and, as our GLI2ΔN studies might suggest, may even show growth advantages upon ciliary loss. Indeed, several reports have described signaling pathways that potentiate Gli transcriptional activity, including KRAS-mediated MAP kinase signaling34, phosphoinositide 3-kinase–Akt signaling35 and transforming growth factor-β signaling34,36. Although it remains unclear whether these pathways are frequently perturbed in BCC, the GLI2ΔN-induced skin tumors provide illustrative support for a tumor-suppressive capacity for cilia during Smo- and ciliary-independent tumorigenesis. As recent studies have reported a lack of correlation between Smo activity and downstream Hh pathway activity in a variety of human cancer cells, it is possible that Smo-independent modulation of the Hh pathway occurs in many different tumor types37.
Our findings advance several general predictions. First, human tumors that rely on upstream activation of Hh signaling, such as many BCCs and medulloblastomas, are predicted to retain cilia. Indeed, we have shown here that both human and mouse skin tumors are ciliated, and others have found that SmoM2-induced medulloblastomas are similarly ciliated and dependent upon ciliary function (Y.-G. Han, H. Kim, A.A.D., D. Ellison, R. Gilbertson and A. Alvarez-Buylla, personal communication). Second, inhibition of ciliogenesis in Smo-dependent tumors is likely to prevent growth. This has major implications for therapy, as the presence of cilia may be a useful biomarker for identifying Hh-dependent cancers responsive to Smo antagonists currently being tested in clinical trials2. Third, given that recent studies have suggested a paracrine role for Hh signaling during tumorigenesis34,37, disruption of cilia in the surrounding tumor mesenchyme may also indirectly affect tumor growth. A final prediction from this study is that loss of cilia may favor progression of tumors initiated by oncogenic events that activate the Hh pathway independently of Smo. Thus, the dual functions of cilia as both a mediator of Smo-dependent tumorigenesis and as a suppressor of Smo-independent tumorigenesis suggest that inhibiting ciliogenesis may be therapeutic in a subset of cancers but may also exacerbate cancer growth in others.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturemedicine/.
Supplementary Material
Acknowledgments
We thank D. Hanahan, O. Nolan-Stevaux, G. Evan and the members of the Reiter lab for critical reading of this manuscript; K. Thorn and the UCSF Nikon Imaging Center for assistance with confocal microscopy; C. Miller and J.D. Fish for assistance with histology; and R. T Bronson for help with pathology We thank T Li, Harvard Medical School, for rabbit antibody to rootletin; K.V. Anderson, Sloan-Kettering Institute, for Ift172−/−- MEFs; S. Scales, Genentech, for mouse antibody to Gli3; J.T Eggenschwiler, Princeton University, for guinea pig antibody to Gli2; B. Yoder, University of Alabama at Birmingham, for Ift88flox mice; L. Goldstein, University of California, San Diego, for Kif3aflox -knockout mice; and L.V. Goodrich, Harvard Medical School, for Ptch1 riboprobe plasmid. This work was funded by grants from the US National Institutes of Health (RO1AR054396), the Burroughs Wellcome Fund, the Packard Foundation and the Sandler Family Supporting Foundation to J.F.R. A.A.D. acknowledges the support of the US National Institutes of Health (RO1CA087837). S.Y.W. acknowledges the support of the A.P. Giannini Foundation, the Herbert W. Boyer Fund and the American Cancer Society.
Footnotes
Author Contributions: S.Y.W. and J.F.R. designed experiments, performed research, analyzed data and wrote the manuscript. A.D.S. assisted with mouse experiments. P.-L.S., A.N.E., C.K.B., E.H.E. and A.A.D. provided reagents and technical advice.
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389:876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- 2.Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5:1026–1033. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 3.Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 4.Xie J, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 5.Reifenberger J, et al. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998;58:1798–1803. [PubMed] [Google Scholar]
- 6.Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997;57:2581–2585. [PubMed] [Google Scholar]
- 7.Tojo M, Kiyosawa H, Iwatsuki K, Nakamura K, Kaneko F. Expression of the GLI2 oncogene and its isoforms in human basal cell carcinoma. Br J Dermatol. 2003;148:892–897. doi: 10.1046/j.1365-2133.2003.05284.x. [DOI] [PubMed] [Google Scholar]
- 8.Oro AE, et al. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276:817–821. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 9.Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates Hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 11.Corbit KC, et al. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 12.Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- 13.Haycraft CJ, et al. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huangfu D, Anderson KV. Cilia and hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.May SR, et al. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 16.Wilson RB, McWhorter CA. Isolated flagella in human skin Election microscopic observations. Lab Invest. 1963;12:242–249. [PubMed] [Google Scholar]
- 17.Mao J, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehman JM, Laag E, Michaud EJ, Yoder BK. An essential role for dermal primary cilia in hair follicle morphogenesis. J Invest Dermatol. 2009;129:438–448. doi: 10.1038/jid.2008.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marszalek JR, et al. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 20.Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA. 1999;96:8551–8556. doi: 10.1073/pnas.96.15.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huangfu D, et al. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 22.St Jacques B, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058–1068. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- 23.Chiang C, et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol. 1999;205:1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- 24.Mill P, et al. Sonic hedgehog–dependent activation of Gli2 is essential for embryonic hair follice development. Genes Dev. 2003;17:282–294. doi: 10.1101/gad.1038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasca di Magliano M, et al. Hedgehog-Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006;20:3161–3173. doi: 10.1101/gad.1470806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider L, et al. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Fuchs E. Skin stem cells: rising to the surface. J Cell Biol. 2008;180:273–284. doi: 10.1083/jcb.200708185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang SH, et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/β-catenin signaling. Nat Genet. 2008;40:1130–1135. doi: 10.1038/ng.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishimura T, et al. Role of the PAR-3–KIF3 complex in the establishment of neuronal polarity. Nat Cell Biol. 2004;6:328–334. doi: 10.1038/ncb1118. [DOI] [PubMed] [Google Scholar]
- 30.Corbit KC, et al. Kif3a constrains β-catenin–dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol. 2008;10:70–76. doi: 10.1038/ncb1670. [DOI] [PubMed] [Google Scholar]
- 31.Haycraft CJ, et al. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134:307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- 32.Hu MC, et al. GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development. 2006;133:569–578. doi: 10.1242/dev.02220. [DOI] [PubMed] [Google Scholar]
- 33.Gerdes JM, et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 34.Nolan-Stevaux O, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23:24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riobó NA, Lu K, Ai X, Haines GM, Emerson CP., Jr Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc Natl Acad Sci USA. 2006;103:4505–4510. doi: 10.1073/pnas.0504337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dennler S, et al. Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007;67:6981–6986. doi: 10.1158/0008-5472.CAN-07-0491. [DOI] [PubMed] [Google Scholar]
- 37.Yauch RL, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–410. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.