

Abstract
Synthetic combinatorial methods now make it practical to readily produce hundreds of thousands of individual compounds, but it is clearly impractical to screen each separately in vivo. We theorized that the direct in vivo testing of mixture-based combinatorial libraries during the discovery phase would enable the identification of novel individual compounds with desirable antinociceptive profiles while simultaneously eliminating many compounds with poor absorption, distribution, metabolism, or pharmacokinetic properties. The TPI 1346 small-molecule combinatorial library is grouped in 120 mixtures derived from 26 functionalities at the first three positions and 42 functionalities at the fourth position of a pyrrolidine bis-cyclic guanidine core scaffold, totaling 738,192 compounds. These 120 mixtures were screened in vivo using the mouse 55°C warm water tail-withdrawal assay to identify mixtures producing antinociception. From these data, two fully defined individual compounds (TPI 1818-101 and TPI 1818-109) were synthesized. These were examined for antinociceptive, respiratory, locomotor, and conditioned place preference effects. The tail-withdrawal assay consistently demonstrated distinctly active mixtures with analgesic activity that was blocked by pretreatment with the non-selective opioid antagonist, naloxone. Based on these results, synthesis and testing of TPI 1818-101 and 1818-109 demonstrated a dose-dependent antinociceptive effect three to five times greater than morphine that was antagonized by mu- or mu- and kappa-opioid receptor selective antagonists, respectively. Neither 1818-101 nor 1818-109 produced significant respiratory depression, hyperlocomotion, or conditioned place preference. Large, highly diverse mixture-based libraries can be screened directly in vivo to identify individual compounds, potentially accelerating the development of promising therapeutics.
Key words: analgesia, in vivo, mixture-based libraries, opioid, testing
INTRODUCTION
Opioid agonists such as morphine are still the “gold standard” for producing a relief of pain (antinociception), but their clinical use is complicated by significant side effects, including respiratory distress, slowing of intestinal motility, tolerance, and abuse (1), spurring the search for novel opioid analgesics with fewer liabilities of use.
Mixture-based synthetic combinatorial libraries are effective tools for generating novel lead compounds in a fraction of the time and cost of equivalent individual compound arrays (see (2,3) for review). These include large libraries of peptides, peptidomimetics, and heterocycles. Such mixture-based combinatorial libraries may be composed of hundreds of thousands of different compounds (4–9). Importantly, it is impractical for the vast majority of laboratories to screen such large numbers of individual compounds, and the harsh reality is that the majority of drug candidates lack desirable drug-like properties at later stages of testing, resulting in a high rate of attrition in the traditional drug discovery process (10).
To circumvent the limitations of existing screening methods, we tested samples from a mixture-based combinatorial library directly in vivo for analgesic properties. Guiding this objective, we initiated our study using the scaffold ranking technique (3). Five small-molecule combinatorial libraries in our collection, TPI 1343, 1344, 1345, 1346, and 1347, were tested as five single mixture-based samples each made up of 738,192 compounds (Fig. 1a). The most active of these sample mixtures in analgesic assays, the 1346 series, was then analyzed using the positional scan approach (11,12) to determine the most active antinociceptive mixtures in the library. The 1346 library is built around a core pyrrolidine bis-cyclic guanidine scaffold (Fig. 1a), with 26 different functionalities at each of the first three diversity positions and 42 functionalities at the fourth position (Table I), resulting in 120 mixtures in this library, varying from 17,576 to 28,392 compounds each, for a total of 738,192 compounds. The most active mixtures in the 1346 series were then used to guide the synthesis of compounds defined in two positions, the 1802 series, and subsequently determine the synthesis of two individual compounds, the 1818 series.
Fig. 1.

a Five different synthetic combinatorial libraries (1343-1347) are derived from the same resin bound N-acylated tetrapeptide 1. The N-acylated tetrapeptide 1 is synthesized on MBHA resin using standard Boc chemistry. a Borane-THF 50°C, 72 h followed by Piperidine at 50°C, 24 h. b HF/anisole 7 h. c Oxalyldiimidazole/DMF, 24 h. d CNBr/DCM, 24 h. e Thiocarbonyldiimidazole/DCM, 24 h. f HF/anisole 1.5 h. Compounds from 1346, 1802, and 1818 libraries all have the same 1346 framework and are synthesized using the same synthetic pathway. b Samples 1818-101 (left) and 1818-109 (right) were synthesized using the synthesis strategy described above. 1818-101 combines the following functionalities from 1346: R1 functionality 14 S-2-hydroxymethyl, R2 functionality 51 S-cyclohexyl, R3 78 R-cyclohexyl and R4 91 2-ethoxybenzyl, while 1818-109 combines the following functionalities from 1346: R1 functionality 14 S-2-hydroxymethyl, R2 functionality 51 S-cyclohexyl, R3 78 R-cyclohexyl, and R4 93 3,4-dichlorobenzyl
Table I.
Identities of 1346 Library Functionality Groups: R1, R2, R3, and R4 Correspond to Positions on the Pyrrolidine Bis-cyclic Guanidine Core Scaffold, 1346 (see Fig. 1)
| R1 | R2 | R3 | R4 | R1,2,3: amino acid R4: carboxylic acid | Functionality |
|---|---|---|---|---|---|
| 1 | 27 | 53 | Boc-L-Ala | S-methyl | |
| 2 | 28 | 54 | Boc-L-Phe | S-benzyl | |
| 3 | 29 | 55 | Boc-Gly | Hydrogen | |
| 4 | 30 | 56 | Boc-L-Ile | S-2-butyl | |
| 5 | 31 | 57 | Boc-L-Leu | S-isobutyl | |
| 6 | 32 | 58 | Boc-L-Ser(Bzl) | R-hydroxymethyl | |
| 7 | 33 | 59 | Boc-L-Thr(Bzl) | (R,R)-1-hydroxyethyl | |
| 8 | 34 | 60 | Boc-L-Val | S-isopropyl | |
| 9 | 35 | 61 | Boc-L-Tyr(BrZ) | S-4-hydroxybenzyl | |
| 10 | 36 | 62 | Boc-D-Ala | R-methyl | |
| 11 | 37 | 63 | Boc-D-Phe | R-benzyl | |
| 12 | 38 | 64 | Boc-D-Ile | R-2-butyl | |
| 13 | 39 | 65 | Boc-D-Leu | R-isobutyl | |
| 14 | 40 | 66 | Boc-D-Ser(Bzl) | S-hydroxymethyl | |
| 15 | 41 | 67 | Boc-D-Thr(Bzl) | (S,R)-1-hydroxyethyl | |
| 16 | 42 | 68 | Boc-D-Val | R-isopropyl | |
| 17 | 43 | 69 | Boc-D-Tyr(BrZ) | R-4-hydroxybenzyl | |
| 18 | 44 | 70 | Boc-L-Phenylglycine | S-phenyl | |
| 19 | 45 | 71 | Boc-L-Norvaline | S-propyl | |
| 20 | 46 | 72 | Boc-D-Norvaline | R-propyl | |
| 21 | 47 | 73 | Boc-L-Norleucine | S-butyl | |
| 22 | 48 | 74 | Boc-D-Norleucine | R-butyl | |
| 23 | 49 | 75 | Boc-L-Naphthylalanine | S-2-naphthylmethyl | |
| 24 | 50 | 76 | Boc-D-Naphthylalanine | R-2-naphthylmethyl | |
| 25 | 51 | 77 | Boc-L-Cyclohexylalanine | S-cyclohexyl | |
| 26 | 52 | 78 | Boc-D-Cyclohexylalanine | R-cyclohexyl | |
| 79 | 1-Phenyl-1-cyclopropanecarboxylic acid | (1-Phenyl-cyclopropyl)-methyl | |||
| 80 | 2-Phenylbutyric acid | 2-Phenylbutyl | |||
| 81 | 3-Phenylbutyric acid | 3-Phenylbutyl | |||
| 82 | m-Tolylacetic acid | m-tolylethyl | |||
| 83 | 3-Fluorophenylacetic acid | 2-(3-Fluoro-phenyl)-ethyl | |||
| 84 | 3-Bromophenylacetic acid | 2-(3-Bromo-phenyl)-ethyl | |||
| 85 | (α-α-α-Trifluoro-m-Tolyl) acetic acid | 2-(3-Trifluoromethyl-phenyl)-ethyl | |||
| 86 | p-Tolylacetic acid | p-tolylethyl | |||
| 87 | 4-Fluorophenylacetic acid | 2-(4-Fluoro-phenyl)-ethyl | |||
| 88 | 3-Methoxyphenylacetic acid | 2-(3-Methoxy-phenyl)-ethyl | |||
| 89 | 4-Bromophenylacetic acid | 2-(4-Bromo-phenyl)-ethyl | |||
| 90 | 4-Methoxyphenylacetic acid | 2-(4-Methoxy-phenyl)-ethyl | |||
| 91 | 4-Ethoxyphenylacetic acid | 2-(4-Ethoxy-phenyl)-ethyl | |||
| 92 | 4-Isobutyl-α-methylphenylacetic acid | 2-(4-Isobutyl-phenyl)-propyl | |||
| 93 | 3,4-Dichlorophenylacetic acid | 3,4-Dichlorophenethyl | |||
| 94 | 3,5-Bis(Trifluoromethyl)-phenylacetic acid | 2-(3,5-bis-trifluoromethyl-phenyl)-ethyl | |||
| 95 | 3-(3,4-Dimethoxyphenyl)-propionic acid | 3-(3,4-Dimethoxy-phenyl)-propyl | |||
| 96 | Phenylacetic acid | Phenethyl | |||
| 97 | 3,4,5-Trimethoxybenzoic acid | 3,4,5-Trimethoxy-benzyl | |||
| 98 | Butyric acid | Butyl | |||
| 99 | Heptanoic acid | Heptyl | |||
| 100 | Isobutyric acid | Isobutyl | |||
| 101 | 2-Methylbutyric acid | 2-Methylbutyl | |||
| 102 | Isovaleric acid | 3-Methylbutyl | |||
| 103 | 3-Methylvaleric acid | 3-Methylpentyl | |||
| 104 | 4-Methylvaleric acid | 4-Methylpentyl | |||
| 105 | p-Toluic acid | 4-Methyl-benzyl | |||
| 106 | Cyclopentanecarboxylic acid | Cyclopentyl-methyl | |||
| 107 | Cyclohexanecarboxylic acid | Cyclohexyl-methyl | |||
| 108 | Cyclohexylacetic acid | Cyclohexyl-ethyl | |||
| 109 | Cyclohexanebutyric acid | Cyclohexyl-butyl | |||
| 110 | Cycloheptanecarboxylic acid | Cycloheptyl-methyl | |||
| 111 | 2-Methylcyclopropanecarboxylic Acid | (2-Methyl-cyclopropyl)-methyl | |||
| 112 | Cyclobutanecarboxylic Acid | Cyclobutyl-methyl | |||
| 113 | 3-Cyclopentylpropionic Acid | 3-Cyclopentyl-propyl | |||
| 114 | Cyclohexanepropionic Acid | Cyclohexyl-propyl | |||
| 115 | 4-Methyl-1-Cyclohexanecarboxylic Acid | 4-Methyl-1-cyclohexyl-methyl | |||
| 116 | 4-tert-Butyl-Cyclohexanecarboxylic Acid | 4-tert-butyl-cyclohexyl-methyl | |||
| 117 | 4-Biphenylacetic Acid | 2-Biphenyl-4-yl-ethyl | |||
| 118 | 1-Adamantanecarboxylic Acid | Adamantan-1-yl-methyl | |||
| 119 | 1-Adamantaneacetic Acid | 2-Adamantan-1-yl-ethyl | |||
| 120 | 2-Norbornaneacetic Acid | 2-Bicyclo[2.2.1]hept-2-yl-ethyl |
We hypothesize that the direct in vivo screening of mixture-based combinatorial libraries will yield therapeutically useful individual analgesics while simultaneously decreasing the failure rate inherent in the traditional drug discovery process. To this end, individual compounds were compared to morphine in a mouse assay of antinociception, the 55°C warm water tail-withdrawal test, while also evaluated for effects on respiration rate, locomotor activity, and conditioned place preference as models for respiratory distress and substance abuse.
MATERIALS AND METHODS
Synthesis of the Libraries TPI 1343, 1344, 1345, 1346, and 1347, with Notes Regarding the TPI 1346 Library
Small-molecule mixture-based libraries 1343 to 1347 were all derived from the same resin bound N-acylated peptide 1 (Fig. 1a) as described earlier (13) utilizing the libraries from libraries approach (8,14). Briefly, the five scaffolds share the same R group functionalities (Table I), but differ in the structure of the core scaffold (Fig. 1a). The R1 through R3 functionalities are derived from 26 amino acids and the R4 functionalities are derived from 42 carboxylic acids such that each of the five libraries contains 738,192 (26 × 26 × 26 × 42) compounds systematically arranged into 120 (26 + 26 + 26 + 42) mixture samples. Utilizing the scaffold ranking method (3), a single sample was made for each library that contained an approximately equal molar mixture of all 738,192 compounds in that library.
The synthesis of the pyrrolidine bis-cyclic guanidine library, 1346, is described in Fig. 1a and elsewhere (13). Notably, this library has been used by our group to identify individual compounds with antimicrobial activity against drug-resistant Gram-positive pathogens (13) as well as affinity for the mu-opioid receptor (MOR) (3). Briefly, the first 26 samples (Table I and Fig. 1a) permit the assessment of the activity of the 26 different functionalities used at the R1 position. In these 26 mixtures, each of the samples has a fixed functionality at the R1 position and an equal molar mixture of the 26 functionalities at R2, 26 functionalities at R3, and 42 functionalities at R4. For example, sample 1 contains an equal molar mixture of 28,392 (26 × 26 × 42) compounds all fixed with S-methyl at the R1 position, whereas sample 2 contains an equal molar mixture of 28,392 compounds all fixed with S-benzyl at the R1 position. In this way, samples 1 to 26 scan the first position by fixing each of the 26 samples with a different functionality at the R1 position. The next 26 samples (samples 27 to 52) assess the functionalities at R2 by fixing the R2 position and having equal molar mixtures at the other three positions. Likewise, samples 53 to 78 assess the R3 functionalities, and samples 79 to 120 assess the R4 functionalities.
Synthesis of the TPI 1802 Library
To confirm the activities of the constituent groups in combination, we used an iterative approach and synthesized 25 mixtures. The 25 mixtures were synthesized using the same synthetic scheme described for 1346 in Fig. 1 (13). The 25 mixtures were designed by fixing the R1 and R2 positions with the combination of the top five functionalities in R1 and R2 from the in vivo screening of 1346. The R3 and R4 positions in these samples contain equal molar mixtures of the 26 R3 functionalities and 42 R4 functionalities present in 1346 (Table II). Each sample in the 1802 series contains an equal molar mixture of 1,092 compounds (26 × 42).
Table II.
Identities of 1802 Library Functionality Groups: R1, R2, R3, and R4 Correspond to Positions on the Pyrrolidine Bis-cyclic Guanidine Core Scaffold, 1346 (see Fig. 1)
| 1802 | R1 | R2 | R3 | R4 |
|---|---|---|---|---|
| 1 | R-benzyl | S-isobutyl | X | X |
| 2 | R-benzyl | R-2-butyl | X | X |
| 3 | R-benzyl | S-2-napthylmethyl | X | X |
| 4 | R-benzyl | S-cyclohexyl | X | X |
| 5 | R-benzyl | S-hydroxymethyl | X | X |
| 6 | S-hydroxymethyl | S-isobutyl | X | X |
| 7 | S-hydroxymethyl | R-2-butyl | X | X |
| 8 | S-hydroxymethyl | S-2-napthylmethyl | X | X |
| 9 | S-hydroxymethyl | S-cyclohexyl | X | X |
| 10 | S-hydroxymethyl | S-hydroxymethyl | X | X |
| 11 | R-butyl | S-isobutyl | X | X |
| 12 | R-butyl | R-2-butyl | X | X |
| 13 | R-butyl | S-2-napthylmethyl | X | X |
| 14 | R-butyl | S-cyclohexyl | X | X |
| 15 | R-butyl | S-hydroxymethyl | X | X |
| 16 | R-cyclohexyl | S-isobutyl | X | X |
| 17 | R-cyclohexyl | R-2-butyl | X | X |
| 18 | R-cyclohexyl | S-2-napthylmethyl | X | X |
| 19 | R-cyclohexyl | S-cyclohexyl | X | X |
| 20 | R-cyclohexyl | S-hydroxymethyl | X | X |
| 21 | S-butyl | S-isobutyl | X | X |
| 22 | S-butyl | R-2-butyl | X | X |
| 23 | S-butyl | S-2-napthylmethyl | X | X |
| 24 | S-butyl | S-cyclohexyl | X | X |
| 25 | S-butyl | S-hydroxymethyl | X | X |
The 1802 series of samples were designed by fixing the first two positions of the pyrrolidine bis-cyclic guanidine scaffold (1346) based on the results of the in vivo screening of the 1346 library. The R3 and R4 positions are equal molar mixtures of the 26 R3 functionalities and 42 R4 functionalities. Each sample in the 1802 series contains an equal molar mixture of 1,092 (26 × 42) compounds
Synthesis of TPI 1818-101 and 1818-109
Based on the data obtained from the in vivo screening of 1802 and 1346, we synthesized the individual pyrrolidine bis-cyclic guanidine compounds TPI 1818-101 and 1818-109 (Fig. 1b) utilizing the same synthetic strategy outlined in Fig. 1 for 1346. TPI 1818-101 combines the following functionalities from 1346; R1 functionality 14 S-2-hydroxymethyl, R2 functionality 51 S-cyclohexyl, R3 78 R-cyclohexyl and R4 91 2-ethoxybenzyl, while TPI 1818-109 combines the functionality 14 S-2-hydroxymethyl (at the R1 position), functionality 51 S-cyclohexyl (at the R2 position), functionality 78 R-cyclohexyl (at the R3 position), and functionality 93, 3,4-dichlorobenzyl (at the R4 position).
Animals
All experiments used male C57Bl/6J mice (20–32 g each, Jackson Laboratories, Bar Harbor, ME). Mice were housed four per cage in a temperature-controlled room. Cages were kept in a room with 12-h light/dark cycle with the lights on from 0700 to 1900 hours and food and water available ad libitum. All procedures with mice were approved by the local IACUC.
Chemicals
In all assays, TPI compounds were dissolved in 10% dimethylsulfoxide, a concentration that did not produce any detectable behavioral effect. Morphine sulfate, naloxone, beta-funaltrexamine (β-FNA), nor-binaltorphimine (NorBNI), and naltrindole were purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in 0.9% sterile saline.
Opioid Receptor Binding to Murine Brain Membranes
Murine brain membranes were prepared as described previously (3). The affinity and selectivity of TPI 1818-101 and TPI 1818-109 were determined by incubating the membranes with radiolabeled ligands and six different concentrations of TPI 1818-101 and TPI 1818-109 as described previously (3). Incubation times of 60 min were used for the MOR selective peptide [3H][d-Ala2,(Me)Phe4,Gly(ol)5]enkephalin ([3H]DAMGO) and 120 min for the delta-opioid receptor (DOR) selective peptide [3H][d-Pen2,Phe4,d-Pen5]enkephalin ([3H]DPDPE) and the kappa-opioid receptor (KOR) selective ligand [3H]U69,593 at final concentrations of 0.87, 2, and 2 nM, respectively.
Antinociceptive Testing Using the 55°C Warm Water Tail-Withdrawal Assay
Male C57Bl/6J mice (n = 8) were used in all antinociceptive testing. A water bath heated to 55°C acted as a nociceptive stimulus with the latency to withdraw the tail taken as the endpoint (15). Mice showing no response within 5 s during the determination of baseline responses were excluded from the experiment. After determining baseline control responses, mice were administered graded doses of morphine or a TPI sample. All compounds were each given as single intraperitoneal (i.p.) injections with tail withdrawal latencies measured 0.5, 1, 2, 3.5, 5, 8, and 24 h post-administration unless otherwise stated. In the receptor selectivity studies, either the MOR-selective antagonist β-FNA (5 mg/kg, i.p.) or the KOR-selective antagonist nor-BNI (10 mg/kg, i.p.) were injected 24 h before TPI sample administration, whereas the DOR-selective antagonist naltrindole (20 mg/kg, i.p.) was administered 20 min prior to agonist administration. A cutoff of 15 s was used to avoid tissue damage; those mice failing to withdraw their tails within this time were assigned a maximal antinociceptive score of 100%.
For scaffold and positional screening studies (1346 and 1802 series results), results are presented as the sum of average responses at each time point across all seven time points tested. For more detailed analysis across time (TPI 1818-101 and 1818-109), antinociception at each time point was calculated as follows: 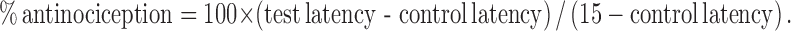
Respiratory Effects
Respiration rates were recorded using the automated, computer-controlled Comprehensive Lab Animal Monitoring System (CLAMS) apparatus (Columbus Instruments, Columbus, OH). Male C57Bl/6J mice were placed in closed apparatus cages (23.5 × 11.5 × 13 cm) and habituated for a 30-min period. Mice were then administered a graded i.p. dose of morphine, saline, TPI 1818-101, or 1818-109. Following administration of the compounds, mice were returned to chambers for 90 min with respiration rate (breaths/min) measured in 30-s intervals. Using a pressure transducer built into the enclosed, sealed CLAMS cage, the respiration rate of each occupant mouse was counted. Each time the mouse breathed, the resultant change in pressure was recorded over the 90-min period to reveal the rate in breaths/min.
Locomotor Activity
Locomotor activity of mice was monitored for 90 min following a 5-min delay after administration of vehicle (10% DMSO in saline, 0.9%, i.p.), morphine (10 mg/kg, i.p.), TPI 1818-101 (5 mg/kg, i.p.), or TPI 1818-109 (10 mg/kg, i.p.). The doses of drug used in the locomotor activity assay were selected on the basis of maximal activity observed in the antinociceptive testing experiments. Locomotor activity was captured and digitized, with the distance traveled calculated by a Noldus Ethovision Pro locomotor tracking system. Before testing, mice were initially administered vehicle and confined to the locomotor chambers for 60 min to habituate the animals to the apparatus.
Conditioned Place Preference
Male C57Bl/6J mice were conditioned using a protocol similar to the previously established biased cocaine-conditioned place preference paradigm (16–18). The testing apparatus (San Diego Instruments, San Diego, CA) consisted of four identical compartmentalized boxes divided into two equal-sized outer compartments (25 × 25 × 25 cm) with distinct visual (horizontal or vertical alternating black and white lines, 1.5 cm wide) or tactile cues (smooth or mottled flooring), each joined to a small central section (8.5 × 25 × 25 cm) accessed through a single doorway (3 cm high). Each unit is fitted with infrared beams, the breaking of which allows an automated measure of the time animals spend in each chamber. Note that the biased place-conditioning protocol involves administration of the test sample (here, morphine, vehicle, TPI 1818-101, or 1818-109) to mice that were then restricted to the outer compartment opposite of their original preference response in an initial preconditioning preference test. The biased conditioned place preference protocol is a sensitive and consistent indicator of conditioned drug reward equivalent to alternative methods (e.g., the counterbalanced design; see (19) for a review).
On day 1, and prior to the start of conditioning, mice were placed inside the apparatus and allowed to move freely between all three chambers for a 30-min testing period. Initial preference was determined by measuring which of the two outer chambers, the left or the right, the mouse spent more time. On day 2, each animal was injected (i.p.) with saline (0.9%) 10 min prior to confinement in the initially preferred outer compartment for 45 min. After six more hours spent in the home cage, each mouse was then administered vehicle (10% DMSO in 0.9% saline), morphine (10 mg/kg), TPI 1818-101 (5 mg/kg), or TPI 1818-109 (10 mg/kg), 10 min prior to confinement in the appropriate opposite outer chamber for 45 min. The doses of drug used in the place conditioning assay were selected on the basis of maximal activity observed in the antinociceptive testing experiments. This conditioning cycle was repeated a second time on day 3, which has been demonstrated to be effective in producing morphine-conditioned place preference (CPP) (20). On day 4, the mice were again allowed to run freely between all three chambers to determine the final drug-conditioned place preference. Data are plotted as the difference in time spent in the eventual drug- and vehicle-paired compartments. By convention, the initial bias represents a negative value, whereas a positive value represents a conditioned preference for the drug-paired side. Note that conditioned place aversion, where animals avoid the drug-paired compartment, is also detectable under this method when animals spend significantly more time in the initially preferred side. However, this was not demonstrated in this study under any conditions.
Statistical Analysis
IC50 values were calculated by least squares fit to a logarithm-probit analysis. The Ki values of TPI 1818-101 and TPI 1818-109 were calculated from the equation 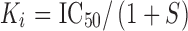 , where S = (concentration of radioligand)/(KD of radioligand) by Prism 5.0 software (GraphPad Software, LaJolla, CA). All dose–response lines were analyzed by regression and D50 (dose producing 50% antinociception) values and 95% confidence limits determined using each individual data point by Prism 5.0 software. Student’s t tests comparing baseline and post-treatment tail-withdrawal latencies were used to determine statistical significance for all tail-withdrawal data. Student’s t tests were also used to determine statistical significance of summarized antinociceptive effects of each individual sample against the same effect of morphine. Ranking of combined library samples (see Fig. 2) was performed with one-way ANOVA, with significant effects further analyzed by Tukey’s multiple comparison post hoc testing using Prism 5.0 software. Data for respiration and locomotor effects were analyzed with one-way ANOVA using Prism 5.0, with significant effects further analyzed by Tukey’s HSD post hoc testing. Data for conditioned place preference experiments were analyzed with two-way ANOVA using SPSS 14.0 (SPSS, Chicago, IL), with analyses examining the main effect of conditioned place preference phase (e.g., pre- or post-conditioning) and the interaction of drug pretreatment (morphine, TPI-1818-101 or 1818-109, or vehicle). Significant effects were further analyzed using Tukey’s HSD post hoc testing. All data are presented as mean ± SEM, with significance set at p ≤ 0.05.
, where S = (concentration of radioligand)/(KD of radioligand) by Prism 5.0 software (GraphPad Software, LaJolla, CA). All dose–response lines were analyzed by regression and D50 (dose producing 50% antinociception) values and 95% confidence limits determined using each individual data point by Prism 5.0 software. Student’s t tests comparing baseline and post-treatment tail-withdrawal latencies were used to determine statistical significance for all tail-withdrawal data. Student’s t tests were also used to determine statistical significance of summarized antinociceptive effects of each individual sample against the same effect of morphine. Ranking of combined library samples (see Fig. 2) was performed with one-way ANOVA, with significant effects further analyzed by Tukey’s multiple comparison post hoc testing using Prism 5.0 software. Data for respiration and locomotor effects were analyzed with one-way ANOVA using Prism 5.0, with significant effects further analyzed by Tukey’s HSD post hoc testing. Data for conditioned place preference experiments were analyzed with two-way ANOVA using SPSS 14.0 (SPSS, Chicago, IL), with analyses examining the main effect of conditioned place preference phase (e.g., pre- or post-conditioning) and the interaction of drug pretreatment (morphine, TPI-1818-101 or 1818-109, or vehicle). Significant effects were further analyzed using Tukey’s HSD post hoc testing. All data are presented as mean ± SEM, with significance set at p ≤ 0.05.
Fig. 2.

Scaffold ranking of libraries 1343 (black bar), 1344 (white bar), 1345 (gray bar), 1346 (red bar), and 1347 (dark gray bar) by in vivo antinociception response in the mouse 55°C warm water tail-withdrawal test. Morphine (10 mg/kg, i.p., right side) was tested as a positive control. Data represent average (±SEM) summed tail-withdrawal latencies calculated by taking the sum of the average tail withdrawal latencies for each animal from each time point over a 24-h period. All samples were administered at a dose of 5 mg/kg, i.p. Bars, 8 mice each. *Significantly different, p < 0.05, Tukey’s test
RESULTS
Ranking the Five Scaffolds for Antinociceptive Potency
The antinociceptive activity of the five library scaffolds (1343, 1344, 1345, 1346, and 1347; see Fig. 1) was evaluated. While the systematic arrangement of each library into 120 (26 + 26 + 26 + 42) mixture samples significantly reduced the screening set size (from 3,690,960 individual samples (5 × 738,192) to 600 mixture-based samples (5 × 120)), this number of samples was still somewhat prohibitive. Therefore, to prioritize the complete library to be screened, we employed the scaffold ranking method (3). For this method, we assayed each library represented by a single sample containing an approximately equal molar mixture of all 738,192 compounds contained in the particular library. Like morphine, administration of all five samples significantly increased the combined time mice required to remove their tails from a noxious stimulus of 55°C warm water (Fig. 2). However, the sample representing the 1346 library (Fig. 2, red bar) produced the greatest magnitude of activity (F(5,42) = 2.7, p < 0.05), establishing this scaffold for further evaluation in this study.
Positional Scanning of the 1346 Series Library OXXX Defined Samples In Vivo
Utilizing the positional scanning format (11,12), the antinociceptive activity of each of the 120 mixture-based samples comprising the 1346 series library was evaluated after administration (5 mg/kg, i.p.) with the 55°C warm water tail-withdrawal assay (Fig. 3). The combined time mice demonstrated to withdraw their tail was summed over the seven time points examined for each sample tested and is reported by substitution position (Fig. 3a–d). Notably, a number of samples defined at each substitution position significantly increased the combined tail-withdrawal time as compared to the effect of morphine (10 mg/kg, i.p.; black bar on right of each graph in Fig. 3).
Fig. 3.

Positional scan screening of 1346-series OXXX samples: summed antinociception produced by 1346 series samples measured in the mouse 55°C warm water tail-withdrawal test across a 24-h period. a 1346 defined at position 1 (red bars). b 1346 defined at position 2 (blue bars). c 1346 defined at position 3 (yellow bars). d 1346 defined at position 4 (green bars). The combined time to withdraw tails (s; y-axis) was calculated by taking the sum of the average tail-withdrawal latencies from each time point. Samples (x-axis; see Table I for full identities) were administered at a dose of 5 mg/kg i.p. for testing. Functionalities of key samples are described in simplified form for convenience; see Table I for complete descriptions. Morphine (10 mg/kg, i.p., far right bar) was tested as a positive control. Data represent average (±SEM) summed tail-withdrawal latencies calculated by taking the sum of the average tail-withdrawal latencies for each animal from each time point over a 24-h period. Samples administered at dose of 5 mg/kg, i.p. Bars = 8 mice each. *Significantly greater than morphine effect, p < 0.05, Student’s t test
Iteration of the 1346 Series Results: 1802 Series Library OOXX Defined Samples In Vivo
Partial iteration of the 1346 library was accomplished with the 1802 library, which shares the scaffold with the 1346 library but which consists of compounds defined at the R1 and R2 substitution positions. In vivo evaluation of the antinociceptive activity of the 25 mixture-based samples in this library was performed with the 55°C warm water tail-withdrawal assay along with morphine as a reference standard (Fig. 4). While four of five samples containing D-Phe in the first substitution position demonstrated statistically greater combined tail withdrawal times than morphine, the greatest magnitude of response was demonstrated by samples defined in the first position with D-Ser and in the second position with D-isoleucine or cyclohexylalanine. In contrast, no other samples showed greater activity than morphine (Fig. 4).
Fig. 4.

Iterative screening of OOXX samples using in vivo antinociception: summed antinociception produced by 1802 series samples (5 mg/kg, i.p.) measured in the mouse 55°C warm water tail-withdrawal test across a 24-h testing period. Samples were defined in the first position (inset key) and second position (see Table II) identified on the x-axis. The combined time to withdraw tails (s; y-axis) was calculated by taking the sum of the average tail withdrawal latencies from each time point. Functionalities of key samples are described in simplified form for convenience; see Table II for complete descriptions. Note the inclusion of morphine (10 mg/kg, i.p., far right bar) as a positive control. Data represent average (±SEM) summed tail-withdrawal latencies calculated by taking the sum of the average tail withdrawal latencies for each animal from each time point over a 24-h period. Samples were administered at a dose of 5 mg/kg, i.p. Bars, 8 mice each. *Significantly greater than morphine effect, p < 0.05, Student’s t test
Testing of Individual Compounds TPI 1818-101 and 1818-109
Based on the data obtained from the in vivo screening of 1346 and 1802, we synthesized a pair of individual pyrrolidine bis-cyclic guanidine compounds, TPI 1818-101 and TPI 1818-109 (Fig. 1b), to determine if individual compounds resulting from the analysis of the data collected from the 1346 and 1802 series samples might demonstrate favorable antinociceptive activity. Using radioligand competition binding assays, TPI 1818-101 and TPI 1818-109 demonstrated poor affinity for the multiple opioid receptors as established by the Ki values for the inhibition of MOR-, DOR-, and KOR binding (Table III).
Table III.
K i Values for the Inhibition of Mu-, Delta-, and Kappa-opioid Receptor Binding to Murine Brain Membrane Protein by TPI 1818-101 and TPI 1818-109
| Radiolabeled ligands | K i (nM) ± SEM | ||
|---|---|---|---|
| [3H]DAMGO (MOR) | [3H]DPDPE (DOR) | [3H]U69,593 (KOR) | |
| TPI 1818-101 | 2,790 ± 741 | 1,930 ± 474 | 3,910 ± 1,640 |
| TPI 1818-109 | 9,470 ± 1,210 | 2,210 ± 932 | 7,750 ± 3,830 |
Murine brain membrane protein was incubated with six different concentrations of each TPI compound in the presence of 0.87 nM [3H]DAMGO, 2 nM [3H]DPDPE, or 2 nM [3H]U69,593 in 50 mM Tris–HCl, pH 7.5, at 25°C as described in “MATERIALS AND METHODS.” Data are listed as the mean K i values ± SEM from at least three experiments
However, both individual small-molecule compounds, TPI 1818-101 and 1818-109, produced a time- and dose-dependent antinociception after i.p. administration in the mouse 55°C warm water tail-withdrawal assay (Fig. 5a, b). TPI 1818-101 and 1818-109 produced maximal antinociception 60 min after administration of 5 mg/kg of TPI 1818-101 and 10 mg/kg of TPI 1818-109 that lasted up to 5 and 2 h, respectively. The antinociceptive D50 values of TPI 1818-101 and 1818-109 (and 95% confidence intervals) were 1.09 (0.06–9.63) mg/kg and 4.63 (2.81–6.40) mg/kg, respectively (Fig. 5c). In comparison, the antinociceptive D50 value of morphine sulfate was 11.24 (2.4–20.1) mg/kg.
Fig. 5.

Antinociception produced by TPI-1818-101 and 1818-109 in the mouse 55°C warm water tail-withdrawal test is dose- and time-dependent. a Time course of TPI 1818-101 antinociception. b Time course of TPI 1818-109 antinociception. c Dose–response lines of morphine, TPI 1818-101, and TPI 1818-109 given by i.p. injection 30 min before testing in the mouse 55°C tail-withdrawal assay. Points, 8–10 mice
Receptor selectivity of both TPI 1818-101 and TPI 1818-109 after i.p. administration was determined by pretreating mice with selective opioid receptor antagonists prior to testing in the 55°C warm water tail-withdrawal assay as shown in Fig. 6a, b. Opioid receptor antagonists were administered at doses and in sufficient advance of TPI compounds to insure inhibition of only one type of opioid receptor. The pretreatment of mice with the MOR-selective antagonist β-FNA before the administration of TPI 1818-101 or 1818-109 significantly reduced the antinociceptive effects of both compounds. In contrast, pretreatment with the KOR-selective antagonist nor-BNI did not alter the antinociception produced by TPI 1818-101 (Fig. 6a), but significantly reduced the antinociception induced by TPI 1818-109 (Fig. 6b). Pretreatment with the DOR-selective antagonist naltrindole had no significant effect on antinociception produced by either compound. These results suggest that TPI 1818-101 produced antinociception through the MOR, whereas TPI 1818-109 induced antinociception through activity mediated by both the MOR and KOR.
Fig. 6.

Antinociceptive effects of a TPI 1818-101 (5 mg/kg, i.p., −30 min) and b TPI 1818-109 (10 mg/kg, i.p., −30 min) in mice with or without pretreatment with β-FNA (5 mg/kg, i.p., −24 h), nor-BNI (10 mg/kg, i.p., −24 h), or naltrindole (20 mg/kg, i.p., −15 min). Bars, 8–10 mice. *Significantly different from baseline latency. †Significantly less than matching 1818 compound alone, p < 0.05
The effects of TPI 1818-101 and TPI 1818-109 on respiration rate and locomotor activity were assessed. Mice were administered TPI compounds at doses corresponding to maximal effects observed in antinociceptive dose–response testing. Additional mice were treated with vehicle (10%DMSO in 0.9% sterile saline, i.p.) and morphine (10 mg/kg, i.p.) for comparison. As expected of a MOR agonist, this dose of morphine significantly reduced the respiration rate 25.1% (F(3,26) = 3.73, p < 0.05; Fig. 7a) while increasing distance traveled fourfold (F(3,36) = 34.77, p < 0.01; Fig. 7b) from the response of saline-treated mice. However, neither TPI 1818-101 (5 mg/kg, i.p.) or TPI 1818-109 (10 mg/kg, i.p.) significantly altered the respiration rate or locomotor activity from the saline-induced response.
Fig. 7.

TPI 1818-101 and 1818-109 do not induce morphine-like liabilities of use. Mice were administered vehicle (10% DMSO in 0.9% saline, gray bar), morphine (10 mg/kg, white bar), TPI 1818-101 (5 mg/kg, green bar), or TPI 1818-109 (10 mg/kg, red bar) and activity determined for 90 min on a respiration rate and b locomotor effects. TPI-treated mice do not show significant a slowed respiratory function (breaths/min) or b excess locomotor activity (cm/min) when compared to vehicle-treated mice, as produced by morphine. Each bar represents eight mice. *Significant difference from the vehicle treated control group (p < 0.05 for A, p < 0.01 for B. †Significant difference from morphine treated mice (p < 0.01)
The rewarding or aversive effects of TPI 1818-101 and TPI 1818-109 were determined by place conditioning mice with either compound at doses corresponding to the maximal effect observed in antinociceptive dose response testing. Morphine (10 mg/kg, i.p.) place conditioning over 2 days (Fig. 8a) resulted in a significant preference for the morphine-paired side (F(3,49) = 6.05, p < 0.01; Fig 8b, white bars), but place conditioning with vehicle, TPI 1818-101, or TPI 1818-109 produced no such conditioned-place preference (Fig. 8b, filled bars). Notably, the TPI compounds also did not demonstrate aversive properties as place conditioning with either TPI 1818-101 or TPI 1818-109 produced final place preference responses that were both statistically similar to matching pre-conditioning responses (Fig. 8b, open bars) and the final saline-conditioned place preference response.
Fig. 8.

TPI 1818-101 and 1818-109 do not induce morphine-like conditioned place preference. a Schematic of testing protocol. b Summary graph of place preference results. Initial preference for either the left or right compartment of the CPP system was determined on day 1 of testing for each group of mice (solid bars) and plotted as the difference in time spent on the eventual drug-paired side (s). On days 2 and 3, individual mice were place conditioned in the initially preferred chamber with vehicle (0.9% saline) followed 6 h later by place conditioning in the initially non-preferred chamber side with morphine (white bars), vehicle (10% DMSO in 0.9% saline, gray bars), TPI 1818-101 (green bars), or 1818-109 (red bars). On the fourth day of testing, mice were again tested to determine final place preference (striped bars). Final conditioned place preference did not significantly differ from the initial preference for mice treated with vehicle, TPI 1818-101, or 1818-109, but was significant for morphine. Each bar represents 8–16 mice. *Significant difference from the initial preference (p < 0.05). †Significant difference from final preference of morphine treated mice (p < 0.05). ‡Significant difference from saline place-conditioned animals (p < 0.05)
DISCUSSION
Agonists activating MOR are generally regarded as the “gold standard” for analgesics (1). However, activation of the MOR produces significant well-established clinical liabilities, notably respiratory depression (21), the slowing of intestinal motility (22), modulation of the immune response (23), antinociceptive tolerance (24,25), and physical and psychological dependence (24,26). Likewise, while agonists selective for both the DOR and KOR also induce potent antinociception, they also produce significant side effects (1), notably respiratory depression (27) and seizures (28) by DOR agonists, and diuresis (29) and dysphoria (30) by KOR agonists. The prevalence of deleterious side effects by the established opioid analgesic agents has encouraged the search for compounds that produce analgesia with fewer or no clinical liabilities.
We have pioneered two of the key methodologies currently utilized to generate and screen very large numbers of low-molecular-weight synthetic compounds. The first is commonly known as the “tea bag” approach (31), and the second is the practical synthesis and deconvolution of mixture-based combinatorial libraries made up of millions of compounds (2–5,8,14,32,33). These approaches enable millions of acyclic and heterocyclic compounds, as well as tens of millions to billions of peptides, to be prepared and screened in readily accessible formats in a fraction of the time and cost of equivalent individual compound arrays.
Two primary approaches are used to prepare and screen the large numbers of compounds produced by combinatorial libraries. These are: (1) the massive parallel synthesis and screening of large individual compound arrays and (2) the generation and screening of extremely large focused, but chemically narrower, mixture-based libraries. Once individual compounds are identified as therapeutically useful, their general target activities are often further improved by classic structure–activity relationship approaches prior to testing in vivo. However, it remains impractical for the majority of academic and small research organizations to make and screen such large numbers of compounds. Moreover, most compounds initially found to be promising are rejected at the in vivo stage of the discovery process. Indeed, the inverse of this concept is also true: an exciting aspect of the present data stems from the promising in vivo activity of both TPI 1818-101 and TPI 1818-109 despite the poor opioid receptor affinity demonstrated by in vitro testing, which might otherwise have lessened enthusiasm for further examination of these compounds.
To circumvent the limitations of existing in vitro screening methods, we sought to directly test samples from a mixture-based combinatorial library in vivo for analgesic properties, thereby simultaneously increasing the evaluation of compounds while decreasing the failure rate inherent in the traditional drug discovery process (10). The in vivo screening and deconvolution of mixture-based combinatorial libraries has previously yielded therapeutically useful individual compounds in a cost-effective manner. Previous screening of a library of 400 separate mixtures each of 130,321 hexapeptides by monitoring blood pressure and heart rate in rats (5) demonstrated the feasibility of this approach, identifying useful therapeutic candidates while simultaneously eliminating compounds with poor absorption, distribution, metabolism, and pharmacokinetic properties. Likewise, a mixture-based combinatorial library containing all d-amino acid hexapeptides was used previously to identify a novel agonist for the MOR, Ac-rfwink-NH2 (34), which induced a potent, long-lasting antinociception in mice that was blocked by administration of the opioid antagonist naloxone (34).
While the present data again suggest the success of this approach, it is notable that the strategy is not without limitations. The evaluation of the 134X scaffolds and the mixture-based samples of the 1346 and 1802 libraries may be susceptible to false negative outcomes resulting from the inclusion of opioid receptor antagonists that reduce or mask the potency of opioid receptor agonists in the tested mixture. It is notable that opioid receptor antagonists have been identified from positional scanning and screening of synthetic peptide combinatorial libraries (11). While the end result of the present search for low-liability analgesic agents was successful, additional detailed screening of libraries based on other scaffolds can be expected to provide enhanced antinociceptive agents. It should be noted that the identification of selective, novel opioid receptor antagonists is itself of potential therapeutic value, especially given findings of the potential antidepressant and anxiolytic effects of KOR antagonists (35). While not within the scope of the present study, the lack of antinociceptive response from select mixture samples in the 1346 library suggests the strong likelihood of finding potential antagonists using this approach.
The potent antinociceptive effect of the individual compounds TPI 1818-101 and 1818-109 are conspicuous for their accompanying lack of respiratory depression, locomotor, or place conditioning effects. A mechanism accounting for this pattern of activity is unknown. It is possible that these effects are due to non-opioid-mediated interactions induced by the TPI compounds. Supporting this, TPI 1818-101 and TPI 1818-109 showed poor affinity for the MOR, with both compounds demonstrating micromolar affinity for the three opioid receptors in competition binding assays in the present study. Likewise, an earlier study of the 1346 library mixture-based samples with competition opioid radioligand binding assays found poor to low affinity for the mu opioid receptor (3). Moreover, it is important to note that the samples found to have higher affinity for the mu-opioid receptor in the previous binding study did not correlate well with the active compounds identified directly using the present in vivo testing approach. However, this possibility is depreciated by the present findings that the opioid-receptor selective antagonists β-FNA and nor-BNI antagonized the antinociceptive effects of the TPI compounds in vivo. Along a similar line of reasoning, it remains possible that the antinociceptive effects observed in vivo are due to a metabolite of either TPI compound tested. As these metabolites might not be expected to occur in the environment of an in vitro binding assay, it is possible that potential metabolites of TPI 1818-101 and 1818-109 could account for the opioid receptor activity, not unlike morphine-6β-glucuronide demonstrating higher potency at the MOR receptor over that of the parent substrate, morphine (36). Alternatively, it is also possible that the administration of TPI 1818-101 or 1818-109 may produce antinociception through the induced release of an endogenous opioid, such as β-endorphin or an enkephalin. A number of compounds induce the release of endorphins to produce opioid-receptor-mediated antinociception, such as cannabinoids (37) or the endothelin A receptor antagonist BQ-123 (38). However, as the endogenous opioids produce all the detrimental effects of opioid agonists (such as the respiratory depression induced by β-endorphin) (39), it is unclear why TPI 1818-101 or 1818-109 would produce opioid-mediated antinociception without the opioid-mediated liabilities. A simpler alternative possibility is that TPI 1818-101 and 1818-109 may not cross the blood–brain barrier, thereby restricting the activity of these compounds to the periphery after intraperitoneal administration. Peripherally restricted opioid agonists such as N-methylmorphine have been shown to produce relief from some types of pain (40,41) and have been generally proposed as novel opioid analgesics with fewer liabilities of use, as they would be expected to lack many of the detrimental clinical effects mediated by mechanisms in the CNS (42). However, additional work to confirm the inability of TPI 1818-101 or 1818-109 to cross the blood–brain barrier, or to produce respiratory and psychostimulant effects after central administration, is required to determine the mechanism producing the effects of these compounds.
CONCLUSION
The in vivo screening of the 120 mixture samples comprising the 738,192 individual compounds making up the 1346 library yielded mixtures that produced robust opioid-mediated antinociception in the 55°C warm water tail-withdrawal assay. Deconvolution of these antinociceptive results resulted in the synthesis of individual compounds, TPI 1818-101 and TPI 1818-109. These compounds produced a dose-dependent antinociception equivalent to morphine that was blocked by MOR- and KOR-selective antagonists, respectively. Importantly, neither of these compounds demonstrated respiratory distress or psychostimulant effects, suggesting they might produce analgesia without many of the clinical liabilities presented by morphine. Moreover, the work demonstrates the validity of using mixture-based combinatorial libraries and deconvolution of in vivo testing results to identify individual compounds with enhanced potential therapeutic value.
Acknowledgments
This work was supported by NIDA grant R21 DA019620 (to RAH) and by the State of Florida, Executive Office of the Governor’s Office of Tourism, Trade, and Economic Development.
Abbreviations
- BBB
Blood–brain barrier
- CNS
Central nervous system
- CPP
Conditioned place preference
- DAMGO
[d-Ala2,(Me)Phe4,Gly(ol)5]enkephalin
- DOR
Delta-opioid receptor
- DPDPE
[d-Pen2,Phe4,d-Pen5]enkephalin
- i.c.v.
Intracerebroventricular
- i.p.
Intraperitoneal
- KOR
Kappa-opioid receptor
- MOR
Mu-opioid receptor
- nor-BNI
Norbinaltorphimine
- s.c.
Subcutaneous
- U50,488
(±)-trans-3,4-Dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide
- U69,593
(+)-(5α,7α,8β)-N-Methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide
References
- 1.Gutstein HB, Akil H. Opioid analgesics. In: Hardman JG, Limbird LE, editors. Goodman and Gilman’s the pharmacological basis of therapeutics. 10. New York: McGraw-Hill; 2001. pp. 569–619. [Google Scholar]
- 2.Houghten RA, Pinilla C, Appel JR, Blondelle SE, Dooley CT, Eichler J, Nefzi A, Ostresh JM. Mixture-based synthetic combinatorial libraries. J Med Chem. 1999;42:3743–78. doi: 10.1021/jm990174v. [DOI] [PubMed] [Google Scholar]
- 3.Houghten RA, Pinilla C, Giulianotti MA, Appel JR, Dooley CT, Nefzi A, Ostresh JM, Yu Y, Maggiora GM, Medina-Franco JL, Brunner D, Schneider J. Strategies for the use of mixture-based synthetic combinatorial libraries: scaffold ranking, direct testing in vivo, and enhanced deconvolution by computational methods. J Comb Chem. 2008;10:3–19. doi: 10.1021/cc7001205. [DOI] [PubMed] [Google Scholar]
- 4.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature. 1991;354:84–6. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 5.Houghten RA. Soluble combinatorial libraries: extending the range and repertoire of chemical diversity. Methods: A companion to Methods in Enzymology. 1994;6:354–60. doi: 10.1006/meth.1994.1035. [DOI] [Google Scholar]
- 6.Thompson LA, Ellman JA. Synthesis and applications of small molecular libraries. Chem Rev. 1996;96:555–600. doi: 10.1021/cr9402081. [DOI] [PubMed] [Google Scholar]
- 7.Nefzi A, Ostresh JM, Houghten RA. The current status of heterocyclic combinatorial libraries. Chem Rev. 1997;97:440–72. doi: 10.1021/cr960010b. [DOI] [PubMed] [Google Scholar]
- 8.Nefzi A, Ostresh JM, Yu Y, Houghten RA. Combinatorial chemistry: libraries from libraries, the art of the diversity-oriented transformation of resin-bound peptides and chiral polyamides to low molecular weight acyclic and heterocyclic compounds. J Org Chem. 2004;69:3603–9. doi: 10.1021/jo040114j. [DOI] [PubMed] [Google Scholar]
- 9.Dolle RE. Comprehensive survey of combinatorial library synthesis: 2004. J Comb Chem. 2005;7:739–98. doi: 10.1021/cc050082t. [DOI] [PubMed] [Google Scholar]
- 10.Carroll FI, Houghten RA. From rapid in vitro screening to rapid in vivo screening in the drug discovery process. Neuropsychopharmacology. 2009;34:251–2. doi: 10.1038/npp.2008.160. [DOI] [PubMed] [Google Scholar]
- 11.Dooley CT, Chung NN, Schiller PW, Houghten RA. Acetalins: opioid receptor antagonists determined through the use of synthetic peptides combinatorial libraries. Proc Natl Acad Sci USA. 1993;90:10811–5. doi: 10.1073/pnas.90.22.10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinilla C, Appel JR, Blondelle SE, Dooley CT, Eichler J, Ostresh JM, Houghten RA. Versatility of positional scanning synthetic combinatorial libraries for the identification of individual compounds. Drug Dev Res. 1994;33:133–45. doi: 10.1002/ddr.430330210. [DOI] [Google Scholar]
- 13.Hensler ME, Bernstein G, Nizet V, Nefzi A. Pyrrolidine bis-cyclic guanidines with antimicrobial activity against drug-resistant Gram-positive pathogens identified from a mixture-based combinatorial library. Bioorganic Medicinal Chem Lett. 2006;16:5073–9. doi: 10.1016/j.bmcl.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 14.Ostresh JM, Husar GM, Blondelle SE, Dörner B, Weber PA, Houghten RA. “Libraries from libraries”: chemical transformation of combinatorial libraries to extend the range and repertoire of chemical diversity. Proc Natl Acad Sci USA. 1994;91:11138–42. doi: 10.1073/pnas.91.23.11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–83. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szumlinski KK, Price KL, Frys KA, Middaugh LD. Unconditioned and conditioned factors contribute to the ‘reinstatement’ of cocaine place conditioning following extinction in C57BL/6 mice. Behav Brain Res. 2002;136:151–60. doi: 10.1016/S0166-4328(02)00102-X. [DOI] [PubMed] [Google Scholar]
- 17.McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C. Prior activation of kappa opioid receptors by U50, 488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsychopharmacology. 2006;31:787–94. doi: 10.1038/sj.npp.1300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carey AN, Borozny K, Aldrich JV, McLaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid antagonist arodyn. Eur J Pharmacol. 2007;569:84–9. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bardo MT, Rowlett JK, Harris MJ. Conditioned place preference using opiate and stimulant drugs: a meta-analysis. Neurosci. Biobehavioral Rev. 1995;19:39–51. doi: 10.1016/0149-7634(94)00021-R. [DOI] [PubMed] [Google Scholar]
- 20.Orsini C, Bonito-Oliva A, Conversi D, Cabib S. Susceptibility to conditioned place preference induced by addictive drugs in mice of the C57Bl/6 and DBA/2 inbred strains. Psychopharmacology (Berl) 2005;181:327–36. doi: 10.1007/s00213-005-2259-6. [DOI] [PubMed] [Google Scholar]
- 21.Shook JE, Watkins WD, Camporesi EM. Differential roles of opioid receptors in respiration, respiratory disease, and opiate-induced respiratory depression. Am Rev Repir Dis. 1990;142:895–909. doi: 10.1164/ajrccm/142.4.895. [DOI] [PubMed] [Google Scholar]
- 22.Burks TF, Fox DA, Hirning LD, Shook JE, Porreca F. Regulation of gastrointestinal function by multiple opioid receptors. Life Sci. 1988;43:2177–81. doi: 10.1016/0024-3205(88)90410-9. [DOI] [PubMed] [Google Scholar]
- 23.Yahya MD, Watson RR. Immunomodulation by morphine and marijuana. Life Sci. 1987;41:2503–10. doi: 10.1016/0024-3205(87)90434-6. [DOI] [PubMed] [Google Scholar]
- 24.Way EL, Loh HH, Shen FH. Simultaneous quantitative assessment of morphine tolerance and physical dependence. J Pharmacol Exp Ther. 1969;167:1–8. [PubMed] [Google Scholar]
- 25.DeLander GE, Portoghese PS, Takemori AE. Role of spinal mu opioid receptors in the development of morphine tolerance and dependence. J Pharmacol Exp Ther. 1984;231:91–6. [PubMed] [Google Scholar]
- 26.Cowan A, Zhu XZ, Mosberg HI, Omnaas JR, Porreca F. Direct dependence studies in rats with agents selective for different types of opioid receptor. J Pharmacol Exp Ther. 1988;246:950–5. [PubMed] [Google Scholar]
- 27.May CN, Dashwood MR, Whitehead CJ, Mathias CJ. Differential cardiovascular and respiratory responses to central administration of selective opioid agonists in conscious rabbits: correlation with receptor distribution. Br J Pharmacol. 1989;98:903–13. doi: 10.1111/j.1476-5381.1989.tb14620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negus SS, Butelman ER, Chang K-J, DeCosta B, Winger G, Woods JH. Behavioral effects of the systemically active delta opioid agonist BW373U86 in rhesus monkeys. J Pharmacol Exp Ther. 1994;270:1025–34. [PubMed] [Google Scholar]
- 29.Leander JD. A kappa opioid effect: increased urination in the rat. J Pharmacol Exp Ther. 1983;224:89–94. [PubMed] [Google Scholar]
- 30.Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–6. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- 31.Houghten RA. General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen–antibody interaction at the level of individual amino acids. Proc Natl Acad Sci USA. 1985;82:5131–5. doi: 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dooley CT, Houghten RA. The use of positional scanning synthetic peptide combinatorial libraries for the rapid determination of opioid receptor ligands. Life Sci. 1993;52:1509–17. doi: 10.1016/0024-3205(93)90113-H. [DOI] [PubMed] [Google Scholar]
- 33.Dooley CT, Ny P, Bidlack JM, Houghten RA. Selective ligands for the mu, delta, and kappa opioid receptors identified from a single tetrapeptide positional scanning combinatorial library. J Biol Chem. 1998;273:18848–56. doi: 10.1074/jbc.273.30.18848. [DOI] [PubMed] [Google Scholar]
- 34.Dooley CT, Chung NN, Wilkes BC, Schiller PW, Bidlack JM, Pasternak GW, Houghten RA. An all d-amino acid opioid peptide with central analgesic activity from a combinatorial library. Science. 1994;266:2019–22. doi: 10.1126/science.7801131. [DOI] [PubMed] [Google Scholar]
- 35.Carlezon WA, Jr, Béguin C, Knoll AT, Cohen BM. Kappa-opioid ligands in the study and treatment of mood disorders. Pharmacol Ther. 2009;123:334–43. doi: 10.1016/j.pharmthera.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paul D, Standifer KM, Inturrisi CE, Pasternak GW. Pharmacological characterization of morphine-6β-glucuronide, a very potent morphine metabolite. J Pharmacol Exp Ther. 1989;251:477–83. [PubMed] [Google Scholar]
- 37.Maldonado R, Valverde O. Participation of the opioid system in cannabinoid-induced antinociception and emotional-like responses. Eur Neuropsychopharmacol. 2003;13:401–10. doi: 10.1016/j.euroneuro.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Quang PN, Schmidt BL. Endothelin-A receptor antagonism attenuates carcinoma-induced pain through opioids in mice. J Pain. 2010; PMID 20071245 (in press). [DOI] [PMC free article] [PubMed]
- 39.Unal CB, Owen MD, Millington WR. Beta-endorphin-induced cardiorespiratory depression is inhibited by glycyl-l-glutamine, a dipeptide derived from beta-endorphin processing. J Pharmacol Exp Ther. 1994;271:952–8. doi: 10.21236/ada283495. [DOI] [PubMed] [Google Scholar]
- 40.Smith TW, Buchan P, Parsons DN, Wilkinson S. Peripheral antinociceptive effects of N-methyl morphine. Life Sci. 1982;31:1205–8. doi: 10.1016/0024-3205(82)90343-5. [DOI] [PubMed] [Google Scholar]
- 41.Oluyomi AO, Hart SL, Smith TW. Differential antinociceptive effects of morphine and methylmorphine in the formalin test. Pain. 1992;49:415–8. doi: 10.1016/0304-3959(92)90249-B. [DOI] [PubMed] [Google Scholar]
- 42.DeHaven-Hudkins DL, Dolle RE. Peripherally restricted opioid agonists as novel analgesic agents. Curr Pharm Des. 2004;10:743–57. doi: 10.2174/1381612043453036. [DOI] [PubMed] [Google Scholar]