

Abstract
Rationale
FK506-binding proteins FKBP12.6 and FKBP12 are associated with cardiac ryanodine receptors (RyR2), and PKA-dependent phosphorylation of RyR2 was proposed to interrupt FKBP12.6-RyR2 association and activate RyR2. However, the function of FKBP12.6/12 and role of PKA phosphorylation in cardiac myocytes are controversial.
Objective
To directly measure in situ binding of FKBP12.6/12 to RyR2 in ventricular myocytes, with simultaneous Ca sparks measurements as a RyR2 functional index.
Methods & Results
We used permeabilized rat and mouse ventricular myocytes, and fluorescently-labeled FKBP12.6/12. Both FKBP12.6 and FKBP12 concentrate at Z-lines, consistent with RyR2 and Ca spark initiation sites. However, only FKBP12.6 inhibits resting RyR2 activity. Assessment of fluorescent FKBP binding in myocyte revealed a high FKBP12.6-RyR2 affinity (Kd = 0.7 ± 0.1 nM) and much lower FKBP12-RyR2 affinity (Kd = 206 ± 70 nM). Fluorescence recovery after photobleach confirmed this Kd difference and showed that it is mediated by koff. RyR2 phosphorylation by PKA did not alter binding kinetics or affinity of FKBP12.6/12 for RyR2. Using quantitative immunoblots, we determined endogenous [FKBP12] in intact myocytes is ∼1 μM (similar to [RyR]), while [FKBP12.6] is ≤ 150 nM.
Conclusions
Only 10-20% of endogenous myocyte RyR2s have FKBP12.6 associated, but virtually all myocyte FKBP12.6 is RyR2-bound (due to very high affinity). FKBP12.6 but not FKBP12 inhibits basal RyR2 activity. PKA-dependent RyR2 phosphorylation has no significant effect on binding of either FKBP12 or 12.6 to RyR2 in myocytes.
Keywords: FKBP12.6, FKBP12, Ca sparks, binding properties, rapamycin, RyR2
Introduction
Cardiac ryanodine receptors (RyR2) are sarcoplasmic reticulum (SR) Ca release channels, crucial in excitation-contraction coupling (ECC)1. Dysfunctional RyR2, exhibiting enhanced Ca leak has been implicated in arrhythmogenesis and heart failure (HF)1. Homotetrameric RyR2s have a transmembrane channel domain, and regulatory and scaffolding cytoplasmic domain.
In heart, FK506 binding protein (FKBP) 12 and 12.6 isoforms are co-expressed, and can bind RyR2 at a stoichiometry of 4 FKBP per RyR tetramer2,3. FKBP12 binds RyR2 with much lower affinity, but is much higher in concentration in heart than FKBP12.64. Nevertheless, FKBP12 & 12.6 share 85% sequence homology and similar 3D structures5. Human and rat FKBP12 differ in only 3 residues, and human and rat FKBP12.6 are identical. This makes study of human FKBP function in rat myocytes reasonable.
Effects of FKBPs on RyR2 activity in myocytes are controversial. Some groups reported that dissociation of FKBP12.6 from RyR2 by immunosuppressants (rapamycin or FK506) activated RyR2 channels and induced sub-conductance states6-8. RyR2 point mutations associated with cardiac sudden death may also exhibit altered FKBP12.6 interaction,9 and FK506 can alter resting Ca2+ spark frequency (CaSpF) and SR Ca2+ content.7,8,10 FKBP12.6 overexpression also increased SR load and enhanced contraction.11 However, others reported that FKBP12.6 removal had no effect on RyR2 activity,3,12 and failed to observe RyR2 subconductance states in channels from FKBP12.6 knockout mice13,14. Some groups suggest that FKBP12 affects RyR2 differently from FKBP12.6.15-17,20 Therefore, FKBP12/12.6 binding and RyR2 effects remain unclear, especially in the cardiomyocyte environment (as studied here).
Altered FKBP-RyR2 interaction is a prominent hypothesis explaining increased SR Ca leak in HF via RyR2 hyperphosphorylation and FKBP dissociation from RyR2.19 However, Li et al. found that PKA-dependent RyR2 phosphorylation did not alter resting RyR2 function in mouse myocytes without phospholamban (PLB) and increased SR Ca load,20 Xiao et al. reported that protein kinase A (PKA) phosphorylation at RyR2-Ser2808 did not dissociate FKBP12.621, and Jiang et al. found that in HF there was neither FKBP12.6 dissociation from RyR2 by PKA hyperphosphorylation nor increased RyR2 channel activity 22. Whether and how PKA phosphorylation of RyR2 affects FKBP-RyR2 interaction requires more study.
Here we characterize in the myocyte environment the interaction between FKBP12/12.6 and RyR2, its functional consequences, and modulation by PKA-dependent phosphorylation. Using fluorescent FKBP12/12.6 (F-FKBP), we simultaneously assessed physical association-dissociation of FKBP12/12.6 with RyR2, and RyR2 activity (via Ca sparks) in permeabilized ventricular myocytes. We found that: 1) both FKBP12.6 and FKBP12 bound to RyR2 (Kd ∼1 nM and 200 nM, respectively); 2) FKBP12.6 but not FKBP12 inhibited CaSpF; 3) the binding properties of FKBP12.6/12-RyR2 were not changed by PKA-dependent phosphorylation of RyR2; 4) endogenous FKBP12 was ∼1 μmol/L cytosol and FKBP12.6 ∼ 100 nmol/L cytosol.
Materials and Methods
Rat and mouse ventricular myocytes were isolated and permeabilized as previously described23 (methodological details in Online supplement). F-FKBP12.6 and F-FKBP12 were characterized using circular dichroism spectroscopy and ligand-binding studies (Online Figure I).24 Consistent with previous reports25, F-FKBP constructs have the same secondary structure, RyR binding affinity and RyR1 effects as unlabeled and WT FKBPs.
Results
Localization of FKBP12.6/12 in permeabilized ventricular myocytes
Saponin-permeabilized rat ventricular myocytes were incubated with F-FKBP12.6 or 12. Figure 1A shows a confocal myocyte image with 20 nM F-FKBP12.6 (note that bath supply of F-FKBP12.6 is inexhaustible). The cross-striated sarcomeric pattern co-localizes with Di-8-ANEPPS (Online Figure III), used to identify transverse tubules at the Z-line. Figure 1B shows that line scan fluorescence intensity peaks at Z-lines exhibit an inter-peak distance (∼2 μm) that matches sarcomere length. The same striation pattern was seen for F-FKBP12 (Online Figure II), but with lower contrast, because of lower RyR2 affinity (i.e. bath F-FKBP12 fluorescence is higher when RyR2 saturates). Pre-incubation with WT-FKBP12.6 (100 nM) completely prevented the appearance of striations, indicating that F-FKBP12.6/12 bind to the same sites as WT-FKBP12.6, and that there is little non-specific binding. Ca sparks can be measured simultaneously using rhod-2, and superimposition of F-FKBP and spark images (Figure 1C-D) shows that Ca spark initiation sites coincide with F-FKBP12.6 localization (Figure 1E). These data indicates that FKBP12.6/12 bind at the Z-line region where SR Ca release initiates during Ca sparks.
Figure 1. F-FKBP12.6 is located at junctional SR.
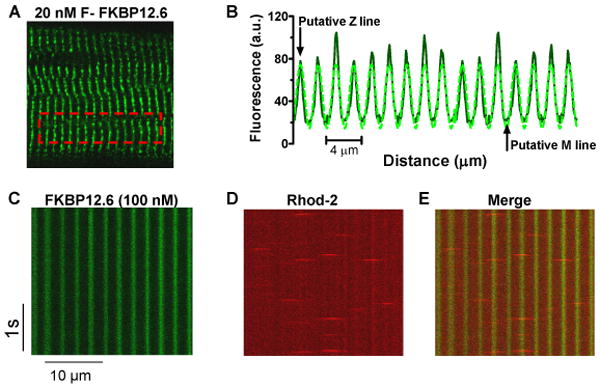
A, Confocal image of saponin-permeabilized rat ventricular myocyte with 20 nM F-FKBP12.6; B, plot profile of striated sarcomeric pattern and sinusoidal fit; C. longitudinal line scan image from myocyte exposed to 100 nM F-FKBP12.6; D, simultaneous Ca spark measurement using rhod-2 as Ca indicator; E, Merged C-D images showing Ca sparks originate from FKBP binding sites.
Effects of cAMP and rapamycin on FKBP binding and resting SR Ca release
Ca sparks are fundamental SR release events during rest and ECC,1 and are a readout of elementary RyR2 function in myocytes. We simultaneously measured Ca sparks and F-FKBP12.6/12 association and dissociation, providing direct online correlation between FKBP-RyR2 interaction and functional consequences.
Figure 2A shows FKBP12 (1 μM) wash-in and Z-line binding (expressed as the difference between Z-line and M-line fluorescence). F-FKBP12 wash-in was rapid, reaching apparent steady state in 2-3 min. Rapamycin (20 μM) caused rapid F-FKBP12 wash-out (rapid decrease of Z-line fluorescence) despite a constant bath [F-FKBP12], but was similar in kinetics to simple washout of F-FKBP12. Neither F-FKBP12 nor rapamycin significantly altered CaSpF (Figure 2B), although rapamycin increased spark duration (Figure 2F). Small apparent decreases in spark amplitude and width were seen with both FKBP12 and rapamycin (Figure 2D-E). These might reflect real FKBP12 actions on RyR2, but the fact that both addition (+FKBP12) and removal (+rapamycin) had identical effects, makes us think these (and any tendency of CaSpF change in Figure 2B) are not simply due to FKBP12. Moreover, wash-out of bath F-FKBP12 caused dissociation of F-FKBP12 from the Z-line (Figure 2A) without any change in CaSpF (not shown). This suggests that dissociation of FKBP12 from RyR2 had little effect on RyR2 activity (consistent with previous reports15,16). Parallel studies using non-fluorescent WT FKBP12 also showed no effect on CaSpF (109±7% of control; Rapamycin 105±8% of control), and both FKBP12 and Rapamycin caused similar slight decreases in Ca spark amplitude and width (12 and 6% respectively; p<0.05), whereas spark duration was unaltered (96-97% of control).
Figure 2. FKBP12-RyR2 association and resting RyR2 activity.

A, Time course of FKBP12 (1μM) wash-in, wash-out, and effects of cAMP and rapamycin on FKBP12 dissociation; B, Normalized effects of FKBP12 & rapamycin on CaSpF and FKBP12-RyR2 binding (n=10-20). Basal CaSpF at 100 nM [Ca]i is 10.1 ± 0.9 sparks (s-1 100μm-1). C, Normalized effects of cAMP-dependent phosphorylation on CaSpF and FKBP12-RyR2 binding (n=10-20). D-F, The effects of FKBP12, rapamycin & cAMP on the Ca spark amplitude, width (FWHM) and duration (FDHM).
Activation of PKA by cAMP to phosphorylate RyR2 (as in 20) did not change FKBP12 binding (Figure 2A), but increased CaSpF by >50%. We previously showed that this CaSpF enhancement was entirely attributed to PLB phosphorylation and consequently increased SR Ca load.20 Therefore, these data suggest that cAMP-dependent PKA phosphorylation does not alter FKBP12 binding to RyR2. Note that bath F-FKBP is an inexhaustible pool, and that RyR2 saturation in intact cells may differ.
Figure 3A shows F-FKBP12.6 (100 nM) wash-in, and Z-line binding also rapidly reached steady state (∼2 min). F-FKBP12.6 binding to RyR2 was paralleled by a small, but highly significant decrease in CaSpF (18%) and a logically consequent increase in SR Ca load (of similar percent, Figure 3B). Spark amplitude and duration were also decreased (Figure 3D, F). These data agree with reports that FKBP12.6 inhibits resting RyR2, thus enhancing SR Ca content.11 Using WT-FKBP12.6 (unlabeled), we found similar results (24±5% decrease in CaSpF, 15±1% decrease in spark amplitude; unaltered width and duration). Addition of cAMP/okadaic acid (OA) did not significantly alter F-FKBP12.6 binding (Figure 3A), but increased both CaSpF and SR Ca load (Figure 3B). Since F-FKBP12.6 binding was unaltered by cAMP, its dissociation cannot explain the enhanced CaSpF. Moreover, a primary enhancement of SR Ca leak should reduce SR Ca load (opposite to observed). We conclude that PKA-dependent phosphorylation does not alter RyR2 binding of FKBP12.6, but enhances CaSpF secondary to PLB phosphorylation-dependent increase in SR Ca content.20
Figure 3. FKBP12.6-RyR2 association and resting RyR2 activity.

A, Time course of FKBP12.6 (100 nM) wash-in and influence of cAMP and rapamycin; B, Normalized FKBP12.6 and cAMP effects on CaSpF, FKBP12.6-RyR2 binding (n=25,17) & SR Ca load (assessed by 10mM caffeine, n=6). Basal control CaSpF is 8.7± 0.6 sparks (s-1 100μm-1). C, Normalized graph of rapamycin effects on CaSpF, FKBP12.6-RyR2 binding, and SR load (n=45,10,7). D-F, FKBP12.6, rapamycin & cAMP effects on Ca spark amplitude, width (FWHM) and duration (FDHM).
Unlike F-FKBP12, wash-out of F-FKBP12.6 was extremely slow upon removal of bath F-FKBP12.6, suggesting much higher affinity for F-FKBP12.6 (see below). Even rapamycin addition after F-FKBP12.6 equilibration only very slowly decreased fluorescence, without significant CaSpF alteration within 10 min. Thus, FKBP12.6 (but not FKBP12) inhibited resting RyR2 in situ, but cAMP-dependent PKA phosphorylation had no acute effect on FKBP12.6/12-RyR2 binding.
FKBP12.6/12 binding kinetics
Three methods were employed to measure FKBP-RyR2 binding affinity and kinetics.
Steady-state Kd and Bmax measurement
To measure FKBP12.6 binding affinity in situ, we incubated permeabilized myocytes with different [F-FKBP12.6]. Figure 4A-B shows that F-FKBP12.6 binding is prevented by 20 μM rapamycin. This also shows that F-FKBP12.6 fluorescence inside myocytes was almost the same as in the bath (providing quantitative control for analysis). Fluorescence intensity at Z-line, M-line, and cell average were plotted vs. bath [F-FKBP12.6] (Figure 4D). The average fluorescence intensity represents the overall bound F-FKBP12.6 in myocytes. FKBP12.6 binding exhibited an apparent dissociation constant (Kd) in ventricular myocytes of ∼1 nM at 100 nM [Ca], higher affinity than a previous report,3 and the linear fluorescence-bath [F-FKBP12.6] relationship (Figure 4C) allowed calibration of the bound [F-FKBP12.6] in myocytes (Figure 4E). The apparent Bmax (∼1 μM) is close to our previous measurements of total concentration of RyR2 monomers in rat ventricular myocytes, based on [3H]-ryanodine binding.26 Therefore, our data agree with RyR2 being the main high-affinity binding site for FKBP12.6, and with a stoichiometry of 4 FKBP12.6 per RyR2 tetramer. Note that additional soluble FKBP binding targets might be lost by myocyte permeabilization. Pretreatment with cAMP/OA did not affect steady-state affinity or Bmax for FKBP12.6 (Figure 4E). Similar Bmax and cAMP results were obtained for steady-state binding of F-FKBP12, except FKBP12 had a much lower affinity (Kd∼200 nM, Figure 4F). Thus, rapamycin prevented FKBP12.6/12 binding to RyR2, but cAMP-dependent PKA phosphorylation had no effect on either apparent Kd or Bmax of FKBP12.6/12-RyR2 binding.
Figure 4. Steady-state Kd and Bmax of F-FKBP in myocytes.
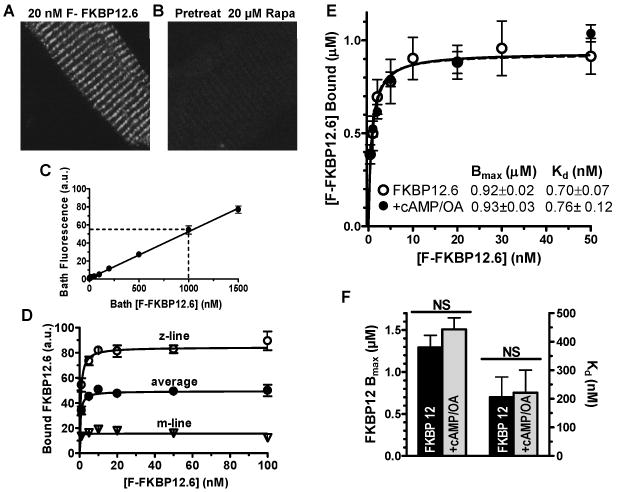
A, Confocal myocyte image with 20 nM bath [FKBP12.6]; B, Same as A but myocyte was pretreated with 20 μM rapamycin; C, Dependence of bath F-FKBP12.6 fluorescence on [F-FKBP12.6]. D, Background-corrected (bath & auto-fluorescence) fluorescence intensity from Z-line, M-line and average, plotted vs. bath [FKBP12.6]. E, Calibrated F-FKBP12.6 binding using known bath concentration and fluorescence (fit with one-site isotherm; n=4 rats). F, Influence of cAMP/OA pretreatment on apparent Bmax and Kd (n=4 rats).
Binding kinetics of FKBP12.6/ 12 measured by FRAP
Even if cAMP doesn't affect Kd of FKBP12.6/12-RyR2, it might change association and dissociation rate constants (kon and koff, respectively). We used fluorescence recovery after photobleaching (FRAP) to investigate binding kinetics of FKBP12.6/12.27
Figure 5A shows a myocyte equilibrated with 1 nM F-FKBP12.6, immediately after photobleaching 3 regions of interest (ROIs), and where striations have partially recovered at 30 min (right). This FRAP indicates replacement of photobleached F-FKBP12.6 with fresh F-FKBP12.6 from the bath, and depends on kon and koff.28 Figure 5B shows exponential fits of FRAP with first order rate constant, kFRAP. This provides an integrated measurement of kon and koff (kFRAP = kon [FKBP12.6] + koff) and measurement at different [F-FKBP12.6] allows determination of kon, koff and Kd (Figure 5C).28 The apparent Kd =1.3 nM for FKBP12.6-RyR2 agrees with our steady-state Kd measurements. Rapamycin (20 μM) completely prevented FRAP (Figure 5C). Inclusion of cAMP/OA caused no significant change in either kon or koff, suggesting that cAMP-dependent PKA phosphorylation does not affect the binding kinetics of FKBP12.6 to RyR2.
Figure 5. FRAP measurement of kon and koff of F-FKBP in myocytes.
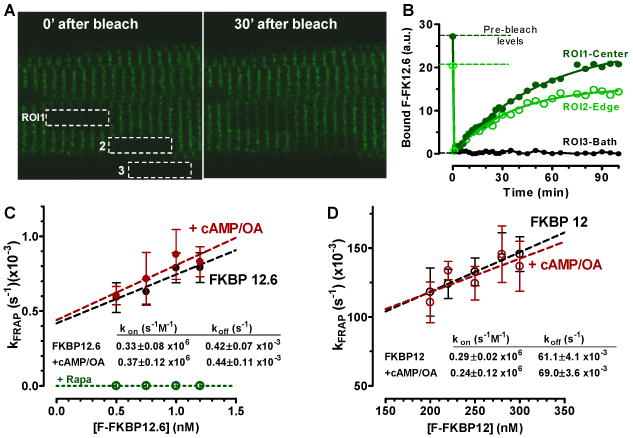
A, Three regions of interest (ROI) from a permeabilized myocyte equilibrated with 1 nM F-FKBP12.6 were photobleached (time 0 and 30 min after bleach). B, Time course of FRAP at 3 ROIs. C, Dependence of FRAP rate constant (kFRAP) on [F-FKBP12.6], and FRAP block by rapamycin. D, Same as in C, but for F-FKBP12.
Diffusion of F-FKBP12.6 is >1000 times faster than FRAP and is thus not rate limiting (e.g. extracellular FRAP in ROI3 was <2 s vs. ∼1000 s in the cell; Figure 5B). FRAP kinetics were also the same at the cell center or edge (ROI1 vs. 2), suggesting that intracellular F-FKBP12.6 diffusion is not rate-limiting in our FRAP measurement (see also below).
FKBP12 has a much lower affinity for RyR2, which accelerates FRAP compared to FKBP12.6. This has two practical consequences. First, FKBP12 FRAP must be measured on a faster time scale, using line-scan mode.27 Second, F-FKBP12 diffusion may contribute to the measured FRAP, so it must be assessed. For line FRAP, we bleach a single line and monitor FRAP in that line at much lower laser power. As diffusion controls, we measured FRAP of F-dextran (MW = 10,000 Da) in the myocyte (Online Figure IV), which recovered with a single time constant (τ ∼1.7 s =1/kFRAP), and F-FKBP12 where binding was blocked by non-fluorescent FKBP12.6 (or rapamycin), giving τ∼1 s. FRAP of FKBP12 (without blocking binding) was always best fit with a double exponential with a fast component (τ ∼1 s; taken as diffusion), and a slower component (τ =6-10 s) attributed to RyR2 binding. The slower kFRAP component was plotted as a function of [F-FKBP12] to extract kon, koff and Kd (Figure 5D). The kon for FKBP12 was similar to that of FKBP12.6, whereas koff was 150 times faster than FKBP12.6, and consequently Kd (210 nM) was ∼150 times higher than for FKBP12.6 and agrees with the steady-state Kd measurements. Similar experiments were done in the presence of cAMP/OA, which again had no effect on kon, koff or Kd of FKBP12-RyR2 binding.
We previously showed that this cAMP/OA treatment in permeabilized ventricular myocytes leads to maximal RyR2 phosphorylation (32P incorporation) within 5 min.20 To test if RyR2 phosphorylation increased here, we assessed RyR2 phosphorylation level at Ser2809 by quantitative western blot. Figure 7C shows results using isolated myocytes treated as in the confocal studies. Basal phosphorylation at Ser2809 is substantial (as in our previous reports20,29). Permeabilization (prior to cAMP/OA exposure) produced a trend toward increase, but cAMP/OA treatment approximately doubled phosphorylation to what may be maximal phosphorylation.
Figure 7. Endogenous FKBP12.6/12 quantification.
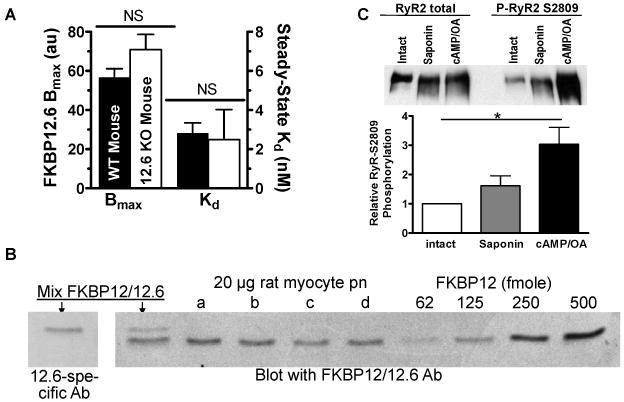
A, Apparent Kd and Bmax measurement in myocytes from WT and FKBP12.6-KO mice (n=3 mice, 50-100 myocyte/ mouse). B, Immunoblot quantification of endogenous total FKBP12/12.6 from intact rat myocytes (n=4 rats). C. RyR2 phosphorylation detected by P-S2808 specific antibody, and normalized to RyR protein, for myocytes treated as in Figure 3A (before and 5 min after saponin exposure, and 30 min after cAMP/OA and FKBP12.6 exposure).
Binding kinetics measured by continuous F-FKBP wash-in/out
Figure 6A shows the wash-in of 1 nM F-FKBP12.6. Higher [F-FKBP12.6] greatly enhances the rate constant of wash-in, kin (e.g. Figure 6B) and is stable during 30 min. Similar to FRAP analysis, kin depends on both kon and koff (kin = kon [F-FKBP] + koff). After steady-state, the superfusate contained 33 nM WT-FKBP12.6 (but lacked F-FKBP to prevent rebinding and have wash-out rate approximate koff). The wash-out rate constant was no different in the absence of WT-FKBP12.6, suggesting that rebinding was negligible. The inferred kon and koff here are more variable, but agree with the FRAP measurements for both FKBP12 and 12.6 (Figure 6C-E and Online Table I). Wash-in/out experiments were also done after cAMP/OA exposure, and again binding characteristics were not altered for FKBP12 or 12.6 (Figure 6C-E & Online Figure V).
Figure 6. Wash-in and wash-out of FKBP.

A. FKBP12.6 wash-in (1 nM) and wash-out (33 nM non-fluorescent FKBP12.6), with exponential fits (rising: ymax(1- exp(-kin t)); falling: y0exp(-koff t), where kin = kon [FKBP12.6] + koff). B, F-FKBP12.6 wash-in (1 and 100 nM). C-D, Neither cAMP nor rapamycin changes the koff of FKBP12.6/12-RyR2 binding (τwashout=1/koff). E, FKBP12.6 and 12 have comparable kon, and cAMP does not alter kon of FKBP12.6/12-RyR2 binding.
Remarkably, rapamycin did not alter F-FKBP12.6 (or F-FKBP12) wash-out kinetics (Figure 6C-E, Online Figure V). We infer that the rapamycin binding site on FKBP is only accessible when FKBP is not RyR2-bound. Thus, rapamycin acts by complexing with free FKBP12.6 to prevent its binding to RyR2, without interfering with RyR2-associated FKBP12.6.
Endogenous FKBP12.6/12 levels in intact rat myocytes
Given the extremely slow koff of FKBP12.6 from RyR2 (τ>30 min; Figures 5-6), we expect some endogenous FKBP12.6 to be retained after myocyte permeabilization, which could complicate our interpretation. So, it was surprising that the measured Bmax (Figure 4) matched the RyR2 monomer concentration in rat myocytes,26 and that there was no discernable slow wash-in phase at high [F-FKBP12.6] (e.g. Figures 3A, 6B) as endogenous FKBP12.6 gradually dissociated. This raises the possibility that endogenous [FKBP12.6] in myocytes is low compared to RyR2, giving a low fractional occupancy of RyR2 with FKBP12.6 in myocytes. That would also explain why RyR2 activity is virtually unaffected in FKBP12.6-KO mice.13 We measured amounts of endogenous FKBP12.6 and FKBP12 using three strategies.
First, we compared total available F-FKBP12.6 binding sites in WT vs. FKBP12.6-KO mouse myocytes (as in Figure 6B with 100 nM F-FKBP12.6). If WT mice exhibit substoichiometric occupancy of RyR2s by endogenous FKBP12.6, the apparent Bmax would be lower in WT vs. FKBP12.6-KO mice. We took advantage of the very slow koff and fast kon of FKBP12.6, washing in saturating [F-FKBP12.6] (100 nM) to rapidly reach steady-state binding (within 10 min), and minimize endogenous FKBP12.6 dissociation. F-FKBP12.6 had the same binding affinity for RyR2 in WT and FKBP12.6-KO myocytes, but Bmax was 20% smaller in WT vs. KO mice (not significant; Figure 7A). Thus, at most ∼20% of RyR2 sites are occupied with native FKBP12.6 in WT myocytes (consistent with 100-200 nM cellular [FKBP12.6], assuming 0.5-1 μmol/L cytosol of RyR2 monomer).26
Second, we intentionally pre-depleted myocytes of native FKBP12.6 by pre-incubating intact myocytes with rapamycin (2 μM) both before (1-2 hr, 37°C) and during permeabilization (followed by washout of rapamycin). In those studies, the apparent Bmax from F-FKBP12.6 experiments was only slightly increased by pre-depletion (10-15%, not shown).
Third, we directly measured endogenous FKBP12.6/12 in intact rat ventricular myocytes by western blot and FKBP concentration standards to establish a calibration curve (Figure 7B). The first two lanes show a mixture of FKBP12/12.6 detected with antibodies that are FKBP12.6-specific (from Dr. Laurent Vignet, France) and to a common epitope in both FKBPs. FKBP12 and 12.6 migrate slightly differently allowing simultaneous detection. The average total FKBP12 was 6 ± 1.5 fmol/μg cell lysate, which implies 1.05 μmol/L cytosol of FKBP12 (based on 168 mg myocyte protein/ml cytosol30). We could not detect FKBP12.6 with either antibody in myocyte homogenates. Therefore the total amount of FKBP12.6 must be lower than the lowest detectable FKBP12.6 standard, i.e. [FKBP12.6] ≤ 100-150 nmol/L cytosol. This agrees with the independent analysis above (at 100 nM, only 10-15% of RyR2 monomers in the myocyte can possibly have FKBP12.6 bound), and the relative predominance of FKBP12 vs. 12.6 observed by many investigators. Thus, FKBP12:FKBP12.6 is >10:1 in cardiac myocytes, and [FKBP12] is roughly stoichiometric with RyR2 monomer concentrations.
Discussion
Using fluorescent FKBP and confocal microscopy of cardiac myocytes, we: 1) made the first in situ characterization of FKBP-RyR2 binding properties (on/off rates & Kd), 2) defined functional consequences of FKBP-RyR2 binding, 3) obtained further evidence that PKA-driven phosphorylation does not affect FKBP-RyR2 binding in the permeabilized myocyte setting, 4) estimated the amount of endogenous FKBP12.6 in mouse and rat myocytes, and 5) clarified the mechanism of rapamycin interference with FKBP/RyR binding.
FKBP12.6/12 binds specifically to RyR2
We used several approaches to ascertain that RyR2 is the predominant myocyte FKBP target. First, the F-FKBP12/12.6 striation patterns overlap Di-8-ANEPPS at T-tubules, where RyR2 is known to localize. Second, Ca sparks originated from F-FKBP labeled sites. Third, preincubation of myocytes with unlabeled FKBP12.6 completely abolished F-FKBP12 striations. Fourth, the FKBP Bmax measured equaled the concentration of myocyte RyR2 monomers. Fifth, we found strong and distinct FRET between F-FKBP12/12.6 donors and acceptor-labeled domains of CaM (F-CaM) in permeabilized myocytes31, which was similar to the FRET reported in skeletal muscle SR and purified RyR124. Thus, in permeabilized cardiac myocytes, FKBP12.6 binds primarily to RyR2.
FKBP12.6/12 binding properties and their functions in heart
A goal here was to measure FKBP-RyR2 interaction kinetics in situ in cardiac myocytes, since previous work used SR vesicles or cell lysates.2-4 Three independent methods demonstrated that FKBP12.6 binds RyR2 with very high affinity (∼1 nM) and slow koff, giving average dwell time of FKBP12.6 on RyR2 of about 20 min, thus unlikely to modulate beat-to-beat. This, and the substoichiometric FKBP12.6:RyR2 and lack of FKBP12 effect on CaSpF, explains why FKBP12.6 overexpression can decrease resting Ca leak and consequently increases SR Ca content and Ca transients11,32. Indeed, FKBP12.6 here inhibited CaSpF and amplitude, while increasing SR Ca load (although the effects were modest).
Rapamycin did not accelerate FKBP12.6 dissociation from RyR2, but instead, prevented rebinding of FKBP12.6 to RyR2. Thus, it is the slow off-rate of FKBP12.6 that determines the velocity of rapamycin effects on RyR2 activity. This clarifies various previous results where rapamycin produced either unaltered RyR2 activity (or heart function) or only small changes in spark duration or width, etc.7 However, lengthy rapamycin treatment indeed reverses FKBP12.6 effects in overexpression models11,32, and in biochemical experiments14,21.
FKBP12 has a similar on-rate as FKBP12.6, but much faster off-rate and thus lower affinity. We could not detect effects of FKBP12 on RyR2 on CaSpF. This agrees with reports showing that co-expressing FKBP12.6 but not FKBP12 decreased Ca release triggered by an RyR2 activator (4-chloro-m-cresol) in CHO cells expressing hRyR216,18. However, FKBP12-deficient mice have normal skeletal muscles but severe dilated cardiomyopathy33. Overexpression of FKBP12 in rabbit cardiomyocytes caused reduced CaSpF, and increased spark amplitude and duration, and Ca transients17. While these imply FKBP12 regulation of RyR2, the 48 hr rapamycin incubation required to reverse the effects, FKBP12 should rapidly dissociate from RyR2 (Figure 2A). Thus indirect pathways cannot be excluded34.
FKBP12.6/12 concentration and its physiological role in heart
We estimate endogenous myocyte FKBP12.6 and FKBP12 at 100 nM and 1μM, respectively (Figure 6). Low total [FKBP12.6] and high affinity for RyR2, indicate that >99% of myocyte FKBP12.6 is bound to RyR2, with subnanomolar free FKBP12.6. Our data also suggest that ≤20% of FKBP binding sites are occupied by native FKBP12.6 (Figure 7), so >80% of RyR2 are available to bind FKBP12. Furthermore, the fast koff of FKBP12 means that permeabilizing myocytes allows endogenous FKBP12 to diffuse out (τ ∼25 s).
Over 80% of endogenous RyR2 are FKBP12.6-free. If FKBP12.6 binding were critical for normal RyR2 function (preventing SR Ca depletion and arrhythmias), then both normal myocytes and FKBP12.6-KO mice would be in an extremely precarious situation. Notably, female FKBP12.6-KO mice have no cardiac dysfunction, myocytes exhibit enhanced Ca transients and larger Ca sparks35 and bilayer experiments showed unaltered RyR2 activity14. While one group found exercise-induced arrhythmias and RyR2 channel hyperactivation in FKBP12.6-KO mice (only after exercise),9 another found no difference in stress-induced arrhythmias.14 This controversy is unresolved, but whether there is zero FKBP12.6 or 10-20% saturation of RyR2 (WT myocytes), SR Ca uptake and release can function relatively normally. Thus, while FKBP12.6 can inhibit diastolic RyR2 activity, the effect is moderate, and in vivo most RyR2 must achieve physiological stability somehow other than FKBP12.6 binding.
These permeabilized myocyte studies are most relevant to diastolic SR Ca release, which can be arrhythmogenic, and SR Ca release during Ca current triggered release may be differently regulated. For example, PKA can accelerate the rate of SR Ca release and inactivation in myocytes when Ca current and SR Ca load are held constant36.
Relevance to HF: PKA-driven phosphorylation & FKBP12.6/12-RyR2 binding
Defective RyR2 regulation is linked to cardiac arrhythmias and contractile dysfunction in HF, via diastolic SR Ca leak via RyR2.9,19,37-39 Extensive publications by Marks and coworkers support the theory that, in HF, RyR2 hyperphosphorylation by PKA disrupts FKBP12.6-RyR2 binding, thereby enhancing SR Ca leak and unloading9,19,37,40,41,42. Some data from others support components of this model,39,43 but key aspects have been challenged by work showing either that RyR2 hyperphosphorylation fail to occur in HF, that PKA-dependent RyR2 phosphorylation does not cause FKBP12.6 dissociation, RyR2 activation, or SR Ca leak, or that FKBP12.6 does not alter RyR2 gating20,21,40. Our results cannot resolve this controversy, but provide new quantitative information about FKBP binding in the normal myocyte environment, with directly parallel functional data. In particular, we did not find any effect of PKA-dependent RyR2 phosphorylation on FKBP12.6 binding (measured 3 different ways). On the other hand, we detected significant diastolic RyR2 inhibition by FKBP12.6 (but not FKBP12). Our finding of low fractional saturation of RyR2 with FKBP12.6 in myocytes, also means that even if FKBP12.6 association is reduced in HF, it could only occur at a minority of RyR2 (which would make those RyR2s like the others, in that respect). Of course, other effects in HF (e.g., redox modulation or CaMKII-dependent phosphorylation39,44,45) may alter RyR2 responsiveness to FKBP or PKA-dependent phosphorylation.
Novelty and Significance.
What is Known?
Small intracellular immunophilins FKBP12 and 12.6 bind to the cardiac ryanodine receptor (RyR2) and modulate its function as the SR Ca release channel.
In some studies PKA-dependent RyR2 phosphorylation (especially during heart failure) has been reported to cause the dissociation of FKBP12.6 t leading to RyR2 leak, and thereby, reduced SR Ca content and arrhythmogenesis. Other investigators have reported opposite results.l.
FKBP12.6 is known to bind RyR2 with higher affinity than FKBP12, but there are no direct measurements of binding in the physiological cardiac myocyte environment
What New Information Does This Article Contribute?
The first direct measurements of FKBP12/12.6 binding kinetics with RyR2 in the adult cardiac myocytes using fluorescent FKBP and confocal microscopy. The Kd for FKBP12.6 is ∼1 nM and for FKBP12 is ∼200 nM.
Simultaneous measurements of RyR2 function (Ca sparks) and FKBP binding measuring FKBP12 binding had no functional effect, while FKBP12.6 inhibited Ca sparks and enhanced SR Ca content.
Measurements of the in situ effects of PKA-driven RyR2 phosphorylation on FKBP association and RyR2 function (there was no detectable effect).
The endogenous myocyte concentrations of FKBP12 is 1 μM and that of FKBP12.6 <100 nM, whereas the concentration of RyR2 is ∼1 μM.
This study was designed in part to address the controversy in this field regarding the role of FKBP12/12.6 and PKA-dependent RyR2 phosphorylation and the relative lack of fundamental data about these issues, especially in the native cardiac myocytes. We developed a novel method to simultaneously measure FKBP binding to RyR2 and RyR2 function, allowing unique physiological insight into this interaction. We used three different ways to measure binding properties (Kd, koff and kon), all of which gave comparable results. This work provides novel and important quantitative constraints. One major constraint is that the number of FKBP12.6 molecules in the normal myocyte is much smaller than the number of RyR2 monomers. Thus, only 10-20% of RyR2 in myocytes can be subject to FKBP12.6 modulation. Other major constraints are that binding of FKBP12.6 (but not FKBP12) inhibits Ca sparks, and that PKA-dependent RyR2 phosphorylation does not alter FKBP12/12.6 binding. Therefore, the increase in SR Ca content and Ca sparks by PKA must be mediated by other pathways (e.g., phospholamban phosphorylation and SR Ca-ATPase stimulation).
Supplementary Material
Acknowledgments
We thank Florentin Nitu for technical support, Drs. Seth Robia, Sanda Despa and Julie Bossuyt for critical comments, and Dr. Susan Hamilton for providing FKBP12.6-KO mice.
Sources of Funding: NIH grants HL30077, HL80101 (DMB); HL92097 (DMB and RC) and HL76433 (BRF).
Non-standard Abbreviations and Acronyms
- τ
time constant
- Bmax
binding maximum
- CaM
calmodulin
- CaMKII
calcium/calmodulin-dependent protein kinase II
- cAMP
cyclic adenosine monophosphate
- CaSpF
calcium spark frequency
- CD
circular dichroism
- Di-8-ANEPPS
Pyridinium, 4-[2-[6-(dioctylamino)-2-naphthalenyl]ethenyl]-1-(3-sulfopropyl)-, inner salt
- ECC
excitation-contraction coupling
- F
fluorescence
- F0
resting fluorescence
- FDHM
full-duration at half maximum
- F-FKBP
fluorescent FKBP
- F-FKBP12
fluorescent FKBP12
- F-FKBP12.6
fluorescent FKBP12.6
- FKBP
FK506 binding protein
- FKBP12.6-KO
Mice lacking FKBP12.6
- FRAP
fluorescence recovery after photobleaching
- FRET
fluorescence resonance energy transfer
- FWHM
full-width at half maximum
- HF
heart failure
- k
rate constant
- Kd
dissociation constant
- koff
dissociation rate
- kon
association rate
- MRE
molar residue ellipticity
- MW
molecular weight
- OA
okadaic acid
- PKA
cAMP-dependent protein kinase A
- PLB
phospholamban
- PLB-KO
PLB knock-out
- Rhod-2
Xanthylium, 9-[4-[bis[2-[(acetyloxy)methoxy]-2-oxoethyl]amino]-3-[2-[2-[bis[2-[(acetyloxy)methoxy]-2-oxoethyl]amino]phenoxy]ethoxy]phenyl]-3,6-bis(dimethylamino)-, 3K+
- RyR2
cardiac ryanodine receptor
- SEM
standard error of the means
- SERCA
sarcoplasmic/endoplasmic reticulum calcium-ATPase
- SR
sarcoplasmic reticulum
- WT
wild type
- WT-FKBP
non-fluorescent wild type FKBP
Footnotes
Disclosures: None.
References
- 1.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 2.Timerman AP, Ogunbumni E, Freund E, Wiederrecht G, Marks AR, Fleischer S. The calcium release channel of sarcoplasmic reticulum is modulated by FK-506-binding protein. Dissociation and reconstitution of FKBP-12 to the calcium release channel of skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1993;268:22992–22999. [PubMed] [Google Scholar]
- 3.Timerman AP, Onoue H, Xin HB, Barg S, Copello J, Wiederrecht G, Fleischer S. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J Biol Chem. 1996;271:20385–20391. doi: 10.1074/jbc.271.34.20385. [DOI] [PubMed] [Google Scholar]
- 4.Jeyakumar LH, Ballester L, Cheng DS, McIntyre JO, Chang P, Olivey HE, Rollins-Smith L, Barnett JV, Murray K, Xin HB, Fleischer S. FKBP binding characteristics of cardiac microsomes from diverse vertebrates. Biochem Biophys Res Commun. 2001;281:979–986. doi: 10.1006/bbrc.2001.4444. [DOI] [PubMed] [Google Scholar]
- 5.Deivanayagam CC, Carson M, Thotakura A, Narayana SV, Chodavarapu RS. Structure of FKBP12.6 in complex with rapamycin. Acta Crystallogr D Biol Crystallogr. 2000;56:266–271. doi: 10.1107/s0907444999016571. [DOI] [PubMed] [Google Scholar]
- 6.Kaftan E, Marks AR, Ehrlich BE. Effects of rapamycin on ryanodine receptor/Ca2+-release channels from cardiac muscle. Circ Res. 1996;78:990–997. doi: 10.1161/01.res.78.6.990. [DOI] [PubMed] [Google Scholar]
- 7.Xiao RP, Valdivia HH, Bogdanov K, Valdivia C, Lakatta EG, Cheng H. The immunophilin FK506-binding protein modulates Ca2+ release channel closure in rat heart. J Physiol. 1997;500(Pt 2):343–354. doi: 10.1113/jphysiol.1997.sp022025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCall E, Li L, Satoh H, Shannon TR, Blatter LA, Bers DM. Effects of FK-506 on contraction and Ca2+ transients in rat cardiac myocytes. Circ Res. 1996;79:1110–1121. doi: 10.1161/01.res.79.6.1110. [DOI] [PubMed] [Google Scholar]
- 9.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D'Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 10.Su Z, Sugishita K, Li F, Ritter M, Barry WH. Effects of FK506 on [Ca2+]i differ in mouse and rabbit ventricular myocytes. J Pharmacol Exp Ther. 2003;304:334–341. doi: 10.1124/jpet.102.041210. [DOI] [PubMed] [Google Scholar]
- 11.Prestle J, Janssen PM, Janssen AP, Zeitz O, Lehnart SE, Bruce L, Smith GL, Hasenfuss G. Overexpression of FK506-binding protein FKBP12.6 in cardiomyocytes reduces ryanodine receptor-mediated Ca2+ leak from the sarcoplasmic reticulum and increases contractility. Circ Res. 2001;88:188–194. doi: 10.1161/01.res.88.2.188. [DOI] [PubMed] [Google Scholar]
- 12.Barg S, Copello JA, Fleischer S. Different interactions of cardiac and skeletal muscle ryanodine receptors with FK-506 binding protein isoforms. Am J Physiol. 1997;272:C1726–1733. doi: 10.1152/ajpcell.1997.272.5.C1726. [DOI] [PubMed] [Google Scholar]
- 13.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 14.Xiao J, Tian X, Jones PP, Bolstad J, Kong H, Wang R, Zhang L, Duff HJ, Gillis AM, Fleischer S, Kotlikoff M, Copello JA, Chen SR. Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J Biol Chem. 2007;282:34828–34838. doi: 10.1074/jbc.M707423200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.George CH, Higgs GV, Mackrill JJ, Lai FA. Dysregulated ryanodine receptors mediate cellular toxicity: restoration of normal phenotype by FKBP12.6. J Biol Chem. 2003;278:28856–28864. doi: 10.1074/jbc.M212440200. [DOI] [PubMed] [Google Scholar]
- 16.George CH, Sorathia R, Bertrand BM, Lai FA. In situ modulation of the human cardiac ryanodine receptor (hRyR2) by FKBP12.6. Biochem J. 2003;370:579–589. doi: 10.1042/BJ20021433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seidler T, Loughrey CM, Zibrova D, Kettlewell S, Teucher N, Kogler H, Hasenfuss G, Smith GL. Overexpression of FK-506 binding protein 12.0 modulates excitation contraction coupling in adult rabbit ventricular cardiomyocytes. Circ Res. 2007;101:1020–1029. doi: 10.1161/CIRCRESAHA.107.154609. [DOI] [PubMed] [Google Scholar]
- 18.George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- 19.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 21.Xiao B, Sutherland C, Walsh MP, Chen SR. Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6) Circ Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- 22.Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- 23.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 24.Cornea RL, Nitu F, Gruber S, Kohler K, Satzer M, Thomas DD, Fruen BR. FRET-based mapping of calmodulin bound to the RyR1 Ca2+ release channel. Proc Natl Acad Sci U S A. 2009;106:6128–6133. doi: 10.1073/pnas.0813010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee EH, Rho SH, Kwon SJ, Eom SH, Allen PD, Kim do H. N-terminal region of FKBP12 is essential for binding to the skeletal ryanodine receptor. JBC. 2004;279(25):26481–8. doi: 10.1074/jbc.M309574200. [DOI] [PubMed] [Google Scholar]
- 26.Bers DM, Stiffel VM. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for E-C coupling. Am J Physiol. 1993;264:C1587–1593. doi: 10.1152/ajpcell.1993.264.6.C1587. [DOI] [PubMed] [Google Scholar]
- 27.Braeckmans K, Remaut K, Vandenbroucke RE, Lucas B, De Smedt SC, Demeester J. Line FRAP with the confocal laser scanning microscope for diffusion measurements in small regions of 3-D samples. Biophys J. 2007;92:2172–2183. doi: 10.1529/biophysj.106.099838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robia SL, Ghanta J, Robu VG, Walker JW. Localization and kinetics of protein kinase C-epsilon anchoring in cardiac myocytes. Biophys J. 2001;80:2140–2151. doi: 10.1016/S0006-3495(01)76187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd. Dordrecht, Netherlands: Kluwer Academic Press; 2001. [Google Scholar]
- 31.Guo T, Cornea R, Fruen B, Bers D. FRET between FKBP and CaM located on the Ryanodine Receptor in the Cardiac Myocyte Environment. Biophys J. 2008;94:2127. [Google Scholar]
- 32.Gomez AM, Schuster I, Fauconnier J, Prestle J, Hasenfuss G, Richard S. FKBP12.6 overexpression decreases Ca2+ spark amplitude but enhances [Ca2+]i transient in rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1987–1993. doi: 10.1152/ajpheart.00409.2004. [DOI] [PubMed] [Google Scholar]
- 33.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 34.Jayaraman T, Marks AR. Rapamycin-FKBP12 blocks proliferation, induces differentiation, and inhibits cdc2 kinase activity in a myogenic cell line. J Biol Chem. 1993;268:25385–25388. [PubMed] [Google Scholar]
- 35.Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, Collier ML, Deng KY, Jeyakumar LH, Magnuson MA, Inagami T, Kotlikoff MI, Fleischer S. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature. 2002;416:334–338. doi: 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- 36.Ginsburg KS, Bers DM. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol. 2004;556:463–80. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 38.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 39.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 40.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, Morley G, Marks AR. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehnart SE, Terrenoire C, Reiken S, Wehrens XH, Song LS, Tillman EJ, Mancarella S, Coromilas J, Lederer WJ, Kass RS, Marks AR. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci U S A. 2006;103:7906–7910. doi: 10.1073/pnas.0602133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase a produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006;98:1506–1513. doi: 10.1161/01.RES.0000227506.43292.df. [DOI] [PubMed] [Google Scholar]
- 44.Zissimopoulos S, Docrat N, Lai FA. Redox sensitivity of the ryanodine receptor interaction with FK506-binding protein. J Biol Chem. 2007;282:6976–6983. doi: 10.1074/jbc.M607590200. [DOI] [PubMed] [Google Scholar]
- 45.Aracena P, Sanchez G, Donoso P, Hamilton SL, Hidalgo C. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003;278:42927–42935. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.